

Summary
The Collaborative Cross (CC) is a panel of reproducible recombinant inbred mouse strains with high levels of standing genetic variation, affording an unprecedented opportunity to perform experiments in a small animal model containing controlled genetic diversity while allowing for genetic replicates. Here, we advance the utility of this unique mouse resource for immunology research because it allows for both examination and genetic dissection of mechanisms behind adaptive immune states in mice with distinct and defined genetic makeups. This approach is based on quantitative trait locus mapping: identifying genetically variant genome regions associated with phenotypic variance in traits of interest. Furthermore, the CC can be utilized for mouse model development; distinct strains have unique immunophenotypes and immune properties, making them suitable for research on particular diseases and infections. Here, we describe variations in cellular immune phenotypes across F1 crosses of CC strains and reveal quantitative trait loci responsible for several immune phenotypes.
Keywords: adaptive immunity, immunogenetics, mouse models, Collaborative Cross, QTL mapping, regulatory T cells
Graphical Abstract
Highlights
-
•
The Collaborative Cross models the phenotypic diversity observed in human immunity
-
•
QTL mapping in the CC reveals candidate genes linked to T cell phenotypes
Graham et al. advance the use of the Collaborative Cross (CC), a panel of reproducible recombinant inbred mouse strains, for immunology research. They demonstrate that the CC better models the phenotypic diversity in human T cell immunity and use quantitative trait locus mapping to reveal candidate genes linked to T cell phenotypes.
Introduction
In seeking to understand the complex interactions and pathways of the human immune response, researchers have long turned to inbred mouse models. 99% of mouse genes are shared with humans (Boguski, 2002), and inbred laboratory mouse strains are well characterized, reproducible, and allow for the use of unique immunological tools such as transgenic or knockout mice. However, given the diverse breadth of clinical outcomes and immune responses observed in the human population, any single traditional inbred mouse model cannot fully capture the range of immune phenotypes expressed across genetically diverse humans. Indeed, although estimates vary, there is a clear contribution of genetic variation in driving diversity throughout human immune responses (Salvetti et al., 2000), and this diversity cannot be fully identified through study of a single gene knockout (KO). To fully investigate these complex networks, additional studies in a model that better captures the genetic diversity of humans is critical.
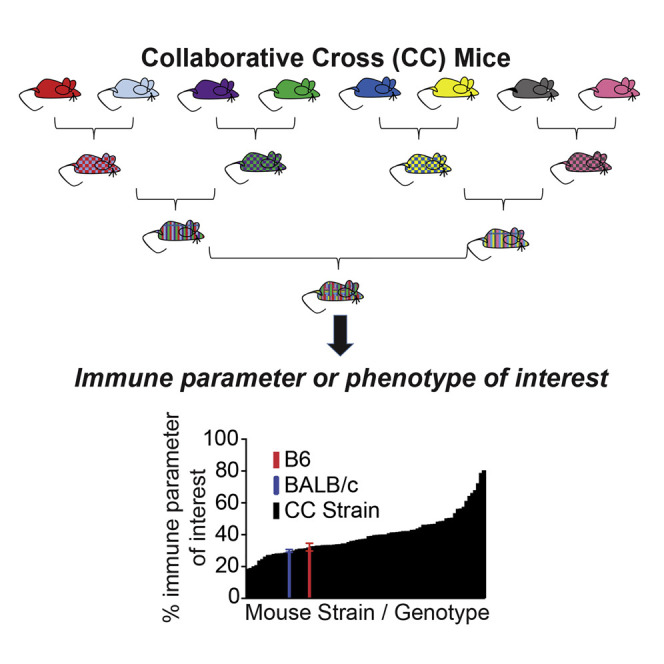
Defining the genetic basis of immune responses and regulation requires approaches and model systems that move beyond classical genetic screens, such as targeted KO mouse strains on a C57BL/6 (B6) background, or random N-ethyl-N-nitrosourea (ENU) mutagenesis studies, which both typically study one gene at a time (Gondo et al., 2010, Mountz et al., 2001, Yates et al., 2009). The Collaborative Cross (CC) mouse genetic reference panel represents a resource that specifically models complex genetic interactions and, therefore, expands classic approaches within traditional (e.g., B6) mouse models. The CC strains are a recombinant inbred (RI) panel and are derived from eight founder strains: five classical inbred strains (C57BL/6J, A/J, 129S1/SvImJ, NOD/ShiLtJ, and New Zealand obese (NZO)/SH1LtJ) and three wild-derived strains (CAST/EiJ, PWK/PhJ, and Watkins star line B (WSB)/EiJ). These eight founder strains represent the three major Mus musculus subspecies (domesticus, musculus, and castaneus) and capture nearly 90% of common genetic variation in laboratory mouse strains, with this variation uniformly distributed across the genome (Churchill et al., 2004, Threadgill et al., 2011, Threadgill and Churchill, 2012). CC-RI strains were created by three generations of funnel breeding to incorporate genomic contributions of all eight founder strains within each CC strain, followed by at least 20 generations of inbreeding (Collaborative Cross Consortium, 2012). The CC therefore provides a reproducible experimental model of many aspects of genetic variation within the human genome. In these studies, F1 progeny from crosses between CC-RI strains (CC-RIX, recombinant intercross) were used (Graham et al., 2015). These RIX lines were heterozygous for the H-2bb major histocompatibility complex (MHC) haplotype, allowing for use of reagents such as tetramers to examine T cell responses in these mice, which is critical to enable study of T cell-mediated immunity.
The CC provides the ability to assess the breadth of phenotypic differences under genetic control and can also provide new mouse models for human diseases because screens have identified variations in immune phenotypes and clinical disease symptoms for infections such as influenza (Ferris et al., 2013), Ebola (Rasmussen et al., 2014), West Nile virus (Graham et al., 2015, Graham et al., 2016), and severe acute respiratory syndrome (SARS) (Gralinski et al., 2015) as well as spontaneous colitis (Rogala et al., 2014), cancer-related phenotypes (Reilly, 2016), and behavioral traits (Chesler, 2014). In our studies as part of the CC Systems Immunogenetics Group, we screened over 110 CC-RIXs for a variety of immune response parameters at steady state in adult male mice of 8–10 weeks of age. Importantly, within the screen we found a wide variation in each immune phenotype measured, and here we fully describe a comprehensive screen of CC-RIXs for cell subsets such as CD3+, CD4+, CD8+, and regulatory T cells (Tregs) as well as activation markers and inflammatory cytokines. Included with these data are the identity of quantitative trait loci (QTLs) or polymorphic host genome regions and potential candidate genes within these regions that affect immune phenotypes. This initial proof-of-concept genetic mapping as well as our extensive dataset pave the way for future use of CC mice for genetic mapping of immune traits as well as targeted CC mouse strain selection for future immunology and immunogenetics studies. Similar to the expansion of immune phenotypes displayed in the so-called “pet store mice” or “dirty mice” compared with mice maintained under specific pathogen-free (SPF) conditions, which has allowed for an improved mouse model that better accounts for the microbial colonization diversity in humans (Masopust et al., 2017), we demonstrate here that use of the CC can address another critical limitation of mouse research by expanding the genetic diversity and resultant immune phenotypes of murine study subjects to more effectively model human immunity.
