

Summary
Single-cell measurements have broadened our understanding of heterogeneity in biology yet have been limited to mostly observational studies of normal or globally perturbed systems. Typically, perturbations are utilized in an open-ended approach wherein an endpoint is assayed during or after the biological event of interest. Here we describe ShootingStar, a platform for perturbation analysis in vivo which combines live imaging, real-time image analysis and automated optical perturbations. ShootingStar builds a quantitative record of the state of the sample being analyzed which is used to automate the identification of target cells for perturbation as well as to validate the impacts of the perturbation. We used ShootingStar to dissect the cellular basis of development, morphogenesis and polarity in the lateral line of Danio rerio and the embryo of Caenorhabditis elegans. ShootingStar can be extended to diverse optical manipulations and enables more robust and informative single-cell perturbations in complex tissues.
Introduction
Perturbation analysis is a central tool in modern molecular, cell and developmental biology. Mutagenesis and RNAi have shed light on many aspects of gene function by directly linking genes with morphological, functional and behavioral phenotypes in development and disease. These approaches have several key limitations, particularly in dissecting the role of essential or ubiquitously expressed genes in specific contexts. A wide variety of approaches to spatially and temporally restricting the impacts of genetic perturbations have been developed yet achieving single-cell resolution control over perturbations using these approaches has proven challenging. Optical methods for inducing targeted perturbations have allowed biologists to more carefully dissect cellular interactions in situ, yet they face practical challenges in deployment. The identification of target cells for perturbation and determining the precise desired timing of perturbation can be challenging in crowded and dynamic tissues. Rare or infrequent phenomena can also prove impractical for manual perturbation and analysis.
We developed ShootingStar to demonstrate a solution to these challenges. ShootingStar readily tracks thousands of cells in real-time on desktop workstations, extracting a variety of phenotypic measurements. Using this information to identify target cells and the timing of specific developmental events, ShootingStar provides precise and automated control over the targeting and timing of perturbations. Since imaging and image analysis begins before and continues after perturbations, a complete and quantitative record of the specimen is maintained allowing experimental outcomes to be directly validated and artifacts induced by off-target effects to be readily detected. We demonstrated the power of this approach by using ShootingStar to automate single-cell perturbations including laser ablation in D. rerio larvae and C. elegans embryos and cell labeling by selective photoconversion in C. elegans embryos. We show that using ShootingStar to automate these types of experiments makes it possible to fully validate and more precisely characterize the outcomes of perturbations. ShootingStar simplifies the study of complex tissues at single-cell resolution. It demonstrates an integrated approach to perturbation analysis in vivo which combines advances in several areas of single-cell analysis to provide a more granular and complete picture of developmental processes.
Design
An ever expanding toolkit of optically responsive reagents and methods for manipulating biological systems at single-cell resolution using light has made it possible to directly interrogate the cellular interactions that underlie processes of development, homeostasis and disease. Several key challenges complicate these types of experiments in vivo and in complex multicellular environments, in particular the reliable identification of target cells, the validation of experimental outcomes and the detection of off-target effects. We developed ShootingStar to address these challenges by integrating the entire experimental pipeline using in toto imaging and real-time image analysis. Flexibility in sample type, target cell definition and perturbation modality were also strong design priorities. While the need for hardware integration makes ShootingStar challenging to deploy to new systems, it demonstrates the power of an integrated approach to perturbation analysis and suggests a route towards more turn-key solutions for single-cell biology.
ShootingStar as a platform comprises three components: a three-dimensional fluorescence microscope, software components for defining and identifying target cells, and an illumination source for cellular perturbation (Figure 1A). The core of ShootingStar's software is a real-time cell-tracking algorithm that feeds into an interface for defining target cells and a visualization tool that can derive lineage identities from tracking results and can also be used to correct tracking errors on-the-fly. The real-time cell-tracking system is designed to balance speed and accuracy in cell tracking, two critical but competing factors in real-time analysis. The tracking system analyzes data across three expanding temporal windows to efficiently achieve high accuracy (Figure 1B). Cell detection is accomplished by segmenting nuclei from local maxima in a difference-of-Gaussians filtered image. Cells are then tracked between time points on the basis of distance. A Bayesian classifier is used to automatically detect and correct errors. Two strategies are used to achieve real-time performance. First, each step of detection and tracking is parallelized. Many computationally expensive steps, such as image filtering, nuclear segmentation (Santella et al., 2010), and cell tracking based on distance, are local to a time point and thus amenable to parallelization. The second key element in achieving real-time performance is the delay of computations dependent on a large temporal context until sufficient information is available. By using a Bayesian classifier to evaluate the semi-local topology of the lineage tree, this approach automatically identifies and corrects detection errors and false divisions (Santella et al., 2014). This step is both the most computationally expensive and the most important for ensuring accurate tracking during long-term imaging over hundreds of time points. Because error correction has non-local impact, this step is not easy to parallelize. ShootingStar evaluates the classifier only at the center of a sliding window, processing the single time point per round of execution that has sufficient forward and backward temporal context to be fully resolved.
Figure 1. ShootingStar platform.
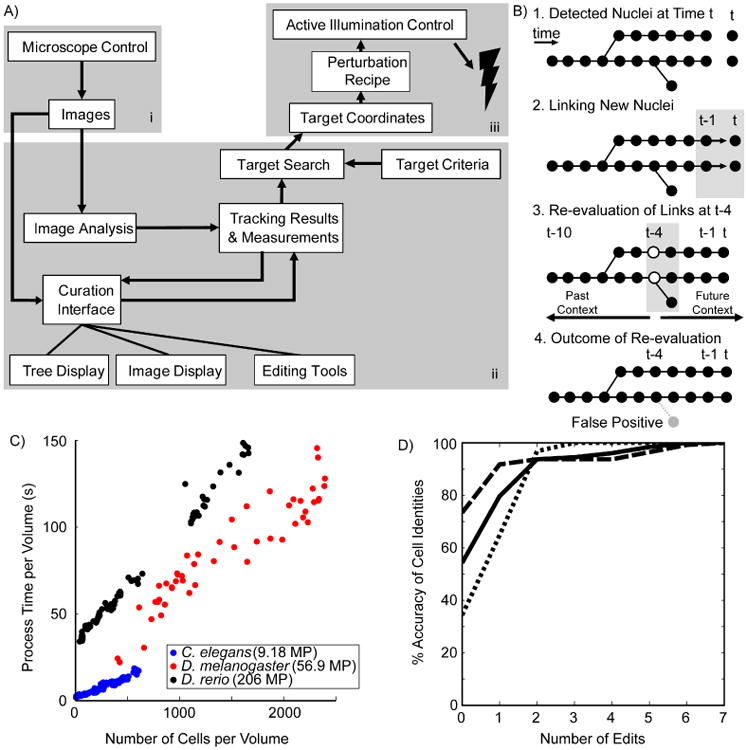
A) A schematic representation of data flow in the ShootingStar pipeline. i) Microscope control; ii) Tracking software and interfaces; iii) Perturbation control. B) Schematic illustration of the four primary steps of cell tracking in ShootingStar. Circles indicate cells detected at a particular time point. C) Per-volume processing times for images acquired of three species; C. elegans (blue), D. melanogaster (red) and D. rerio (black). MP stands for megapixels. D) Cumulative accuracy of cell identities in tracking each of three C. elegans embryos (solid, dashed and dotted lines. N= 3, 256 cells each).
