

Key Points
IAP expression in hosts regulates GVHD.
IAP expression in nonhematopoietic host targets is critical for mitigating GVHD damage.
Abstract
Inhibitors of apoptosis proteins (IAPs) regulate apoptosis, but little is known about the role of IAPs in the regulation of immunity. Development of IAP inhibition by second mitochondria-derived activator of caspase (SMAC) mimetics is emerging as a novel therapeutic strategy to treat malignancies. We explored the role of IAPs in allogeneic immunity with 2 distinct yet complementary strategies, namely, chemical and genetic approaches, in clinically relevant models of experimental bone marrow transplantation (BMT). The small-molecule pan-IAP inhibitor SMAC mimetic AT-406 aggravated gastrointestinal graft-versus-host disease (GVHD) in multiple models. The role of specific IAPs in various host and donor cellular compartments was explored by utilizing X-linked IAP (XIAP)– and cellular IAP (cIAP)–deficient animals as donors or recipients. Donor T cells from C57BL/6 cIAP1−/− or XIAP−/− animals demonstrated equivalent GVHD severity and allogeneic responses, both in vivo and in vitro, when compared with B6 wild-type (B6-WT) T cells. By contrast, when used as recipient animals, both XIAP−/− and cIAP1−/− animals demonstrated increased mortality from GVHD when compared with B6-WT animals. BM chimera studies revealed that cIAP and XIAP deficiency in host nonhematopoietic target cells, but not in host hematopoietic-derived cells, is critical for exacerbation of GVHD. Intestinal epithelial cells from IAP-deficient animals showed reduced levels of antiapoptotic proteins as well as autophagy-related protein LC3 after allogeneic BMT. Collectively, our data highlight a novel immune cell–independent but target tissue–intrinsic role for IAPs in the regulation of gastrointestinal damage from GVHD.
Visual Abstract
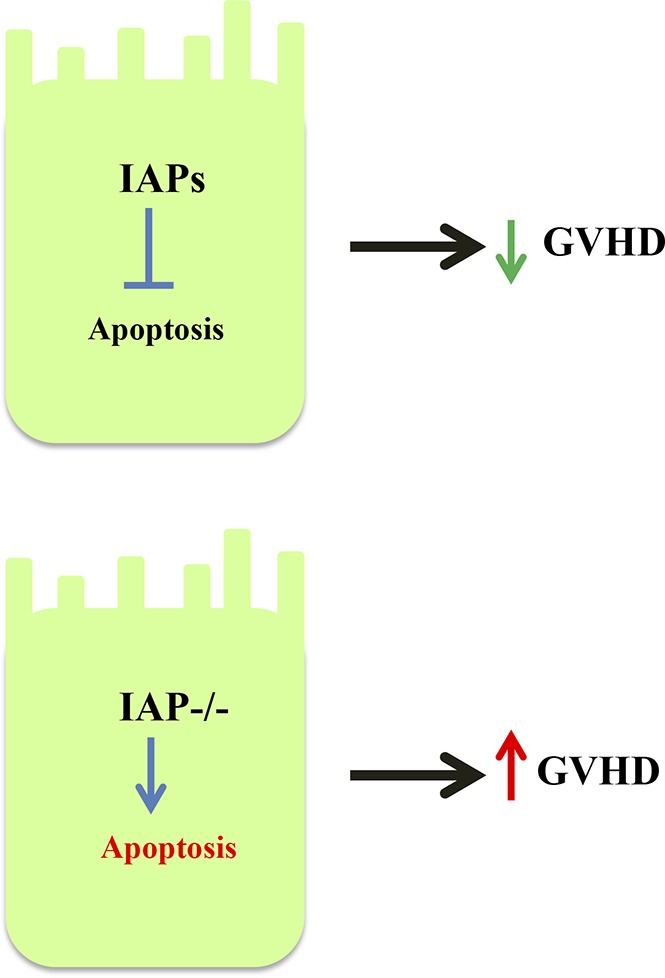
Introduction
Inhibitors of apoptosis proteins (IAPs) regulate apoptosis, and recent data suggest that they play an important role in both innate and adaptive immunity1-3 through diverse mechanisms beyond the inhibition of caspases.4 Among IAPs, X-linked IAP (XIAP), cellular IAP1 (cIAP1), and cIAP2 regulate the production of proinflammatory cytokines and the activation of T cells and antigen-presenting cells (APCs) through the modulation of MAPK and nuclear factor κB (NF-κB) signaling pathways.1,5 In addition, genetic mutations and IAP deficiencies are associated with congenital immunodeficiency disorders, such as X-linked lymphoproliferative disease type 2 and familial hemophagocytic lymphohistiocytosis in humans.6,7 XIAP deficiency has also been associated with inflammatory bowel disease (IBD).8
Because IAPs are highly expressed in solid cancers and associated with tumor progression, resistance to chemotherapy, and poor prognosis, small-molecule IAP antagonists are under clinical development for the treatment of solid and hematological malignancies.9-11 These are known as second mitochondria-derived activator of caspase (SMAC) mimetics.9-11 All IAPs contain baculovirus IAP repeat (BIR) domains and a carboxy-terminal RING finger domain and inhibit caspases.12-15 Cell-intrinsic SMACs bind to IAPs at BIR3 domains. SMAC mimetics interfere with this interaction and thus modulate IAP function and apoptosis. SMAC mimetics also promote degradation of IAPs (especially cIAP), resulting in activation of noncanonical NF-κB pathway.12,13,16-18 The cytotoxic killing induced