Results
Screening CC-RIX Lines for Immune Phenotypes
We conducted a comprehensive screen of 113 CC-RIX lines for immune response phenotypes to gauge the diversity of responses resulting from natural genetic variation. CC-RIX lines were bred to ensure that lines were heterozygous for the H-2b locus, having one copy of the H-2bb haplotype and one copy of the other various haplotypes at the MHC locus. This design was selected so that we could examine antigen-specific T cell responses for our parallel studies of immunogenetics of virus infection while concurrently maintaining genetic variation throughout the rest of the genome. Through our screen, three to six adult, 8- to 10-week-old male mice were examined for each CC-RIX line, and we measured and cataloged an extensive list of T cell phenotypes within the spleen at steady state with no experimental manipulations. The range of phenotypes included frequency of T cell subsets, proportion of cells expressing various activation markers, frequency of cells producing inflammatory cytokines, and quantity of cells expressing tissue migration markers (Table S1; Figure S1). For each phenotype examined, we measured abundant variability between RIX lines (detailed below), as observed upon examination of any immune parameter in a human population. Indeed, even body weight varied extensively between RIX lines, despite careful age-matching (Table S2; Figure S2A). We present an extensive array of steady-state immunity data for each animal in the screen, garnered from our three thirteen-color flow cytometry panels as well as clinical observations. Each immune phenotype examined resulted in a high degree of variability by genetic background of the host and exceeded the breadth of responses observed in the most commonly used inbred strains, C57BL/6J and BALB/cJ (Table S3). Importantly, because our dataset is included (Table S4) and available on ImmPort, researchers can select from baseline phenotypes of interest for a particular infection or disease to perform a small subset of experiments rather than a large, time- and resource-consuming phenotypic screen. We anticipate that our dataset, along with these accompanying proof-of-concept studies detailing examples of genetic mapping of immune traits of interest, will advance the use of the CC in immunology and genetic mapping studies.
Variation in T Cell Frequencies across CC-RIX Lines Presents Mouse Models for Immunological Studies
Previous studies have demonstrated variation in T cell frequencies based on host genetics, although these studies have largely been restricted to a limited number of commonly used inbred strains of mice (Chen et al., 2005, Feuerer et al., 2007, Holler et al., 2007, Mostafavi et al., 2014, Paula et al., 2011, Petkova et al., 2008, Bogue and Grubb, 2004, Grubb et al., 2004). To put this previously described variation into context of a larger group of mice with increased genetic variation, we examined the frequency of T cell populations in the spleens of 113 CC-RIX lines compared with the commonly used BALB/cJ and C57BL/6J model strains. We found a high degree of variability in the frequency of CD3+ T cells based on host genetics, with average frequencies in CC-RIX lines ranging from 16%–62% of gated lymphocytes, whereas C56BL/6J mice had an average frequency of 36.96%, and BALB/cJ had an average frequency of 46.4% (Figure 1 A). Within these splenic CD3+ T cell populations, we further examined the frequencies of CD8+ T cells, CD4+ T cells, and also CD4+Foxp3+ Tregs. Notably, CD8 T cell frequencies in CC-RIX lines ranged from 14.1%–65.3% of CD3+ T cells, with C57BL/6J and BALB/cJ average frequencies of 36.6% and 32.1%, respectively (Figure 1B). The variability in CD4 T cell frequency in the spleen in CC-RIX mice ranged from 27%–73.1% of CD3+ T cells, with C57BL/6J and BALB/cJ average frequencies of 58.96% and 64.5%, respectively (Figure 1C). As expected because of this variability in CD8 and CD4 T cell frequency, there is also extensive diversity in the CD4/CD8 T cell ratio across CC-RIX lines (Figures S2B and S2C). Finally, we quantified the frequency of Tregs as a percentage of CD4+ T cells and found that CC-RIX lines had a range of 1.8%–25.5% Tregs compared with C57BL/6J and BALB/cJ average frequencies of 10% and 12.5%, respectively (Figure 1D). Importantly, the variability in Treg frequency modeled by the CC-RIX is similar to that observed in a large human cohort (Figure 1E), suggesting that the CC improves greatly upon the ability of two common laboratory strains to represent the diversity in T cell frequency observed in humans. In summary, we find that CC-RIX lines display greatly enhanced variation in the frequency of total T cells as well as CD8+, CD4+, and Foxp3+ subsets compared with traditional inbred models. Further, we propose that this variation will be key to extend immunological mouse models as well as QTL mapping studies to identify genes regulating homeostatic T cell population dynamics and better understand the role variant immune homeostasis plays in the resultant immunological responses.
Figure 1.

Variation in Frequency of T Cell Populations at Steady State
(A–D) Frequencies of CD3+ cells (A), CD8+ cells (B), CD4+ cells (C), and Treg+ cells (D) are plotted for the 110 cohorts of CC mice screened during the discovery phase of the experiment. The average value for the indicated phenotype of each CC-RIX line (n = 3) is plotted in black in ascending order on the x axis. B6 (red) and BALB/c (blue) baseline values are plotted for comparison. All values represent uninfected controls.
(E) Frequency of Tregs in human blood from 244 individuals.
Error bars show ± SEM.
Variation in Conventional T Cell Phenotypes across CC-RIX Lines
In addition to examining T cell frequencies in mice with distinct CC-RIX genetic backgrounds, we performed an extensive phenotypic analysis of conventional T cells to classify them further into subtypes as well as catalog their activation and functional profiles (Tables S1 and S3). Although the full dataset is available on ImmPort (ImmPort: SDY1176), we here focus on the frequency of Ki67+ CD8 T cells, which indicates recently or actively proliferating cells; CD44+ CD8 T cells, indicating cells that have experienced antigen; CD62L- CD8 T cells or activated CD8 T cells; as well as the frequency of CD8 T cells that secrete interferon γ (IFNγ) or interleukin-17 (IL-17) upon polyclonal anti-CD3/CD28 stimulation ex vivo (Figures 2 A–2E). For reference, although C57BL/6J mice had an average of 10.8% of CD8 T cells that were Ki67+ and BALB/cJ had 7.2%, CC-RIX lines ranged from averages of 2.5%–22.5% (Figure 2A), again providing a much more diverse response that can be used downstream for unique mouse immunology model studies or for QTL mapping. In addition to this heterogeneous response based on host genetics, we additionally identified, in CC-RIX lines, a wide range of antigen-experienced CD8 T cell frequencies (Figure 2B), activated CD8 T cells (Figure 2C), IFNγ-secreting CD8 T cells (Figure 2D), and IL-17-secreting CD8 T cells (Figure 2E). Importantly, all mice used within these studies were bred in the same facility, and experiments were similarly conducted in a common environment. Together, these data suggest that homeostatic immune responses and the propensity to initiate adaptive immunity are under strong genetic control, and this dataset may thus inform further studies of autoimmunity and tolerance.
Figure 2.

Variation in Conventional T Cell Phenotypes Across CC-RIX Lines
(A–K) Further analysis of cells subtypes, activation status, and functional profiles across CC-RIX at steady state. Frequencies of CD8+ cells that are Ki67+ (A), CD44+ (B), CD62L− (C) IFNγ+ (D), or IL-17+ (E) are shown at the top; frequencies of CD4+ cells that are Ki67+ (F), Tbet+ (G), IFNγ+ (H), IL-17+ (I), CD44+ (J), and CD62L− (K) are shown at the bottom. The average value for the indicated phenotype of each CC-RIX line (n = 3) is plotted in black in ascending order on the x axis. B6 (red) and BALB/c (blue) baseline values are plotted for comparison. All values represent uninfected controls.
Error bars show ± SEM.