While fully automated perturbations can be validated post hoc to ensure that only correctly targeted experiments are retained, ShootingStar also supports real-time data curation when absolute accuracy is needed (Boyle et al., 2006). A double-buffering architecture ensures that both the cell-tracking pipeline and the user are always presented with the most up-to-date results. Each pipeline maintains a working copy of the tracking results, an architecture that allows hierarchical synchronization. Before processing each new data sample, the tracking pipeline searches the user copy for new edits and incorporates them into its copy to ensure that tracking decisions are based on the most accurate information. When new tracking results are generated by the tracking pipeline, they are synchronized to the user copy while preserving user edits. The user interface offers a suite of tools to facilitate rapid error correction on-the-fly. A 4D image browser allows visual inspection of the tracking history of any selected cell, using the keyboard arrow keys to follow the cell backwards and forwards in time as well as up and down through image planes. With some practice, it typically takes a few minutes to visually track a cell over 100 time points and several divisions to examine the automated tracking results. Independent of a priori knowledge of a particular sample's lineage topology, many visual features such as mitotic figures can thus be used to identify tracking errors.
A core challenge in studying multicellular processes in a crowded tissue is the robust identification of target cells. Target cells are identified by ShootingStar on the basis of a user-specified combination of cell lineage, gene expression, or cellular behaviors measured from tracking results. Example implementations derived from the experiments described below are provided in the source repository. Once a target cell has been identified, the tracking pipeline implements a user-defined recipe of perturbation that specifies the number of repetitions, the time interval between repetitions, and the delay between the first detection of the target cell(s) and the initiation of perturbation. ShootingStar executes this recipe by generating an output text file containing the target's X, Y, and Z coordinates and a status flag to trigger illumination. Because the targeting data is written in a simple format, they can easily be passed to user-designed or commercially available illumination tools and thus adapted to any perturbation modality. Examples are provided along with the source distribution for integrating a galvanometer-steered dye laser to conduct ablations and for integrating a digital micromirror device (DMD) to conduct photoconversion experiments. This modular design, by separating perturbation control from the core tracking software, ensures that ShootingStar can be easily extended to support new perturbation and microscopy modalities. In addition to our implementations using MetaMorph, the open source Micro-Manager software (Edelstein et al., 2014), and Zen software with the Open Application Development tools (Carl Zeiss), offer the features required in order to support ShootingStar integration.
Results
Tracking Throughput and Accuracy
ShootingStar exhibits a linear relationship between the total processing time required per volume and the number of cells in each volume (Figure 1C). Tracking throughput was profiled on a Hewlett-Packard Z620 workstation with dual Intel Xeon E5-2643v2 processors at 3.5 GHz with 32 GB of RAM and a 512 GB Samsung 840 Pro SSD. We profiled ShootingStar by analyzing embryonic development in three species—C. elegans, Drosophila melanogaster (Keller et al., 2010), and D. rerio (Keller et al., 2008)—of successively greater image and physical size. The imaging periods were 60 s, 180 s and 90 s, respectively. The linear increase of processing time over this range of sample sizes demonstrates the efficiency of the underlying algorithms and implementation, and suggests that faster hardware could be used to scale throughput for still larger samples and images. Furthermore, since processing time per volume is shorter than the imaging time, the pipeline can also process multiple samples in parallel to increase experimental throughput. In the C. elegans experiments described below two embryos were typically processed simultaneously, and for the zebrafish experiments six specimens were imaged and processed in parallel.
ShootingStar maintains the high accuracy necessary for reliable cell tracking and target cell detection while tracking large numbers of cells over many rounds of cell division. Tracking accuracy was benchmarked in C. elegans embryos, whose invariant cell lineage (Sulston et al., 1983) provides an objective standard. Cells were imaged and tracked through six rounds of cell division and dramatic tissue-level rearrangements such as gastrulation, ending with about 350 cells in the embryo. Across three embryos, between 34 % and 73 % of cells were correctly tracked to the end with no errors in cell identity (Figure 1D). In all three cases, over 90 % of all cells' identities could be correctly determined with two or fewer edits of the automated tracking results. For the 768 cells validated across these three embryos, an average of 0.82 errors occurred per cell over 160 time points of tracking, yielding an average accuracy of 99.5 %. This metric represents an underestimate of the accuracy of ShootingStar, because any error in lineage-tracing affects the correct identification of at least two cells. Thus, a minimal amount of human editing during imaging readily yields a 100 % success rate. The above results in throughput and accuracy show that ShootingStar is well-suited for tracking hundreds to thousands of cells with minute-level temporal resolution.
Systematic Monitoring of Experiments
A major challenge in performing targeted optical manipulations in a complex tissue or organism is the assessment of off-target effects. Standard protocols for performing laser ablations in C. elegans, for example, recommend a visual check for apoptosis in nearby cells after an ablation (Fang-Yen et al., 2012). Given the dose required to cause cell death by laser ablation, a visual screen for the appearance of apoptosis is an insensitive approach to assessing off-target damage in surrounding cells. The live imaging component of ShootingStar makes it possible to conduct a systematic and quantitative assessment of off-target effects and thus identify potential sources of confounding errors.
To illustrate this capability, we monitored collateral damage during the optimization of laser-ablation parameters. We screened for prolonged cell-cycle length as a marker for damage in two C. elegans embryos after target cells were ablated, and compared the results to the cell-cycle lengths measured from 72 unperturbed embryos (Figure 2A) (Moore et al., 2013). In one ablated embryo, three cells exhibited prolonged cell cycles (Figure 2A.i-iii). The most severely delayed cell exhibited visible signs of damage in the form of anaphase bridging during its division (Figure 2A.iii). This division was delayed by only 16.6 min, an interval that, while quantitatively substantial, represents a subtle phenotype that would prove challenging to identify manually. Careful examination of the relative 3D positions of the delayed cells shows that damage is most likely to occur along the illumination axis (Figure 2B). Since ShootingStar continues to image and analyze the same after a perturbation has occurred, the targeted cell itself can also be tracked so that the final outcome of the perturbation can be fully validated. This is particularly important in the study of cellular interactions to verify that an ablated cell was cleared from the sample and thus no longer physically capable of affecting its neighbors (Figure 2C). We also conducted ablations of neurons and their progenitors in 12 C. elegans embryos and used the known patterns of axonal outgrowth in nearby neurons (Figure 2D-F, Santella et al., 2015) to assess off-target damage. These experiments showed that targeted cells could be reliably eliminated, while closely related and neighboring neurons were unaffected and able to generate normal patterns of axonal outgrowth (Figure 2G-I).
Figure 2. Systematic monitoring of automated cell ablations.