Similarly, we examined a wide range of conventional CD4 T cell phenotypes based on cellular expression of activation markers and homing and effector molecules (Tables S1 and S3). As examples of the phenotypic diversity measured in CD4 T cells across different CC-RIX lines, we demonstrate a large range of Ki67+ CD4 T cells, from 3.8%–46.9% of total splenic CD4 T cells, whereas C57BL/6J and BALB/cJ mice had similar average frequencies of 8.9% and 8.5%, respectively (Figure 2F). Similarly, the fraction of CD4 T cells that were positive for the transcription factor Tbet, and thus likely to be Th1 cells, ranged from nearly 0% to 35.1% across CC-RIX lines, but, again, C57BL/6J and BALB/cJ mice had similar average frequencies of 2.5% and 2.3%, respectively (Figure 2G), underscoring the utility of CC-RIX mice to establish unique mouse models of immune phenotypes beyond what can be modeled using C56BL/6J or BALB/cJ mice. Additionally, there are large differences in the frequencies of splenic CD4+ T cells able to secrete IFNγ or IL-17 following a polyclonal ex vivo stimulus (Figures 2H and 2I) as well as antigen-experienced CD44+ or activated CD62L− CD4 T cells (Figures 2J and 2K). In conclusion, both this dataset and the CC-RIXs are an invaluable resource for investigators looking for an improved mouse model in which to study phenotypic diversity in T cells as well as for use in genetic mapping studies of T cell phenotypes.
Genetic Variation Results in Differential Immunoregulation
Tregs, defined by the lineage marker forkhead box P3 (Foxp3), promote self-tolerance and limit autoimmunity (Belkaid and Tarbell, 2009, Campbell and Koch, 2011, Kim et al., 2007), and altered Treg function is implicated in the manifestation of many autoimmune syndromes (Buckner, 2010). Most severely, in the complete absence of Tregs, as exemplified by Treg-deficient scurfy mice or individuals carrying non-functional versions of the Foxp3 gene, life-threatening multi-organ autoimmunity and lymphoproliferative diseases manifest in early development (Brunkow et al., 2001, Gambineri et al., 2003). Given the importance of Tregs in regulating conventional T cells, we extensively examined activation and homing molecule expression patterns on Tregs within CC-RIX lines. Most notably, there is a full range of Treg expression levels of the immunosuppressive marker CD73 across CC-RIX mice, from 3.5%–97.8% (Figure 3 A). Similarly, the average frequency of Tregs from CC-RIX lines that expressed CTLA-4, a critical suppressive mechanism, ranged from 11.2%–86.2%. Finally, there were wide ranges of Tregs expressing CD44 and homing molecule β1 integrin (CD29) across CC-RIX lines (Figures 3C and 3D). Thus, our thorough analysis demonstrates the variation in Treg-mediated immunoregulation dependent on host genetics, thus providing a rich resource for additional immunoregulatory and immunogenetics research.
Figure 3.

Differential Treg Activation across CC-RIX Lines
(A–D) Frequencies of Tregs that are CD73+ (A), CTLA-4+ (B), CD44+ (C), and CD29+ (D) across CC-RIX at steady state. The average value for the indicated phenotype of each CC-RIX line (n = 3) is plotted in black in ascending order on the x axis. B6 (red) and BALB/c (blue) baseline values are plotted for comparison. All values represent uninfected controls.
(E–G) Correlation between the number of Tregs and the frequency of Tregs expressing CTLA-4 (E), CXCR3 (F), or ICOS (G).
(H) Comparison of CC-RI lines (colored data points) that served as dams or sires in matings that resulted in RIX lines at the extreme ends of Treg frequency (Figure 2D) (black data points).
Error bars show ± SEM.
Because of the large variation in frequency of Tregs we observed in overtly healthy and age-matched mice, similar to what has been observed in humans (Figure 1E), we further interrogated for a possible relationship between the number of Tregs and their suppressive capacity to address the question of whether Treg function could compensate for altered Treg frequency to maintain the overall health of the host. First we performed a linear regression analysis of Treg number by CD4+ and CD8+ T cell proliferation, as indicated by Ki67 expression, or activation, indicated by CD44 or absence of CD62L expression. There was not a statistically significant correlation between Treg number and T cell proliferation or activation (Figure S3), suggesting that mice with varying numbers of Tregs are equally able to control T cell proliferation and activation. This finding supported our hypothesis that mice with lower Treg numbers may compensate for this by having increased Treg activation or Treg-suppressive capacity to control immune activation, similar to mice with a higher number of total Tregs. Thus, we tested for correlations between Treg number and the frequency of various Treg activation or suppression markers and found that there was indeed a statistically significant inverse correlation between the number of Tregs and the frequency of Tregs expressing CTLA-4, CXCR3, or inducible T cell costimulator (ICOS) (Figures 3E–3G). Thus, our data support a model in which mice with lower numbers of Tregs may compensate for this by having increased activation or suppressive capacity of Tregs to maintain tolerance and overall health. Alternatively, mice with less functional or suppressive Tregs may have a compensatory expansion, perhaps because reduced suppression of T cells, mediated by CTLA-4, for example, results in abundance of IL-2 that can thus support expansion of the Treg population. Together, our data demonstrate that mice on different genetic backgrounds support a wide range of Treg frequencies while maintaining health, at least in part by altering the suppressive function of Tregs.
Mouse Model Development Using the CC
As demonstrated above, there is tremendous phenotypic diversity generated within T cell subsets dependent on the natural standing genetic variation available within the CC. To provide an example of the utility of the CC for development of new mouse models for immunology research, we focused on the extreme diversity observed in the frequency of Tregs (Figure 1D). The vast majority of research on Tregs over the past 15 years has used C57BL/6J mice, which have a population of Tregs comprising approximately 10% of the total CD4 T cell compartment within the spleen. To identify potential mouse models with different Treg frequencies (e.g., akin to the diversity observed in human cohorts; Figure 1E), we further examined the frequency of Tregs in CC-RI strains that served as dams or sires in matings that resulted in CC-RIX lines at the extreme ends of Treg frequency (Figure 1D). We found that CC-RI strains CC030/GeniUnc, CC061/GeniUnc, CC051/TauUnc, and CC003/Unc had average Treg frequencies of 12.03%, 7.05%, 8.47%, and 4.66%, respectively (Figure 3H). Notably, none of these RI lines had Treg frequencies that were elevated or decreased to the extremes observed in the CC-RIX screen, although CC030 does have a consistently lower frequency of Tregs and might represent a model of decreased Treg activity. These results highlight the potential for emergent phenotypes within heterozygous animals (e.g., because of epistatic genetic interactions) and further underscore the utility of assessing F1 crosses to increase both the genetic and phenotypic diversity assessed.
QTL Mapping Using the CC
The CC was initially conceived of as a powerful resource for genetic mapping studies and integrated systems genetics approaches (Churchill et al., 2004). To measure the amount of phenotypic variation attributable to genetic differences, we calculated the intraclass correlation (ICC) (Gelman and Hill, 2007) measures as an estimate of heritability (Table S4). In addition, to demonstrate the overall immune system phenotypic diversity among CC-RIX lines, we performed principal-component analysis (PCA) using the flow cytometry measures listed in Table S4 (using strain average values) and then plotted the top two principal components (PCs) to show strain clustering within these composite immune phenotypes (Figure S4). This analysis demonstrates that there is tremendous overall immune system phenotypic diversity across the CC-RIX and not just diversity within single immune measures, as demonstrated in Figures 1, 2, and 3. Further, although there is diversity among the lines with regard to multiple different cell populations, there are also a number of lines with quite extreme phenotypes that may thus be suitable for follow-up studies.
Next, to demonstrate the utility of the CC for identifying QTLs contributing to population-wide variations in steady-state immune phenotypes, we performed QTL mapping on select T cell phenotypes using our flow cytometry data in combination with haplotype reconstructions of the CC strains generated from the mouse universal genotyping array (Mega-MUGA) platform (Morgan et al., 2015). Through these proof-of-concept studies, we identified several QTL candidates for follow-up, and below we highlight examples chosen because of high logarithm of the odds ratio (LOD) scores and clear founder effects. First, we found a QTL (Host Immunity 1, HI1) driving the frequency of CD73+ Tregs within the X chromosome near position 160 Mb-telomere (Figure 4 A) that appears to be driven largely by a PWK/PhJ founder effect (Figure 4B). Confirming this locus, CC-RIX lines with a PWK/PhJ haplotype at position 166 Mb of the X chromosome have frequencies of CD73+ Tregs that are on the upper end of the range we observed across all CC-RIXs examined (Figures 3A and 4C). This has clear implications on host immunoregulation at steady state because there is an inverse correlation between CD73+ Treg frequency and the frequency of activated CD8 or CD4 T cells (Figure 4D).
Figure 4.

QTL Mapping Using the CC
(A and B) A QTL driving the frequency of CD73+ Tregs found within the X chromosome near position 160 MB-telomere that appears to be driven largely by a PWK/PhJ founder effect (B). In (A), the solid red line indicates the LOD score threshold for p = 0.05, and the dashed line indicates p = 0.01.
(C) With the frequency of CD73+ Tregs on the x axis, we see that CC-RIX lines with PWK/PhJ variants at position 166Mb of the X chromosome have CD73+Treg frequencies at the upper end of the range observed across all CC-RIX examined.
(D) Implications on host immunoregulation at the basal state because there is an inverse correlation between CD73+ Treg frequency and the frequency of activated CD8 or CD4 T cells.
To narrow our QTL regions to likely candidate genes, we utilized whole-genome sequence information from the 8 founder strains of the CC (Keane et al., 2011). Guided by the allele effects underlying the QTL analysis (estimates of in which founder haplotype(s) causative genetic variants occur), we sought to identify genetic variants within the QTL regions that were consistent with allele effects underlying the QTL and also had an effect on the amino acid sequence and/or splice forms of a gene because these variants would be the highest-priority candidates for altering protein function. Accordingly, within the QTL, a total of 102,596 SNPs and 20,432 insertions or deletions (indels) segregate across the CC population. Of these, 248 SNPs and 14 indels cause coding or splice differences across 43 genes. Because a PWK/PhJ allele was the sole allele contributing to phenotypic differences, we focused on private PWK/PhJ variants. We identified 36 private PWK/PhJ SNPs and 2 indels within 22 genes (Table 1 ) as potential causal candidates.
Table 1.
Candidate Genes Driving Select Phenotypes of Interest
| Phenotype | QTL Region | Founder Effects | Candidate Genes in Region |
|---|---|---|---|
| Frequency of CD73+ Tregs | ChrX: 160-Mb telomere | PWK high | Arhgap6, Asb11, Ctps2, Egfl6, Figf, Frmpd4, Gemin8, Gja6, Gp464, Map3k15, Mospd2, Nhs, Ofd1, Phka2, Piga, Reps2, Rnf138rt1, Syap1, Tceanc, Tlr7, Tlr8, Zsr2, Zrsr2 |
| Frequency of CXCR3+ Tregs | ChrX: 100–106 Mb | PWK low | 1700011 M02Rik, 8030474 K03Rik, Abcb7, Atrx, Awat1, C77370, Cxcr3, Dgat2l6, Dmrtc1b, Gm5166, Gm9112, Itgb1bp2, Kif4, Magee1, Otud6a, Phka1, Rps4x, Taf1, 170031F 05Rik, Zrsr2 |
| Frequency of CXCR3+ Tregs | ChrX: 100–106 Mb | NZO high | Nhsl2, Kif4, Rgag4, Nhsl2, Asmt |
| Frequency of ICOS+ Tregs | ChrX: 140–145 Mb | PWK high | Ammercr1, Col4a5, Col4a6, E230019M04Rik, Gucy2f, Irs4, Nxt2, Rgag1, Tmem164, Vsig1, Prps1 |
In addition to this QTL, we also identified a highly significant QTL (HI2) within the X chromosome at position 100–106 Mb driving the frequency of CXCR3+ Tregs (Figure 5 A), CXCR3+ CD4+, and CD8+ T cells (data not shown). When we examined the founder effects, we found that PWK/PhJ again was a significant driver of this QTL for all three phenotypes (Figure 5B and data not shown), with a clear trend toward low levels of CXCR3 expression on all three subsets of T cells of CC-RIX lines when there were PWK/PhJ variants at position 105.5 Mb on the X chromosome (Figures 5C and 5D and data not shown). However, this QTL had a more complex pattern of allele effects depending on the phenotype. Although, in all cases, a PWK/PhJ allele drives a decreased frequency of CXCR3+ T cells for any of the three subsets of T cells, there is additional evidence for phenotype-specific allele effects leading to increases in frequency of each of these specific cell types. Specifically, there is a WSB/EiJ haplotype associated with increased CXCR3+ CD8+ T cells and also an NZO/HILtJ haplotype increasing CXCR3+ Tregs (Figures 5B and 5E). In this 6-Mb region, a total of 55,082 SNPs and 12,377 indels segregate within the CC. This includes 149 nonsynonymous SNPs and 11 indels across 42 genes. PWK has 35 private SNPs and 5 private indels in 26 genes; NZO has 4 SNPs and 1 indel in 4 genes (Table 1). WSB/EiJ has no private SNPs or indels within this region, suggesting that the effects of the WSB/EiJ haplotype may be due to regulatory variation. Of note, genes within this X chromosome QTL driving the low frequency of CXCR3+ T cells include Cxcr3 itself (Table 1), likely demonstrating novel gene variants that result in altered expression levels within T cells. CXCR3 is a critical chemokine receptor used by T cells and other immune cells to migrate to tissue sites of infection and sites of autoimmune reactions, and Tbet+ CXCR3+ Tregs have been shown to provide protection from autoimmune diabetes (Tan et al., 2016). Further study of CC-RI or CC-RIX with the PWK/PhJ haplotype in this region could better model human disease associated with altered T cell migratory activity and also be used to study the importance and function of this chemokine receptor itself.
Figure 5.

A Highly Significant QTL within the X Chromosome Drives the Frequency of CXCR3+ Tregs
(A–E) A QTL was identified within the X chromosome at position 100–106 Mb (A), driven by PWK/PhJ (B) in all cases with a trend toward lower levels of CXCR3 expression on T cell subsets of CC-RIX lines with PWK/PhJ variants at position 105.5 Mb on the X chromosome (C and D). Phenotypic-specific allele effects lead to increases in a single phenotype because there is an NZO/HILtJ effect associated with increased frequency of CXCR3+ Tregs (B) and a WSB/EiJ effect associated with increased frequency of CXCR3+ CD8+ T cells (E). In (A), the solid red line indicates the LOD score threshold for p = 0.05, and the dashed line indicates p = 0.01.
Error bars show ± SEM.
Finally, we identified a third candidate QTL within the X chromosome at position 140–145 Mb (HI3) driving the frequency of ICOS+ Tregs in the spleen (Figure 6 A). Although this ICOS+ Treg result does not meet the threshold for statistical significance in our analysis, the allele effects are clear, and we include it here as an example of the diverse allele effects seen in the RIX population. An examination of the founder allele effects at this locus revealed that the PWK/PhJ haplotype was associated with an increased frequency of ICOS+ Tregs (Figures 6B and 6C), accounting for the extreme frequency at the higher ranges (Figure 6D). However, there also appeared to be a second, NZO/HILtJ allele that had an intermediate increase in ICOS+ Tregs relative to the other six haplotypes (Figure 6B). Within this region, there are a total of 51,137 SNPs and 10,884 indels segregating in the CC. Of these, 58 SNPs and 5 indels cause coding or splice differences in 18 genes. PWK/PhJ has 17 private SNPs and 2 private indels across 11 genes (Table 1). In contrast, NZO/HILtJ has no SNPs or indels, either private or shared, with PWK/PhJ in this region, suggesting the potential for regulatory variants to play a role in this locus. In summary, we highlight examples of QTL analysis for immunoregulatory phenotypes at homeostasis, with several potential candidate genes under the QTL identified for follow-up studies.