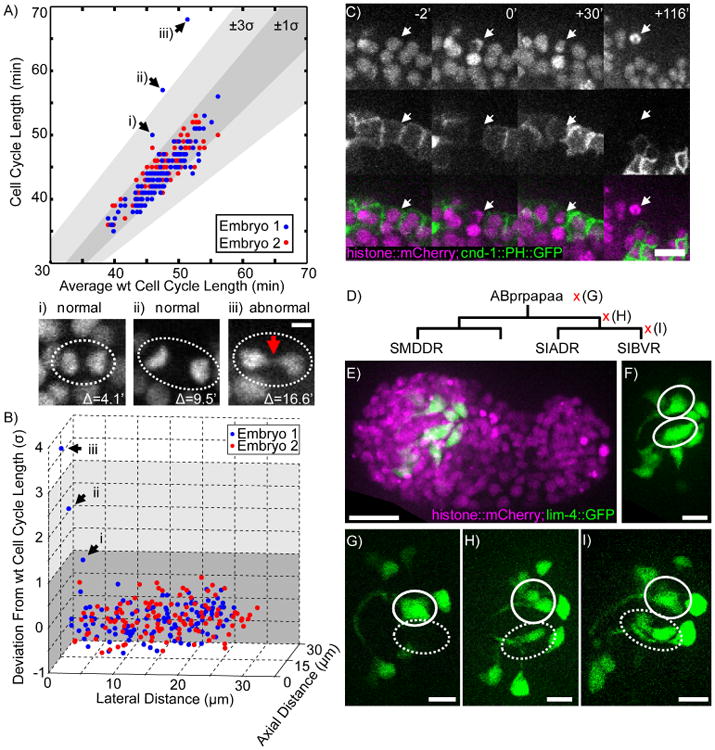
A) Cell cycle length of 127 remaining AB cell descendents in ablated C. elegans embryos (N= 2) plotted against the average cell cycle length of the corresponding cell in wild-type embryos (N= 71). (i-iii) Max projections of mitosis in three cells from Embryo 1 (dashed ovals). Red arrow indicates anaphase bridging (iii). The scale bar represents 2 μm. B) Scatterplot of cell cycle lengths shown in Figure 3 plotted in 3D against the lateral distance between every cell and the target cell and the axial distance between every cell and the target cell. Arrows (i-iii) highlight the same cells as in panel A. C) Time series showing the dynamics of cell death after laser ablation as visualized by a fluorescent histone reporter (white arrow) and membrane marker for a subset of neurons (cnd-lp∷PH∷GFP). The scale bar represents 10 μm. D) Lineage tree showing ablations performed across three generations. Left is more anterior, right more posterior. E) Max projection of a wild-type embryo at the comma stage. The scale bar represents 10 μm. F) GFP channel from (E) showing the neurons named in (D) as left-right symmetric pairs (white ovals). G) Representative embryo where ABprpapaa was ablated (N=4). Dashed oval shows loss of neurons on ablated side. H) Representative embryo where ABprpapaap was ablated (N=4). Dashed oval shows surviving neuron. I) Representative embryo where SIBVR was ablated (N=4). Dashed oval shows two surviving neurons. The scale bars in G-I represent 5 μm. See also Supplemental Movie 1.
Phenotypic Selection of Target Cells
ShootingStar supports a flexible definition of target cell identity that can incorporate factors such as marker expression and cellular behaviors. To demonstrate this capability we applied ShootingStar to investigate polarity establishment in the lateral line of D. rerio with automated cell ablations.
The lateral line of D. rerio comprises sensory organs called neuromasts. Each neuromast contains a cluster of hair cells, which are the receptor cells of the auditory, vestibular, and lateral-line organs. The apical surface of a hair cell bears a hair bundle of mechanosensitive stereocilia. Hair cells are born as sibling pairs that polarize in opposite directions along the anteroposterior axis. The polarity of a hair cell can be observed as a displacement of the kinocilium, the hair cell's primary cilium, relative to the center of the stereociliary bundle. In hair cells expressing a fluorescent β-actin reporter, the kinocilium appears as a dark spot surrounded by the bright actin-rich stereocilia (Figure 3A-C). Because each hair cell senses motion from a specific direction, the opposing polarity between pairs of hair cells is functionally critical. Although signaling between the sister hair cells is thought to establish this opposing polarization, the precise mechanism and function of this signaling remains unknown (López-Schier and Hudspeth, 2006; Mirkovic et al., 2012; Wibowo et al., 2011).
Figure 3. Automated ablation of hair cells in zebrafish neuromasts.
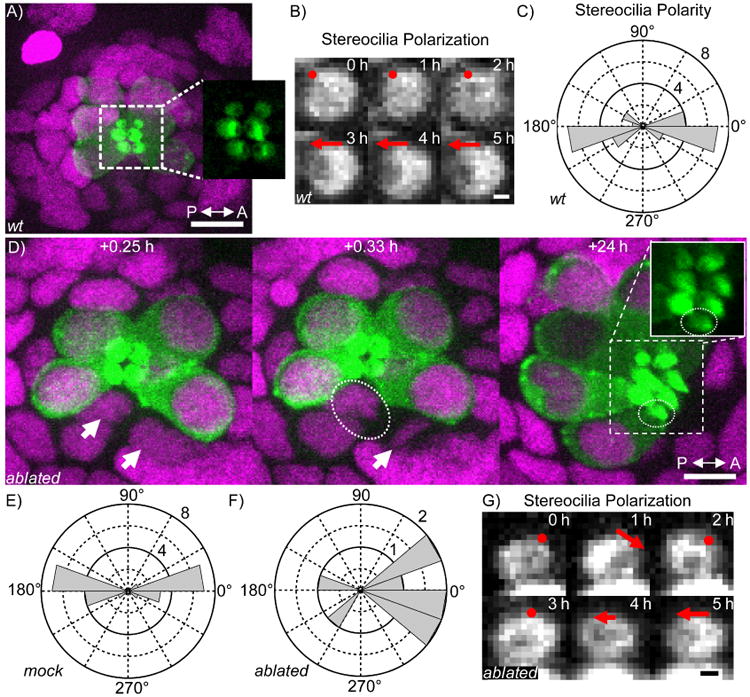
A) Max projection of a single neuromast in a 3 dpf D. rerio larva expressing cldnb∷H2B∷mCherry (magenta) and myo6b∷β-actin∷GFP (green). Inset shows expanded view of the GFP-labeled hair cell stereocilia. wt, wild-type. The scale bar represents 10 μm. B) Time series of polarity establishment in wild-type hair cell visualized by β-actin∷GFP. Red dot indicates no evident polarity, red arrow indicates polarity. The scale bar represents 0.5 μm. C) Distribution of hair cell polarity in wild-type neuromasts (N=26). D) Example sequence from the automated ablation of a hair cell (white arrow) and the subsequent maturation of the surviving sister (dashed ovals). The scale bar represents 10 μm. E) Distribution of hair cell polarity in mock-ablated neuromasts where a neighboring mature hair cell was ablated instead of one of the two newly born hair cells. (N=24). F) Distribution of polarity in hair cells whose siblings were ablated soon after birth (N=9). G) Time series of early polarization in a hair cell whose sibling was ablated soon after birth. Annotations as in (B). The scale bar represents 0.5 μm.
In order to probe the role of signaling between hair cell siblings in polarity establishment, we used ShootingStar to automatically detect dividing hair cell progenitors and ablate one of the two sibling hair cells immediately after division. The rarity and slow kinetics of divisions by hair-cell precursors, typically one in each neuromast per 24 hours, render manual identification and ablation of newly born hair cells challenging. Further complicating this task, not all divisions in neuromasts give rise to hair cells.
Based on these constraints, we defined three criteria in ShootingStar to identify newly born hair cells with two-color imaging. First, we used the expression of a β-actin∷GFP marker to detect precursors of hair cells. This marker is expressed in hair cell progenitors shortly before division. Second, we used a ubiquitously expressed histone∷mCherry to detect cell divisions. Third, we required the persistent detection of both daughters from the division of a hair cell progenitor for three subsequent time-points or 15 minutes. This requirement served to eliminate false detection of cell divisions and thus incorrect targeting of ablations. Over 24 hours of cell tracking, hair cell progenitor divisions are approximately a 1-in-10,000 event.
Using ShootingStar, we tracked a total of 14 neuromasts for at least 48 hours each over the course of three experiments. Eleven newly born hair cells were ablated in total. In nine of these ablations, the surviving sister formed a mature hair bundle (Figure 3D). The remaining two ablations resulted in the immediate death of both sisters. As in wild-type neuromasts and mock-ablated neuromasts where a neighboring mature hair cell was killed, the surviving hair cells in ablated neuromasts were able to polarize along the anteroposterior axis. The polarity deviation from the anteroposterior axis of both mock-ablated (Figure 3E, N=24, p=0.54) and sister-ablated hair cells (Figure 3F, N=9, p=0.18) was not different from that of wild-type hair cells (Figure 3C). These results suggest that sustained signaling between newly born hair cell siblings is not required for their survival, maturation, or polarization.