Figure 6.

A QTL Driving the Frequency of ICOS+ Tregs in the Spleen
(A) A third QTL within the X chromosome at position 140–145 Mb drives the frequency of ICOS+ Tregs in the spleen. The solid red line indicates the LOD score threshold for p = 0.05, and the dashed line indicates p = 0.01.
(B–D) An examination of founder effects revealed that the PWK/PhJ allele at position 141.71 Mb drives a high frequency of ICOS+ Tregs (B and C), accounting for the extreme frequency at the higher ranges (D).
Error bars show ± SEM.
Discussion
The highly genetically diverse CC was conceived as an experimental system that could be used to understand how genetic variation could affect a variety of complex traits while still maintaining the benefit of reproducibility and the legion of other advantages of a small animal model, such as a controlled environment, diet, age, sex, and pathogen exposure. An early study using individual mice from 66 strains from incipient lines of the CC or pre-CC demonstrated diversity in the steady-state frequency of subsets of lymphocytes and antigen-presenting cells that was larger than that detected in the eight founder strains (Phillippi et al., 2014); here, we use the completed CC to extend these initial observations using replicate animals and a wider and comprehensive range of phenotypic analyses. A number of studies have shown that genetic variants circulating within the CC population can lead to divergent responses to pathogens (Elbahesh and Schughart, 2016, Ferris et al., 2013, Graham et al., 2015, Graham et al., 2016, Gralinski et al., 2015, Leist et al., 2016, Lorè et al., 2015, Rasmussen et al., 2014), inflammatory diseases (Rogala et al., 2014), and drug responses (Nachshon et al., 2016). Here we advance and expand this growing body of work with a demonstration of the unique benefits for immunology research. CC-RI and CC-RIX lines can provide exciting new and reproducible models for cellular immunology research and provide us with a critical resource for mapping immune phenotypes to genetic loci and genes.
Importantly, each cellular immune phenotype we examined as part of our screen resulted in a wide range of responses at homeostasis, with increased diversity beyond the most commonly used laboratory inbred strains of C56BL/6J and BALB/cJ. This consistent range of differences between CC recombinant inbred lines extends our ability to model human diversity in T cell frequency and phenotype and, potentially, the unique resultant infection or disease outcomes, such as autoimmunity. For example, CC-RIX lines or CC-RI strains at the tails of the phenotypic distribution plots are useful for elucidating the underlying mechanisms of the high or low phenotype of interest. For example, as identified in Figure 3H, CC-RI strain CC003/Unc and CC-RIX lines CC003xCC062 and CC070xCC003 all have splenic Treg frequencies that are about half of that measured in C57BL/6J mice (Figure 1D), although these lines are all overtly healthy and display no signs of autoimmunity within the 8- to 10-week age range. It has been previously noted that BALB/c mice have more CD4+CD25+ Tregs compared with C57BL/6 mice, including greater suppression of their CD4+CD25− responder T cells (Chen et al., 2005), but the genetic loci regulating this difference were not identified. Here we confirm this finding of differential Treg frequency between Balb/cJ and C57BL/6J as well as within the CC-RIX (Figure 1D), although, because of the large and high-throughput nature of our screen, we cannot draw conclusions about the suppressive capacity of the Tregs across the CC-RIX lines because in vitro suppression assays were not performed for each of the lines screened. Important follow-up studies include an examination of Treg suppressive capacity through use of standard suppression assays to determine whether any of the CC-RIX lines represent unique mouse models of extreme immunoregulation mediated by Treg suppression. Further, such studies could be used to map the gene(s) responsible for differential Treg activity, which could have critical downstream implications for susceptibility to infection, autoimmunity, and tumorigenesis.
In addition to provision of mouse models for cell type- and disease-specific research, we demonstrate that the T cell phenotypic diversity present within the CC affords us an unprecedented ability to perform genetic mapping to identify loci controlling adaptive immune phenotypes and responses. Through the proof-of-concept QTL analyses performed by our group (Figures 4, 5, and 6), we identified 3 QTLs underlying distinct immunoregulatory phenotypes at steady state. Our ability to identify effects on the X chromosome was increased compared with autosomes because our mapping population consists of males only. In this case, significance thresholds for the X chromosome are somewhat lower than for autosomes because of the hemizygous genotypes of males because there are no heterozygotes. Notably, a candidate gene under the X chromosome QTL driving very low expression of CXCR3 on Tregs, CD4, and CD8 T cells is Cxcr3 itself, suggestive of a deleterious allele resulting in lowered levels of protein expression. Because CXCR3 is a chemokine receptor important for lymphocyte migration toward chemokines CXCL9, CXCL10, and CXCL10, which are generally expressed in tissue regions of inflammation (Groom and Luster, 2011), mouse lines with this allele may represent a unique model in which to study the importance of this signaling pathway in various disease or infection states. In humans, CXCR3 polymorphisms have been associated with the risk of asthma (Cheong et al., 2005), pointing to a potential future use of the CC-RIX to improve mouse models of asthma research to better recapitulate human disease states. Additionally, the list of candidate genes provided in Table 1 requires follow-up studies to narrow in on the particular gene or genes causing the phenotypes of interest.
Genetic variants driving phenotypic differences can be due to both coding and regulatory differences. Indeed, many of the most significant human genome-wide association study (GWAS) hits occur in regulatory regions (Zhang and Lupski, 2015). Here we present an initial analysis where we instead focused on variants affecting protein sequence. Many studies in the CC have shown that differences in coding sequence can have large effects on phenotypic outcomes (Ferris et al., 2013, Gralinski et al., 2015). Furthermore, regulatory variants are notoriously difficult to identify because of their condition and cell type-specific effects on gene expression, which itself does not always lead to differences in protein levels (Chick et al., 2016). Nevertheless, differentiating between protein functional and expression level differences in phenotypic responses provides critical hypotheses to address in future studies of the functions of variant regions on immune homeostasis.
Our study and the power of our dataset have several limitations. First, we cannot determine a role for distinct microbiota within CC-RIX lines or animals in driving some of the divergent immune phenotypes. Mice of different lines were not co-housed or co-fostered, although they were all bred in the same room of the same facility by the same technician across the entire study. Similarly, experiments were conducted in a consistent facility, so together, these factors should minimize the effect of a variety of environmental factors. However, the microbiome could explain some of the observed variation, and although a study of this magnitude, performed over the course of 4 years, did not allow for an extensive examination of the microbiome as well, future studies will likely investigate the role of host genetics on microbiome colonization as well as downstream effects of distinct microbial communities on immune phenotypes. An additional limitation of our study is that haplotype blocks are fairly large within this CC-RIX population. However, in contrast to classical 2-allele mapping populations, the multiple founder haplotypes do allow us to reduce large haplotype blocks to much fewer numbers of candidate features, which we did here as an illustrative example of the use of this resource and dataset.
In conclusion, we advance that the CC is an invaluable resource for many types of immunology, immunogenetics, and disease research, as we have specifically demonstrated in the context of steady-state T cell phenotypes and frequencies. This dataset will be useful for individuals seeking to identify new mouse models with increased diversity of T cell phenotypes more akin to human diversity as well as for additional QTL mapping studies. Finally, the data presented here provide a gateway for CC strain selection for future studies of cancer immunity, autoimmune conditions, and various infections because investigators can tailor selection of strains based on the frequencies of particular T cell subsets as well as their activation status, steady-state cytokine expression, and other phenotypic selection parameters (Document S1. Figures S1–S4 and Tables S1–S3, Table S4. CC-RIX Immunophenotype Dataset, Related to Figures 1, 2, 3, 4, 5, and 6). As the need for mechanistic studies of immunity and disease persists, including the genetic mechanisms underlying such states, we propose that the CC represents an outstanding and unique model that incorporates genetic diversity while at the same time retaining beneficial features of mouse models, such as reproducibility, low cost, and ubiquity and availability of reagents and tools.
Experimental Procedures
Mice
CC RI mice were obtained from the Systems Genetics Core Facility at the University of North Carolina, Chapel Hill (UNC) (Welsh et al., 2012). For the screen, CC-RIX lines were bred at UNC under SPF conditions. 6- to 8-week-old F1 hybrid male mice were transferred from UNC to the University of Washington and housed directly in a BSL-2+ laboratory within an SPF barrier facility. Age- and sex-matched 8- to 10-week-old mice were used for all experiments. All animal experiments were approved by the University of Washington Institutional Animal Care and Use Committee. The Office of Laboratory Animal Welfare of the NIH approved UNC (A3410-01) and the University of Washington (A3464-01), and this study was carried out in strict compliance with the Public Health Service (PHS) Policy on Humane Care and Use of Laboratory Animals. Genotypes of interest in this study, H2b, were obtained from the Systems Genetics Core Facility at UNC (Welsh et al., 2012).
Cell Preparation for Flow Cytometry Assays
Following euthanasia, mice were perfused with 10 mL PBS to remove any residual intravascular leukocytes. Spleens were homogenized, treated with ammonium chloride potassium (ACK) lysis buffer to remove red blood cells, washed, and resuspended in fluorescence-activated cell sorting (FACS) buffer (1× PBS, 0.5% fetal bovine serum [FBS]). Cells were counted by hemacytometer using trypan blue exclusion.
Flow Cytometry Analysis
Following preparation of single-cell suspensions, cells were plated at 1 × 106 cells/well and stained for surface markers for 15 min on ice. Cells were subsequently fixed, permeabilized (Foxp3 fixation/permeabilization concentrate and diluent, eBioscience) and stained intracellularly with antibodies for 30 min on ice. Flow cytometry was performed on a BD LSRII machine using BD FACSDiva software. Analysis was performed using FlowJo software. The following directly conjugated antibodies were used: CD3-ECD (145-2C11), CD4-BV605 (RM4-5), CD8-BV650 (53-6.7), Foxp3-Alexa 700 (FJK-16S), CD44-FITC (IM7), CD62L-Alexa 700 (MEL-14), IFNγ-PerCP eFluor710 (XMG1.2), IL-17-fluorescein isothiocyanate (FITC) (TC11-18H10.1), Tbet-PECy7 (4B10), CD73-BV421 (TY/11.8), CTLA-4-antigen presenting cell (APC) (UC10-4B9), ICOS-PECy5 (7E.17G9), CD29-APC Cy7 (HMb1-1), CXCR3-PerCP eFluor710 (CXCR3-173), and Ki67-FITC (SolA15). AmCyan live/dead stain (Invitrogen) was used in all panels for identification of live cells. Gating schemes and trees used for flow cytometry analysis are shown in Figure S1. To measure the amount of phenotypic variation attributable to genetic differences, ICC values were calculated for all flow measures (Gelman and Hill, 2007).
Measurement of Human Treg Frequency
Cryopreserved peripheral blood mononuclear cells (PBMCs) were obtained from individuals participating in the Partners PrEP Study (ClinicalTrials.gov number NCT00557245). Study procedures have been described previously (Baeten et al., 2012). For the present analysis, samples were selected from men and women who were HIV-negative. The procedures of the Partners PrEP Study, including collection of samples for immunologic assays, were approved by the institutional review boards of the University of Washington and collaborating site institutions; participants provided written informed consent.
PBMCs from 244 individuals were thawed and cultured in R10. Counts and viability were acquired using the TC-20 automated cell counter (Bio-Rad). Tregs were stained with the Live/Dead Fixable Aqua Dead Cell Stain Kit from Molecular Probes (OR, USA), followed by cell surface staining with the appropriate cell panel. Surface markers examined for phenotype staining protocol included CD3 (OKT3 and HIT3a), CD4 (OKT4), CD25 (BC96), and CD127 (A019D5) from BioLegend (CA, USA) and CD8 (SK1) from eBioscience(CA, USA). Intracellular markers examined included FoxP3 (236 A/E7) from eBioscience (CA, USA). Immediately following staining, samples were analyzed using a LSRII flow cytometer (BD Biosciences, CA, USA) with FlowJo software (Tree Star OR). Tregs were considered to be CD3+CD4+CD25hiFoxp3+CD127dim cells, as published previously (Pattacini et al., 2016).
Statistical Analysis
When comparing groups, two-tailed unpaired Student’s t tests were conducted, with p < 0.05 considered significant. Error bars show ± SEM or ± SD.
Principal Component Analysis
PCA was performed using all measures listed in Table S4. For all measures, average values for each CC-RIX line were calculated. The proportion of missing values was examined for each CC-RIX line and each flow cytometry measure. CC-RIX lines and measures with more than 15% missing values were excluded from the analysis. The remaining missing values were replaced with the average value across all lines. Finally, each measure was scaled and centered before performing PCA.
Calculation of ICC (Estimate of Heritability)
A random effects model was fit for each flow variable,
where yij is the i-th observation for the j-th group, μ is the overall mean, αj is a random effect, and εij is the error. ICC is defined below, where σ2α is the variance explained by the grouping factor (RIX line), and σ2ε is the variance not explained by the grouping factor:
QTL Mapping
QTL mapping was done as described previously (Aylor et al., 2011, Ferris et al., 2013). Briefly, the haplotypic makeup of each CC-RIX was determined based on the consensus most recent common ancestor (MRCA) for each CC-RI line. Because each RIX is an F1, the haplotype probabilities were averaged between the two strains for each of the autosomes, whereas the X chromosome for each RIX was determined solely with the mother strain’s haplotypic makeup (because these CC-RIX were males, they all have one copy of the X chromosome inherited from their dam). The DOQTL package conducts an eight-variable (the probability of each of the eight founder haplotypes at a locus) regression on probabilities framework at each marker of interest, looking to find significant associations between the founder haplotype probabilities at a marker with the associated phenotypes present within the RIX. Genome-wide significance is determined by permutation testing (randomly shuffling the phenotypic values across the CC-RIX and asking for the most significant response value). Allele effects at each locus are determined by taking the effect estimate for each of the eight founder haplotypes (the β within a linear regression framework).
To determine likely causative variants at a locus, we used the allele effects to identify the largest split between effects at a QTL. When there were two splits of roughly equal magnitude, then we assumed that there were 3 allele groups with a more complex SNP pattern. We took all of the called genetic variants within the QTL based on the Sanger whole-genome sequences of the eight founder strains and looked at the variant distribution pattern. As described in the results, we filtered these variants based on potential effect on protein sequence to derive lists of higher-priority candidate genes.
An additive haplotype regression model implemented in the DOQTL R (Team, 2016) package was used for QTL mapping analyses (Gatti et al., 2014). Briefly, QTL genome scans are performed by regressing the phenotype on genotype probabilities for each of the eight founder strains. A random-effect term is included in the model to account for kinship among animals. The DOQTL software also requires sex to be included as a covariate in the regression model, although all of our samples are male. An LOD score for each marker is calculated from the likelihood ratio comparing the regression model described above to a regression model without the founder genotype probabilities. The statistical significance of LOD scores is determined via a permutation test. A threshold of p ≤ 0.05 was used to select significant associations.