In a significant fraction of the sister-ablations (three of nine, p = 0.013 by Fisher's exact test), instability was observed during the early establishment of polarity in the surviving cell. In these hair cells the stereocilia polarized along the anteroposterior axis, then transitioned through a phase with no clear polarity before re-establishing a polarity that remained stable for at least 30 hours (Figure 3G). All 24 newly born hair cells in mock ablated animals matured normally and exhibited no instabilities in early polarity establishment. These results suggest that signaling between the siblings plays a role in stabilizing polarity, but further investigation will be necessary to determine the precise nature of this signaling.
In addition, we observed a qualitative anterior bias in the polarity of the surviving hair cell (Figure 3F) that accords with the results of genetic perturbations (Mirkovic et al., 2012). A larger number of ablations will be required to conclusively establish that the observed bias is different from a randomization of polarity, in which half of the surviving hair cells would be expected to adopt each polarity.
This application demonstrates ShootingStar's adaptability to different model systems, its flexibility in defining the criteria for selecting target cells, the accuracy of its real-time image analysis to target rare (1-in-10,000) events, as well as its efficiency in analyzing multiple samples in parallel.
Multi-Target Ablation
We employed ShootingStar for an experiment in which multiple targets needed to be identified and subjected to simultaneous ablation among more than 400 cells after being tracked for 3 hours and through six rounds of cell division. Specifically, we perturbed the six homologous glia-like GLR cells in C. elegans in order to investigate their role in the formation of the nerve ring. The nerve ring is the primary neuropil of C. elegans and is considered the brain of the organism.
Based on their strategic positions and morphology, the GLR cells have long been postulated as a critical tissue scaffold for the nerve ring. In the embryo, these cells are positioned circumferentially around the pharynx shortly before the nerve ring forms, and border the future nerve ring from the posterior side (Figure 4A). Testing the role of the GLR cells in nerve ring formation, however, has been technically difficult. These cells are derived from diverse branches of the mesodermal (MS) lineage (Figure 4B), making it challenging to trace all of them simultaneously by eye over an extended period of time. In one previous effort of manual laser ablations (Andrew Chisholm, personal communication), it was reported that the nerve ring shifted anteriorly and defasciculated. Surrounding neurons and skin cells showed large vacuoles. However, because it was difficult to repeat the experiment and assess off-target effects, these results remain unpublished. As a consequence, the precise function of the GLR cells in nerve ring formation remains an open question.
Figure 4. Ablation of multiple targets.
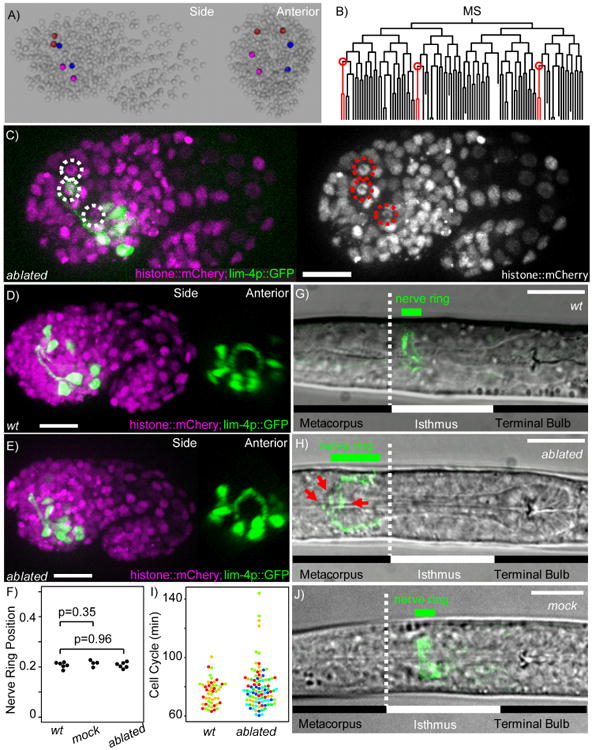
A) 3D model of a lineaged C. elegans embryo showing the final positions of the 6 GLR cells derived from MSaaaaaa (red), MSapaaaa (blue) and MSppaaaa (magenta). B) The MS lineage showing the three mothers of the GLR cells (circled in red). C) Max projection of representative embryo after ablation of GLR progenitors showing the location of the dead cells' nuclei (dashed circles). D) Max projection of a wild-type embryo expressing a lim-4p∷GFP reporter at the 1.5-fold stage showing normal nerve ring position and morphology. E) Max projection of an embryo where all three GLR progenitors were ablated. F) Normalized nerve ring position in embryos at the 1.5-fold stage. 0 represents the position of the sensory dent, 1 represents the eggshell's posterior pole. G) Image of a wild-type L1 showing the normal position of the nerve ring. Cell bodies were cropped from the green channel for clarity. H) Image of a L1 after GLR ablations. Red arrows point to defasciculated processes in the nerve ring. The scale bars represent 10 μm. I) Distribution of cell cycle lengths of 16 nearby cells (within 2 nuclear diameters of the targeted cell) in unablated embryos (N=3) and embryos where all 3 GLR progenitors were ablated (N=6). Each color denotes an embryo. J) Image of an L1 after mock ablation where neighboring cells were ablated instead of the 3 GLR progenitors. The scale bars represent 10 μm.
Using ShootingStar, we were able to reliably and repeatedly locate the three GLR mothers (Figure 4B) by automated lineage tracing, ablate them soon after their birth and image nerve ring formation with a neuronal marker to characterize GLR function. We repeated the experiment in 6 embryos. The ablated cells exhibited a fixed pattern of speckled histone fluorescence over time, failed to divide, and did not migrate to their correct circumpharyngeal positions, indicating a successful ablation (Figure C).
In comparison to wild-type animals, the nerve ring in GLR-ablated embryos formed in time by the 1.5-fold stage and showed no qualitative anatomical defects (Figure 4D,E). There was also no change in nerve ring position when it initially formed in GLR-ablated embryos (N=6) or mock ablated embryos (N=4) where a cell neighboring each of the GLR progenitors was ablated, compared to unablated embryos (N=6) (Figure 4F). After hatching, the nerve ring in GLR-ablated animals was shifted anteriorly as reported previously. In wild-type L1 animals the nerve ring lies near the anterior end of the isthmus, between the two pharyngeal bulbs (Figure 4G). In GLR-ablated animals, the nerve ring lies on top of the anterior bulb (Figure 4H). In 2 of the 6 GLR-ablated embryos, 2 cells nearby the ablated GLR progenitors showed cell cycle delay, however none exhibited visible signs of damage such as anaphase bridging or incomplete cytokinesis (Figure 4I). The nerve ring phenotypes in these 2 embryos are indistinguishable from the other 4 that showed no obvious off-target damages. Furthermore, mock ablated larvae showed normal positioning of the nerve ring (Figure 5J). These results suggest that the anterior shift of the nerve ring is due to ablation of GLR's and not off-target damages. Also as reported before, the nerve ring in GLR-ablated animals exhibited signs of defasciculation, and the mock-ablated animals did not. However, we did not observe signs of vacuolation in neurons or skin cells in these animals as previously noted, suggesting that these phenotypes may have been the consequence of subtle off-target effects in previous ablations. Taken together, our results show that the GLR's are not required for nerve ring formation, but for the maintenance of its position and structural integrity afterwards.