Author Contributions
Conceptualization, J.B.G. and J.M.L.; Methodology, J.B.G., M.T.F., and J.M.L.; Formal Analysis, J.B.G., M.M., G.C., S.J., S.M., M.T.F., and J.M.L.; Investigation, J.B.G. and J.S.; Resources, D.R.M. and M.T.F.; Data Curation, J.B.G., M.M., G.C., and S.M.; Writing – Original Draft, J.B.G. and J.M.L.; Writing – Review & Editing, J.B.G., J.L.S., M.M., G.C., S.J., D.R.M., M.T.F., S.M., and J.M.L.; Visualization, J.B.G. and M.M.; Supervision, J.B.G., S.M., and J.M.L.
Acknowledgments
Funding for this study was provided by NIH grants U19AI100625 (Project 4 and Cores C and D) (to M.T.F., S.M., and J.M.L.) and R01 AI096968 and AI087657 (to J.M.L.). We wish to thank our collaborators in the Systems Immunogenetics Group for helpful discussions and generation of mice. In particular, we wish to thank Ginger Shaw for generating the RIX mice used in this study.
Published: November 21, 2017
Footnotes
Supplemental Information includes four figures and four tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2017.10.093.
Data and Software Availability
The data that support the findings of this study are included in Tables S2, S3, and S4 and are available via ImmPort: SDY1176.
Supplemental Information
References
- Aylor D.L., Valdar W., Foulds-Mathes W., Buus R.J., Verdugo R.A., Baric R.S., Ferris M.T., Frelinger J.A., Heise M., Frieman M.B. Genetic analysis of complex traits in the emerging Collaborative Cross. Genome Res. 2011;21:1213–1222. doi: 10.1101/gr.111310.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeten J.M., Donnell D., Ndase P., Mugo N.R., Campbell J.D., Wangisi J., Tappero J.W., Bukusi E.A., Cohen C.R., Katabira E., Partners PrEP Study Team Antiretroviral prophylaxis for HIV prevention in heterosexual men and women. N. Engl. J. Med. 2012;367:399–410. doi: 10.1056/NEJMoa1108524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkaid Y., Tarbell K. Regulatory T cells in the control of host-microorganism interactions (∗) Annu. Rev. Immunol. 2009;27:551–589. doi: 10.1146/annurev.immunol.021908.132723. [DOI] [PubMed] [Google Scholar]
- Bogue M.A., Grubb S.C. The Mouse Phenome Project. Genetica. 2004;122:71–74. doi: 10.1007/s10709-004-1438-4. [DOI] [PubMed] [Google Scholar]
- Boguski M.S. Comparative genomics: the mouse that roared. Nature. 2002;420:515–516. doi: 10.1038/420515a. [DOI] [PubMed] [Google Scholar]
- Brunkow M.E., Jeffery E.W., Hjerrild K.A., Paeper B., Clark L.B., Yasayko S.A., Wilkinson J.E., Galas D., Ziegler S.F., Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- Buckner J.H. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat. Rev. Immunol. 2010;10:849–859. doi: 10.1038/nri2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell D.J., Koch M.A. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat. Rev. Immunol. 2011;11:119–130. doi: 10.1038/nri2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Oppenheim J.J., Howard O.M. BALB/c mice have more CD4+CD25+ T regulatory cells and show greater susceptibility to suppression of their CD4+CD25- responder T cells than C57BL/6 mice. J. Leukoc. Biol. 2005;78:114–121. doi: 10.1189/jlb.0604341. [DOI] [PubMed] [Google Scholar]
- Cheong H.S., Park C.S., Kim L.H., Park B.L., Uh S.T., Kim Y.H., Lym G.I., Lee J.Y., Lee J.K., Kim H.T. CXCR3 polymorphisms associated with risk of asthma. Biochem. Biophys. Res. Commun. 2005;334:1219–1225. doi: 10.1016/j.bbrc.2005.07.019. [DOI] [PubMed] [Google Scholar]
- Chesler E.J. Out of the bottleneck: the Diversity Outcross and Collaborative Cross mouse populations in behavioral genetics research. Mamm. Genome. 2014;25:3–11. doi: 10.1007/s00335-013-9492-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chick J.M., Munger S.C., Simecek P., Huttlin E.L., Choi K., Gatti D.M., Raghupathy N., Svenson K.L., Churchill G.A., Gygi S.P. Defining the consequences of genetic variation on a proteome-wide scale. Nature. 2016;534:500–505. doi: 10.1038/nature18270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill G.A., Airey D.C., Allayee H., Angel J.M., Attie A.D., Beatty J., Beavis W.D., Belknap J.K., Bennett B., Berrettini W., Complex Trait Consortium The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat. Genet. 2004;36:1133–1137. doi: 10.1038/ng1104-1133. [DOI] [PubMed] [Google Scholar]
- Collaborative Cross Consortium The genome architecture of the Collaborative Cross mouse genetic reference population. Genetics. 2012;190:389–401. doi: 10.1534/genetics.111.132639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbahesh H., Schughart K. Genetically diverse CC-founder mouse strains replicate the human influenza gene expression signature. Sci. Rep. 2016;6:26437. doi: 10.1038/srep26437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris M.T., Aylor D.L., Bottomly D., Whitmore A.C., Aicher L.D., Bell T.A., Bradel-Tretheway B., Bryan J.T., Buus R.J., Gralinski L.E. Modeling host genetic regulation of influenza pathogenesis in the collaborative cross. PLoS Pathog. 2013;9:e1003196. doi: 10.1371/journal.ppat.1003196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerer M., Jiang W., Holler P.D., Satpathy A., Campbell C., Bogue M., Mathis D., Benoist C. Enhanced thymic selection of FoxP3+ regulatory T cells in the NOD mouse model of autoimmune diabetes. Proc. Natl. Acad. Sci. USA. 2007;104:18181–18186. doi: 10.1073/pnas.0708899104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambineri E., Torgerson T.R., Ochs H.D. Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked inheritance (IPEX), a syndrome of systemic autoimmunity caused by mutations of FOXP3, a critical regulator of T-cell homeostasis. Curr. Opin. Rheumatol. 2003;15:430–435. doi: 10.1097/00002281-200307000-00010. [DOI] [PubMed] [Google Scholar]
- Gatti D.M., Svenson K.L., Shabalin A., Wu L.Y., Valdar W., Simecek P., Goodwin N., Cheng R., Pomp D., Palmer A. Quantitative trait locus mapping methods for diversity outbred mice. G3 (Bethesda) 2014;4:1623–1633. doi: 10.1534/g3.114.013748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman A., Hill J. Cambridge University Press; Cambridge, New York: 2007. Data analysis using regression and multilevel/hierarchical models. [Google Scholar]
- Gondo Y., Fukumura R., Murata T., Makino S. ENU-based gene-driven mutagenesis in the mouse: a next-generation gene-targeting system. Exp. Anim. 2010;59:537–548. doi: 10.1538/expanim.59.537. [DOI] [PubMed] [Google Scholar]
- Graham J.B., Thomas S., Swarts J., McMillan A.A., Ferris M.T., Suthar M.S., Treuting P.M., Ireton R., Gale M., Jr., Lund J.M. Genetic diversity in the collaborative cross model recapitulates human West Nile virus disease outcomes. MBio. 2015;6 doi: 10.1128/mBio.00493-15. e00493-e004915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham J.B., Swarts J.L., Wilkins C., Thomas S., Green R., Sekine A., Voss K.M., Ireton R.C., Mooney M., Choonoo G. A mouse model of chronic West Nile virus disease. PLoS Pathog. 2016;12:e1005996. doi: 10.1371/journal.ppat.1005996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gralinski L.E., Ferris M.T., Aylor D.L., Whitmore A.C., Green R., Frieman M.B., Deming D., Menachery V.D., Miller D.R., Buus R.J. Genome wide identification of sars-cov susceptibility loci using the Collaborative Cross. PLoS Genet. 2015;11:e1005504. doi: 10.1371/journal.pgen.1005504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groom J.R., Luster A.D. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol. Cell Biol. 2011;89:207–215. doi: 10.1038/icb.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb S.C., Churchill G.A., Bogue M.A. A collaborative database of inbred mouse strain characteristics. Bioinformatics. 2004;20:2857–2859. doi: 10.1093/bioinformatics/bth299. [DOI] [PubMed] [Google Scholar]