Figure 5. Automated single cell photoconversion.
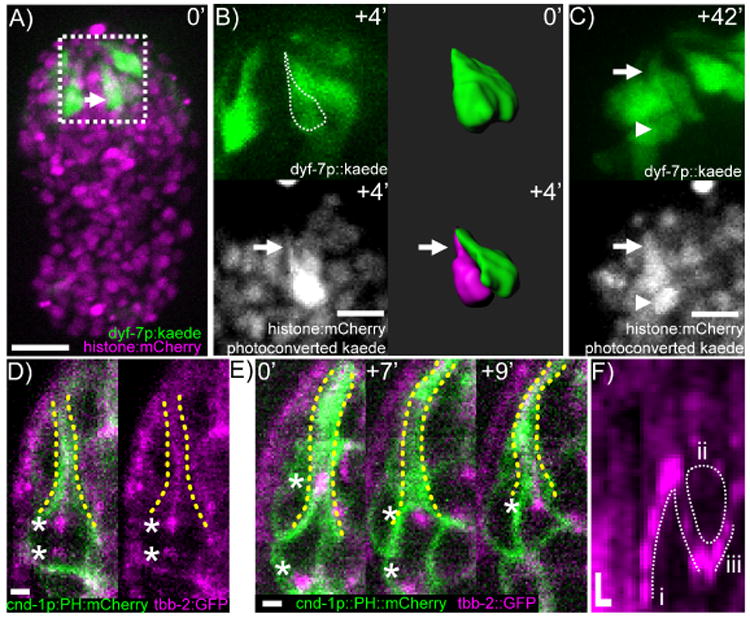
A) Max projection of a C. elegans embryo expressing dyf-7p∷Kaede. White arrow points to cell body of neural progenitor. Scale bar is 10 μm. B) Two-color imaging after photoconversion. White arrow points to cell process. Dashed outline shows tracing of photoconverted cell outline superimposed on unconverted Kaede fluorescence. Accompanying 3D reconstructions illustrates resolution of adjoining cells before (green) and after photoconversion (magenta). The scale bar represents 5 μm. C) Terminal division of the mother of CEPV. Arrow points to cell process, arrowhead points to posterior sister cell of CEPV. The scale bar represents 5 μm. D) β-tubulin localization in the mother of CEPV shortly after centrosome duplication. The scale bar represents 1 μm. E) β-tubulin dynamics in the mother of CEPV before and during its division. Asterisks mark centrosomes. The scale bar represents 1 μm. F) Kymograph of β-tubulin dynamics. (i) The movement of the anterior centrosome into the proximal dendrite. (ii) Depletion of β-tubulin from the proximal dendrite. (iii) Cytokinesis. Horizontal scale bar represents 2 minutes. Vertical scale bar represents 1 μm.
This application demonstrates the ability of ShootingStar to automate complex, multi-target manipulations, the precision of targeting via automated laser control to minimize off-target effects, as well as the value of our integrated approach in assessing transient phenotypes in development.
Single-Cell Labeling through Targeted Photoconversion
Recent advances have produced a large variety of optically responsive reagents. To demonstrate the utility of ShootingStar with these tools, we used it to selectively label individual neurons in the late C. elegans embryo through the green-to-red photoconversion of the fluorescent protein Kaede (Ando et al., 2002). A dyf-7p∷Kaede marker labels a large set of densely packed sensory neurons (Heiman and Shaham, 2009), hindering the ability to resolve individual neuron shapes and neurites (Figure 5A). Using ShootingStar, we converted a single cell during the outgrowth of sensory dendrites to disambiguate its structure from that of surrounding cells (Figure 5B). This cell, named ABplpaapppp, divides once to give rise to a single cephalic neuron (CEPV) and a cell that undergoes apoptosis (Sulston et al., 1983). The ABplpaapppp cell was identified by its lineage identity after about 4 hours of real-time lineage tracing.
Converting the ABplpaapppp cell body unexpectedly revealed that the cell, before differentiating into a post-mitotic neuron, extends a process to the sensory dent at which sensory dendrites are anchored in the head (Figure 5B). This process is maintained during and after its final division and is apparently the precursor of the dendrite for the anterior daughter cell, CEPV (Figure 5C). Given the peculiar challenges involved in a cell maintaining a process during mitosis, we characterized the dynamics of microtubule organization in ABplpaapppp using GFP-labeled β-tubulin (TBB-2) and a membrane-bound mCherry label broadly expressed in head neurons. We observed two microtubule structures with unexpected interactions during mitosis. The first structure is the mitotic spindle, represented by the centrosomes (asterisks in Figure 5D,E). The second and unexpected structure is the cell's process, that is, the precursor of CEPV's dendrite. The process is filled with microtubules before the onset of mitosis (yellow dashed lines in Figure 5D). This structure interacts with the centrosome during mitosis and exhibits drastic reorganization in three phases (Figure 5E-F, N=4). In the first phase, after centriole duplication and the two centrosomes become visible, one centrosome moves toward the base of the tubulin-rich dendrite and often moves deep into the dendrite (Figure 5E,F.i). The other centrosome moves to the opposite/posterior side of the cell. In the second phase, the anterior centriole moves back into the cell body, close to the base of the dendrite. The lower half of the dendrite is depleted of tubulin (dashed circle, Figure 5F.ii). The cell then proceeds to divide. In the third phase, shortly after cytokinesis, microtubules are re-assembled in the dendrite (Figure 5E,F.iii). The centrosome can be seen at the base of the dendritic microtubule bundle before it becomes invisible, presumably because it has shed the pericentriolar matrix. It is worth noting that CEPV is a ciliated neuron, so one expects the centrioles to eventually migrate to the dendrite tip and become the basal body of the sensory cilia.
The unexpected dynamics of the dendritic microtubules and their interaction with the centrosome during mitosis suggest that there may be mechanisms yet to be discovered that coordinate different microtubule structures depending on the cell's needs. For example, it is not clear why only one of the centrosomes moves into the dendrite, or which one. It is also not clear what triggers the disassembly of the dendritic microtubules and the release of the trapped centrosome, or how this process is related to spindle assembly. Furthermore, it is interesting to postulate that trapping by the dendrite may serve as a novel mechanism to orient the spindle and the direction of asymmetric cell division. Inasmuch as a similar phenomenon is observed in vertebrate radial glial cells, whose anchoring processes are maintained during asymmetric cell divisions (Noctor et al., 2004), this behavior might be conserved in neural progenitors.
Discussion
We have demonstrated an integrated approach to perturbation analysis with live imaging, real-time image analysis and automated optical perturbations to track and probe individual cells in vivo in response to their dynamic cellular behaviors and interactions. We developed ShootingStar as a software platform for real-time image analysis to coordinate with imaging and perturbation. Central to these types of experiments is the ability to reliably identify target cells in real-time, based on visible indicators of cell identity and behavior. ShootingStar can simultaneously track hundreds of cells, target multiple cells per sample and process multiple samples in parallel. It also provides users with flexibility to specify criteria for target selection. The automated control permits precise spatial and temporal control of optical perturbations, while the in toto imaging allows systematic assessment of off target effects. Both the precision and the systematic assessment are difficult to achieve traditionally in manual experiments. Finally, as demonstrated in the applications, this integrated approach can be applied to different biological samples with diverse optical methods to investigate a wide range of questions in tissue biology.
Limitations
In its current implementation ShootingStar can reliably track samples containing a few thousand cells faster than typical imaging intervals used for in toto imaging. We have shown that ShootingStar exhibits a linear relationship between cell number and throughput, suggesting that even larger samples or more parallel specimens could readily be accommodated with faster hardware. The primary bottleneck in further increasing ShootingStar's throughput is currently in our cell tracking pipeline, where aspects of its automated error correction algorithm have non-local impact and are thus challenging to parallelize. However, we reason that the capacity to track thousands of cells is largely sufficient for most single-cell resolution studies.
ShootingStar was designed to detect and track cellular-scale events in real-time. Some phenomena, such as sub-cellular transport, action potentials and cellular protrusions, exhibit dynamics on sub-minute to sub-second timescales and are thus too fast to readily image in an in toto context while tracking large numbers of cells in real-time. A multi-scale approach for identifying target cells and subsequently imaging and perturbing local high-speed phenomena in real-time could make these types of phenomena amenable to automated perturbation using ShootingStar.
While ShootingStar simplifies the targeting of perturbations, off-target effects remain a fundamental challenge in optical systems. In general, our experiments demonstrate that systematic optimization of laser ablation parameters is able to minimize off-target damage when certain constraints are taken into consideration. First, orient the sample so that the target cell is as close to the surface of the sample as possible. Second, off-target damage is most likely to occur along the beam-path. Finally, systematic optimization and careful controls are always necessary. ShootingStar can be used both to more reliably detect off-target damage in laser ablation experiments as well as to guide the optimization of ablation parameters to minimize its occurrence.
Perhaps the most significant limitation of ShootingStar is that it remains challenging for the non-technical user to deploy. For potential users with similar hardware available (a spinning disk confocal run using MetaMorph software with a Micropoint dye laser for ablations) ShootingStar may be used effectively turn-key with some minor configuration that is described in the accompanying manual. For many users who might use other microscope control software or who might have laser systems from other vendors, some degree of modification will be necessary to achieve integrated control using ShootingStar. Two features are required for any microscope control software to support integration with ShootingStar. First, images must be externally accessible after acquisition. Second, the software must support a scripting interface that can read the text-based target file from ShootingStar and use the target coordinates to trigger and target perturbation. For perturbation hardware, any illumination system which can be triggered externally or controlled directly by the microscope control software can be easily controlled including commercial or custom-built modules for fluorescence recovery after photobleaching (FRAP) and femtosecond lasers for ablation or 2-photon excited photoactivation. For users with experience in MATLAB scripting, ShootingStar in its current form will be a powerful starting point as it integrates a broad set of capabilities in a modular fashion. Support for alternative imaging and/or perturbation modalities can be readily implemented.
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Zhirong Bao (baoz@mskcc.org).
Experimental Model and Subject Details
Worm Strains and Culture
Strain JIM113 (ujlsll3 [Ppie-l∷mCherry∷H2B; Pnhr-2∷mCherry∷fflS-24-let-858UTR; unc-119(+)] II) was a gift of J. Murray. BV258 (ujlsll3 [Ppie-l∷mCherry∷H2B; Pnhr-2∷mCherry∷fflS-24-let-858UTR; unc-119(+)] II; mglsl9 [Plim-4∷GFP; rol-6(sul006)]) was generated by crossing JIM113 with OH96 (mglsl9), which was a gift of O. Hobert. BV354 (ujlsll3 [Ppie-l∷mCherry∷H2B; Pnhr-2∷mCherry∷fflS-24-let-858UTR; unc-119(+)] H; nsIs96[Pdyf-7∷Kaede]) was generated by crossing JIM113 with nsls96 (Heiman and Shaham, 2009), a gift of S. Shaham. Transgenic worms were maintained in standard conditions at 22 °C on 60 mm plates filled with nematode growth medium seeded with a 250 μL suspension of OP50 bacteria.
Zebra fish Lines and Maintenance
Tg(cldnb∷H2B∷mCherry) zebrafish, a gift of D. Nogare, were outcrossed to Tg(myo6b∷actin∷GFP) fish (Kindt et al., 2012) and cldnb∷H2B∷mCherry;myo6b∷actin∷GFP larvae were imaged. Experiments were conducted in accordance with the standards of Rockefeller University's Institutional Animal Care and Use Committee. Zebrafish were maintained under standard conditions (Westerfield, 2000). Embryos were raised at 28 °C in E3 medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgS04) containing 1 μg/mL methylene blue. All experiments used larvae at 2-4 days post fertilization (dpf).
Method Details
Microscopy
Ablation experiments were conducted on a spinning disk confocal microscope (Quorum Technologies Inc.) comprising a Zeiss AxioObserver Zl frame, a Yokogawa CSU-X1 spinning-disk unit, and two Hamamatsu C9100-13 EM-CCD cameras. A 1.6× magnifying tube lens (Quorum Technologies Inc.) was inserted between the spinning-disk unit and the microscope body. An Olympus UPLSAPO60XS objective was used with a thread adapter to mount on the Zeiss body (Thorlabs). The microscope stage used for multi-position imaging was an ASI MS2000 XY stage with a piezoelectric top plate capable of 150 μm travel. Images were acquired with 0.75 μm spacing between slices, resulting in a voxel size of 0.16×0.16×0.75 μm (X,Y,Z). Photoconversion experiments were conducted on a spinning-disk confocal microscope in Rockefeller University's Bio-Imaging Resource Center (BIRC). The microscope was a Zeiss Axiovert 200, the camera was an Andor iXon Ultra 897 EM-CCD, and the voxel size of images acquired for the photoconversion experiments was 0.25×0.25×0.75 μm (X,Y,Z). Tubulin dynamics were imaged using an instant structured illumination microscope (Visitech iSIM) using an Olympus IX73 body, an Olympus UPLSAPO60XS objective and acquired every 60 s using a Hamamatsu Flash 4.0v2 sCMOS camera with a voxel size of 0.108×0.108×0.75 μm (X,Y,Z). With the exception of a linear contrast adjustment for display, no post-processing was performed on these images.
For all C. elegans ablation experiments, volumes were acquired with 35 slices every 60 s with an exposure of 150 ms per slice. Zebrafish larvae were imaged every 5 min with 35 slices spaced 1 μm apart using an exposure of 250 ms per slice. Both image acquisition and ablations were controlled with the MetaMorph microscopy automation software (Molecular Devices); the multi-dimensional acquisition module and custom journals are provided for reference in Supplementary Software.
C. elegans embryos were mounted under light compression for imaging in a suspension of 20 μm polystyrene beads in M9 buffer (3 g KH2PO4, 6 g Na2HPO4, 5 g NaCI, and 1 mL 1 M MgSO4 per liter of water) according to a previously published protocol (Bao and Murray, 2011). In brief, gravid hermaphrodites were picked into a droplet of M9 buffer and cut. Embryos at two-or four-cell stages were transferred to a 1 μL droplet of M9 containing 50-100 beads on a #1.5 coverslip. Prior to imaging, a smaller coverslip was laid on top of the droplet and sealed with melted petroleum jelly.
Zebrafish larvae at 2-4 dpf were anesthetized in 600 μM 3-aminobenzoic acid ethyl ester methanesulfonate in E3 medium and mounted in a 35 mm glass-bottomed dish in 1 % low-melting-point agarose.
Software
Real-time detection and tracking were performed across a set of sliding windows during a time-lapse acquisition (Figure 1B). First, cells were detected in the most recently acquired timepoint using a parallelized adaptation of a hybrid 2D/3D blob-detection algorithm.(Santella et al., 2010) By parallelizing per-object computations, performance scaled with the availability of more physical threads on a workstation. New detections were then assigned to pre-existing tracks based on distance and overlap. Unclaimed new detections were putatively assigned as cell divisions. A classifier was applied to the track topology to identify detection errors and false divisions. The classifier was evaluated at a single timepoint for which four future and six past timepoints were available.