- Holler P.D., Yamagata T., Jiang W., Feuerer M., Benoist C., Mathis D. The same genomic region conditions clonal deletion and clonal deviation to the CD8alphaalpha and regulatory T cell lineages in NOD versus C57BL/6 mice. Proc. Natl. Acad. Sci. USA. 2007;104:7187–7192. doi: 10.1073/pnas.0701777104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane T.M., Goodstadt L., Danecek P., White M.A., Wong K., Yalcin B., Heger A., Agam A., Slater G., Goodson M. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011;477:289–294. doi: 10.1038/nature10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.M., Rasmussen J.P., Rudensky A.Y. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- Leist S.R., Pilzner C., van den Brand J.M., Dengler L., Geffers R., Kuiken T., Balling R., Kollmus H., Schughart K. Influenza H3N2 infection of the collaborative cross founder strains reveals highly divergent host responses and identifies a unique phenotype in CAST/EiJ mice. BMC Genomics. 2016;17:143. doi: 10.1186/s12864-016-2483-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorè N.I., Iraqi F.A., Bragonzi A. Host genetic diversity influences the severity of Pseudomonas aeruginosa pneumonia in the Collaborative Cross mice. BMC Genet. 2015;16:106. doi: 10.1186/s12863-015-0260-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masopust D., Sivula C.P., Jameson S.C. Of mice, dirty mice, and men: using mice to understand human immunology. J. Immunol. 2017;199:383–388. doi: 10.4049/jimmunol.1700453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan A.P., Fu C.P., Kao C.Y., Welsh C.E., Didion J.P., Yadgary L., Hyacinth L., Ferris M.T., Bell T.A., Miller D.R. The mouse universal genotyping array: from substrains to subspecies. G3 (Bethesda) 2015;6:263–279. doi: 10.1534/g3.115.022087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafavi S., Ortiz-Lopez A., Bogue M.A., Hattori K., Pop C., Koller D., Mathis D., Benoist C., Immunological Genome Consortium Variation and genetic control of gene expression in primary immunocytes across inbred mouse strains. J. Immunol. 2014;193:4485–4496. doi: 10.4049/jimmunol.1401280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mountz J.D., Van Zant G.E., Zhang H.G., Grizzle W.E., Ahmed R., Williams R.W., Hsu H.C. Genetic dissection of age-related changes of immune function in mice. Scand. J. Immunol. 2001;54:10–20. doi: 10.1046/j.1365-3083.2001.00943.x. [DOI] [PubMed] [Google Scholar]
- Nachshon A., Abu-Toamih Atamni H.J., Steuerman Y., Sheikh-Hamed R., Dorman A., Mott R., Dohm J.C., Lehrach H., Sultan M., Shamir R. Dissecting the effect of genetic variation on the hepatic expression of drug disposition genes across the Collaborative Cross mouse strains. Front. Genet. 2016;7:172. doi: 10.3389/fgene.2016.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattacini L., Baeten J.M., Thomas K.K., Fluharty T.R., Murnane P.M., Donnell D., Bukusi E., Ronald A., Mugo N., Lingappa J.R., Partners PrEP Study Team Regulatory T-cell activity but not conventional HIV-specific T-cell responses are associated with protection from HIV-1 infection. J. Acquir. Immune Defic. Syndr. 2016;72:119–128. doi: 10.1097/QAI.0000000000000919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paula M.O., Fonseca D.M., Wowk P.F., Gembre A.F., Fedatto P.F., Sérgio C.A., Silva C.L., Bonato V.L. Host genetic background affects regulatory T-cell activity that influences the magnitude of cellular immune response against Mycobacterium tuberculosis. Immunol. Cell Biol. 2011;89:526–534. doi: 10.1038/icb.2010.116. [DOI] [PubMed] [Google Scholar]
- Petkova S.B., Yuan R., Tsaih S.W., Schott W., Roopenian D.C., Paigen B. Genetic influence on immune phenotype revealed strain-specific variations in peripheral blood lineages. Physiol. Genomics. 2008;34:304–314. doi: 10.1152/physiolgenomics.00185.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillippi J., Xie Y., Miller D.R., Bell T.A., Zhang Z., Lenarcic A.B., Aylor D.L., Krovi S.H., Threadgill D.W., de Villena F.P. Using the emerging Collaborative Cross to probe the immune system. Genes Immun. 2014;15:38–46. doi: 10.1038/gene.2013.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen A.L., Okumura A., Ferris M.T., Green R., Feldmann F., Kelly S.M., Scott D.P., Safronetz D., Haddock E., LaCasse R. Host genetic diversity enables Ebola hemorrhagic fever pathogenesis and resistance. Science. 2014;346:987–991. doi: 10.1126/science.1259595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly K.M. Using the Collaborative Cross to study the role of genetic diversity in cancer-related phenotypes. Cold Spring Harb. Protoc. 2016;2016 doi: 10.1101/pdb.prot079178. pdb prot079178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogala A.R., Morgan A.P., Christensen A.M., Gooch T.J., Bell T.A., Miller D.R., Godfrey V.L., de Villena F.P. The Collaborative Cross as a resource for modeling human disease: CC011/Unc, a new mouse model for spontaneous colitis. Mamm. Genome. 2014;25:95–108. doi: 10.1007/s00335-013-9499-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvetti M., Ristori G., Bomprezzi R., Pozzilli P., Leslie R.D. Twins: mirrors of the immune system. Immunol. Today. 2000;21:342–347. doi: 10.1016/s0167-5699(00)01658-3. [DOI] [PubMed] [Google Scholar]
- Tan T.G., Mathis D., Benoist C. Singular role for T-BET+CXCR3+ regulatory T cells in protection from autoimmune diabetes. Proc. Natl. Acad. Sci. USA. 2016;113:14103–14108. doi: 10.1073/pnas.1616710113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team R.C. R Foundation for Statistical Computing; Vienna, Austria: 2016. R: A language and environment for statistical computing. [Google Scholar]
- Threadgill D.W., Churchill G.A. Ten years of the collaborative cross. G3 (Bethesda) 2012;2:153–156. doi: 10.1534/g3.111.001891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threadgill D.W., Miller D.R., Churchill G.A., de Villena F.P. The collaborative cross: a recombinant inbred mouse population for the systems genetic era. ILAR J. 2011;52:24–31. doi: 10.1093/ilar.52.1.24. [DOI] [PubMed] [Google Scholar]
- Welsh C.E., Miller D.R., Manly K.F., Wang J., McMillan L., Morahan G., Mott R., Iraqi F.A., Threadgill D.W., de Villena F.P. Status and access to the Collaborative Cross population. Mamm. Genome. 2012;23:706–712. doi: 10.1007/s00335-012-9410-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates L., McMurray F., Zhang Y., Greenfield A., Moffatt M., Cookson W., Dean C. ENU mutagenesis as a tool for understanding lung development and disease. Biochem. Soc. Trans. 2009;37:838–842. doi: 10.1042/BST0370838. [DOI] [PubMed] [Google Scholar]
- Zhang F., Lupski J.R. Non-coding genetic variants in human disease. Hum. Mol. Genet. 2015;24(R1):R102–R110. doi: 10.1093/hmg/ddv259. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.