For applications in which absolute accuracy in target cell identification was necessary, the curation interface was launched by the tracking pipeline from the MATLAB Java virtual machine (JVM). Critical to on-line acquisition and curation, we implemented bi-directional synchronization of tracking results between the automated pipeline and the curation interface. For the curation interface we integrated an existing application, AceTree (Boyle et al., 2006), into ShootingStar. AceTree was modified to implement a semaphore that was set by the automated pipeline during synchronization every time a user edit was committed to prevent race conditions. AceTree supported all the features necessary to display and curate datasets, track cells, and compute systematic cell names for the C. elegans data.
Cellular Ablation
Ablations were carried out using a Photonics Instruments (now Andor Technology Ltd.) MicroPoint dye laser equipped with a dye cell filled with 5 mM coumarin-440 in methanol. For ablations, 35 pulses with separations of 0.5 s were targeted at the nuclear centroid of the target cell. The neutral-density filter integrated into the MicroPoint system was adjusted empirically such that the pulse sequence would result in clearly visible damage to the targeted nucleus but no apparent damage to neighboring nuclei. After the power level had been optimized, performance remained stable as long as the dye solution was exchanged regularly, every two to three weeks during periods of heavy usage. The same ablation procedure was found to be effective for ablations in the MS and AB lineages beginning with the AB128 stage and until the onset of twitching.
Two criteria were used to define ablation success. First, the targeted cell failed to divide and, if the ablated cell was near the outer surface of the embryo, it was extruded from the embryo. Second, the nucleus of the ablated cell appeared speckled in a fixed pattern, suggesting that chromatin had condensed or aggregated and was no longer moving freely within the nucleus. In all cases, the ablated cell was tracked until the onset of twitching in order to validate the ablation. See also: Supplemental Movies 1 and 2.
ShootingStar is provided with the identity of the target cell(s) in a comma-separated text file. To allow the completion of cytokinesis, ShootingStar was configured to trigger ablation in C. elegans samples 10 min after a target's birth. To permit multi-target ablations, a simple queue was implemented in the target-identification code so that one cell per embryo would be ablated in a single time point to ensure that ablations would occur during the interval between the acquisition of successive images.
Ablation of GLRs
Born inside the embryo, the GLRs were not extruded upon ablation. An ablation was therefore scored as a success if the ablated cells did not divide subsequently and failed to migrate to their correct positions prior to the 1.5-fold stage. The position of the nerve ring at the 1.5-fold stage was measured manually with ImageJ as the distance from the sensory dent normalized against the length of the embryo. The post-embryonic position of the nerve ring was assessed by heat-killing L1 larvae by passing the coverslip quickly through a flame two or three times and scoring the position of the GFP-labeled nerve ring relative to the isthmus of the pharynx.
Ablation of Zebrafish Neuromasts
As with C. elegans embryos, ablations were conducted with 35 laser pulses spaced 0.5 s apart. The laser power was adjusted such that the β-actin∷GFP marker was bleached only in the targeted hair cell. This power varied between sample preparations, presumably owing to the relatively large variations in the gap between the coverglass and the larvae after mounting in agarose. To compensate for this variation, the laser energy was coarsely calibrated on neuromasts in a mounted larva that was not used for ablation experiments. We used the highest laser energy that did not visibly bleach off-target hair cells.
Tracking was restricted to hair-cell progenitors and hair cells and by filtering local maxima prior to cell detection on the basis of an empirical threshold applied to the β-actin∷GFP channel after smoothing with a median filter over a 13×13-pixel neighborhood. Detected cells were evaluated for two behavioral criteria in order to flag target cells for ablation: the detection of a cell division and the persistence of both daughters for 15 min. In the mock ablations a neighboring mature hair cell was ablated instead of one of the newly born hair cells.
After ablation, the surviving hair cell was tracked for at least 30 hr and scored for survival and polarity. Up to six neuromasts were tracked in parallel per experiment, each of which lasted for 48 hr of imaging. No cells were ablated in neuromasts treated as the wild-type control population. The final polarity of hair cells was scored 24 hr after birth and confirmed 6 hr later. The angle of polarity was quantified as the angle between the anteroposterior axis of the animal and the vector from the center of the kinocilium to the center of the stereociliary bundle as measured manually with Fiji.
Single-Cell Photoconversion
Single-cell photoconversion was achieved by targeting 405 nm illumination with a Photonics Instruments (now Andor Technology Ltd.) Mosaic system, which uses a digital micromirror device (DMD) to project an arbitrary illumination pattern onto the sample. Individual cells were converted by exposure for 2 s. The illumination region was defined as a five-pixel-diameter circle centered on the target nucleus. The laser power during photoconversion was measured to be 8 μW before the objective lens.
Example Parameters for Automated Target Identification
ShootingStar supports a flexible text-based system for defining target cells. Currently five parameters are supported: target name (Target), nuclear diameter (Size), marker expression level (Marker), z-position in the image (Z) and the delay between observing a cell and perturbing it (Persistence). Each of these can be defined separately for any number of targets. Below is an example for a 3-target ablation based on the GLR ablation experiments.
%Target definition for GLR ablations
%Three cells are defined by their lineage names
%Any size, marker expression or position
%Ablate 7 time points after detection
Definitions = 3;
Target_1 = ‘MSaaaaaa’;
Size_1 >= 0;
Marker_1 >= 0;
Z_1 >= 0;
Persistence_1 = 7;
Target_2 = ‘MSapaaaa’;
Size_2 >= 0;
Marker_2 >= 0;
Z_2 >= 0;
Persistence_2 = 7;
Target_3 = ‘MSppaaaa’;
Size_3 >= 0;
Marker_2 >= 0;
Z_3 >= 0;
Persistence_3 = 7;
Here is an example file based on the zebrafish hair cell ablations, which uses marker expression level to define target cells.
%Target definition example for zebrafish hair cell ablations
%Any cell with a hair cell specific GFP marker expression greater than 1500
%Ablate 3 time points after detection
Definitions = 1;
Target_1 = ‘Any’;
Size_1 >= 0;
Marker >= 1500;
Z_1 >= 0;
Persistence_1 = 3;
Quantification and Statistical Analysis
Assessment of Tracking Accuracy
The accuracy of cell tracking was assessed from the perspective of a typical experiment in which the validity of an individual target cell's identity was the essential metric. Images of three different C. elegans embryos were processed off-line using ShootingStar and lineage accuracy was assessed at the 256-cell stage in the anterior daughter of the C. elegans zygote (AB256). The number of independent errors in the lineage of each of the 256 cells was scored manually for each embryo.
Quantification of Off-Target Damage
Detection of off-target damage was performed by tracking all cells in the AB lineage from the AB128 stage until their division. For all 127 non-targeted cells, the cell cycle length was plotted against the mean cell cycle length for the same cell measured in a previously published dataset containing a large number of wild-type embryos (Moore et al., 2013). Cells that exhibited more than one standard deviation from the wild-type distribution of cell-cycle length were examined for signs of damage such as anaphase bridging.
Statistical Methods
Data were analyzed as described above. The mean and standard deviation for cell cycle lengths in the wild-type data from Figure 3 were calculated from published data (Moore et al., 2013). The upper and lower limits for the distribution about the mean were estimated in MATLAB by linear regression from the mean cell-cycle length plus or minus one or three standard deviations. The frequency of unstable polarity in zebrafish ablations (3/9) was compared to the frequency observed in wild-type larvae (0/26) and in the mock ablated larvae (0/24) by a 2×2 Fisher's exact test. The mean nerve-ring positions in Figure 5G were compared by a two-sample, one-tailed Student's t-test.
Data and Software Availability
ShootingStar source code and documentation are freely available for modification and redistribution under the GNU general public license; a MATLAB license is required. Updated documentation, source code and binary executables are available at: https://github.com/RealTimeLineaging/ShootingStar
Supplementary Material
Supplemental Movie 1. Dynamics of laser-induced cell death, related to Figure 2 and STAR Methods: Cellular Ablation. Timelapse of an ablation experiment showing a thin section (top, max projection through 5 slices) and a max projection of the entire embryo. The target cell's lineage is tracked with a magenta circle. Ablation is marked by a cyan circle.
Supplemental Movie 2. Example ablation experiment using ShootingStar, related to STAR Methods: Cellular Ablation. Screen capture with live recording of an ablation experiment being performed with ShootingStar. Main window shows screen capture recording during ablation experiment, lower right corner shows synchronized recording of the microscope showing when user intervention is occurring and when images are being acquired. Long periods without user intervention are shown at 40× or 80× real-time as shown for brevity. Overlaid annotations describe user interface elements and key phases of automated and user-assisted operation of ShootingStar.
Highlights.
ShootingStar is a systematized platform to perform perturbation analysis in vivo
Real-time targeting of perturbations from measured cell identity and behaviors
Quantitative experimental records for outcome validation and artifact detection
Adaptability to different perturbation modalities and sample types
Acknowledgments
The authors would like to thank O. Hobert and B. Iglesias for suggested experiments; A. Chisholm for sharing unpublished observations about GLR's; H. Shroff, D. Colon-Ramos and W. Mohler for helpful discussions; and A. North and The Rockefeller University Bio-Imaging Resource Center for equipment used in the photoconversion experiment. This work was supported by NIH grants (GM097576 and HD075602 to ZB, and the MSK Cancer Center Support/Core P30 CA008748). Some strains were provided by the CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440). AJH is an Investigator of Howard Hughes Medical Institute.
Footnotes
Author Contributions: PKS, AS, and ZB designed the ShootingStar system. PKS developed the software, performed the C. elegans experiments, and processed the data. PKS, AJ, KS, and AJH designed the zebrafish experiments and PKS, AJ, and KS performed the zebrafish experiments and interpreted the results. PKS and ZB drafted the manuscript and AS, AJ, KS, and AJH edited it. All authors reviewed the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ando R, Hama H, Yamamoto-Hino M, Mizuno H, Miyawaki A. An optical marker based on the UV-induced green-to-red photoconversion of a fluorescent protein. Proc Natl Acad Sci. 2002;99:12651–12656. doi: 10.1073/pnas.202320599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Z, Murray JI. Mounting Caenorhabditis elegans Embryos for Live Imaging of Embryogenesis. Cold Spring Harb Protoc. 2011;2011 doi: 10.1101/pdb.prot065599. pdb.prot065599-prot065599. [DOI] [PubMed] [Google Scholar]
- Boyle TJ, Bao Z, Murray JI, Araya CL, Waterston RH. AceTree: a tool for visual analysis of Caenorhabditis elegans embryogenesis. BMC Bioinformatics. 2006;7:275. doi: 10.1186/1471-2105-7-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, Stuurman N. Advanced methods of microscope control using μManager software. J Biol Methods. 2014;1:10. doi: 10.14440/jbm.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang-Yen C, Gabel CV, Samuel ADT, Bargmann CI, Avery L. Laser microsurgery in Caenorhabditis elegans. Methods Cell Biol. 2012;107:177–206. doi: 10.1016/B978-0-12-394620-1.00006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman MG, Shaham S. DEX-1 and DYF-7 Establish Sensory Dendrite Length by Anchoring Dendritic Tips during Cell Migration. Cell. 2009;137:344–355. doi: 10.1016/j.cell.2009.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller PJ, Schmidt AD, Wittbrodt J, Stelzer EHK. Reconstruction of Zebrafish Early Embryonic Development by Scanned Light Sheet Microscopy. Science. 2008;322 doi: 10.1126/science.1162493. 80. [DOI] [PubMed] [Google Scholar]
- Keller PJ, Schmidt AD, Santella A, Khairy K, Bao Z, Wittbrodt J, Stelzer EHK. Fast, high-contrast imaging of animal development with scanned light sheet-based structured-illumination microscopy. Nat Methods. 2010;7:637–642. doi: 10.1038/nmeth.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindt KS, Finch G, Nicolson T. Kinocilia Mediate Mechanosensitivity in Developing Zebrafish Hair Cells. Dev Cell. 2012;23:329–341. doi: 10.1016/j.devcel.2012.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Schier H, Hudspeth AJ. A two-step mechanism underlies the planar polarization of regenerating sensory hair cells. Proc Natl Acad Sci U S A. 2006;103:18615–18620. doi: 10.1073/pnas.0608536103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkovic I, Pylawka S, Hudspeth AJ. Rearrangements between differentiating hair cells coordinate planar polarity and the establishment of mirror symmetry in lateral-line neuromasts. Biol Open. 2012;1 doi: 10.1242/bio.2012570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JL, Du Z, Bao Z. Systematic quantification of developmental phenotypes at single-cell resolution during embryogenesis. Development. 2013;140 doi: 10.1242/dev.096040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noctor SC, Martínez-Cerdeño V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–144. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- Santella A, Du Z, Nowotschin S, Hadjantonakis AK, Bao Z, Bao Z, Murray J, Boyle T, Ooi S, Sandel M, et al. A hybrid blob-slice model for accurate and efficient detection of fluorescence labeled nuclei in 3D. BMC Bioinformatics. 2010;11:580. doi: 10.1186/1471-2105-11-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santella A, Du Z, Bao Z, Bischoff M, Parfitt D, Zernicka-Goetz M, Keller P, Schmidt A, Wittbrodt J, Stelzer E, et al. A semi-local neighborhood-based framework for probabilistic cell lineage tracing. BMC Bioinformatics. 2014;15:211. doi: 10.1186/1471-2105-15-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santella A, Catena R, Kovacevic I, Shah P, Yu Z, Marquina-Solis J, Kumar A, Wu Y, Schaff J, Colon-Ramos D, et al. WormGUIDES: an interactive single cell developmental atlas and tool for collaborative multidimensional data exploration. BMC Bioinformatics. 2015;16:189. doi: 10.1186/s12859-015-0627-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- Westerfield M. The Zebrafish Book A Guide for the Laboratory Use of Zebrafish (Danio rerio) Eugene: University of Oregon Press; 2000. [Google Scholar]
- Wibowo I, Pinto-Teixeira F, Satou C, Higashijima S, López-Schier H. Compartmentalized Notch signaling sustains epithelial mirror symmetry. Development. 2011;138:1143–1152. doi: 10.1242/dev.060566. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Movie 1. Dynamics of laser-induced cell death, related to Figure 2 and STAR Methods: Cellular Ablation. Timelapse of an ablation experiment showing a thin section (top, max projection through 5 slices) and a max projection of the entire embryo. The target cell's lineage is tracked with a magenta circle. Ablation is marked by a cyan circle.
Supplemental Movie 2. Example ablation experiment using ShootingStar, related to STAR Methods: Cellular Ablation. Screen capture with live recording of an ablation experiment being performed with ShootingStar. Main window shows screen capture recording during ablation experiment, lower right corner shows synchronized recording of the microscope showing when user intervention is occurring and when images are being acquired. Long periods without user intervention are shown at 40× or 80× real-time as shown for brevity. Overlaid annotations describe user interface elements and key phases of automated and user-assisted operation of ShootingStar.