by SMAC mimetics might be associated with a simultaneous secretion of autocrine/paracrine tumor necrosis factor α (TNFα) or TNF receptor 1 (TNFR1)–induced cell signaling.16,17,19 These findings are consistent with the notion that SMAC mimetics may promote inflammation because both XIAP and cIAP negatively regulate inflammation through the inhibition of TNFR1-dependent proinflammatory cytokine production or NLRP3 inflammasome activation in myeloid cells.20-22 In contrast, SMAC mimetic treatment has also been shown to attenuate inflammatory responses by blunting leukocyte infiltration23 or by inhibiting macrophages.24 These data indicate that SMAC mimetics may have conflicting effects on the modulation of inflammatory responses under different circumstances.
Allogeneic hematopoietic stem-cell transplantation (allo-HCT) is a potentially curative treatment modality for a number of malignant hematologic disorders. It is characterized by conditioning-related as well as allogeneic reactivity–induced inflammations that cause its major complication, acute graft-versus-host disease (GVHD).25 The proinflammatory cytokines activate both host- and donor-derived APCs that stimulate allogeneic reactive donor T cells, which migrate to the gastrointestinal (GI) tract, liver, and skin and cause GVHD that is characterized by target-cell apoptosis.26-29 Despite the increasing appreciation of the role of targeting IAPs in immunity and malignancies, their role in T-cell allogeneic immunity, APC responses, and response of target tissues to immune-mediated attack in GVHD remains unknown.
In the present study we determined the role of IAPs in GVHD by utilizing clinically relevant, well-characterized murine models of allogeneic bone marrow transplantation (allo-BMT). To chemically target IAPs, we used AT-406 (SM-406), an SMAC mimetic that actively antagonizes all IAPs (XIAP and cIAPs)30 in vitro and in vivo.31 AT-406 is an orally bioavailable SMAC mimetic and is being currently investigated in phase I clinical trials for the treatment of human solid cancers and hematological malignancies.32,33 In addition, as a genetic approach, we used cIAP1/XIAP knockout (cIAP1−/− or XIAP−/−) animals, which develop normally without any inflammation in steady state.1,2,15 We found that global inhibition of IAPs exacerbated experimental GVHD, and the critical cellular component that aggravated the severity of GVHD resulted from the absence of IAPs in target tissues and not from their effects on donor or host immune cells. Our data thus suggest a role for target tissue–intrinsic programs that can mitigate GVHD without directly altering the functionality of immune cells.
Materials and methods
Mice
C57BL/6 (B6, H-2b, CD45.2+), B6D2F1 (H-2b/d), and C3H.sw (H-2b) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). BALB/c (H-2d) mice were purchased from Charles River Laboratories (Wilmington, MA). B6Ly5.2 (H-2b, CD45.1+) mice were purchased from National Cancer Institute Frederick (Frederick, MD). B6-background cIAP1−/− and XIAP−/− animals were provided by C.S.D. and have been described previously.15 All animals were cared for according to regulations reviewed and approved by the University Committee on Use and Care of Animals of the University of Michigan, based on University Laboratory Animal Medicine guidelines.
Reagents
The SMAC mimetic SM-164 (AT-406)30 was a kind gift from Shaomeng Wang (University of Michigan).
Generation of BM chimeras
BM chimeras (B6→B6 Ly5.2, cIAP1−/− →B6 Ly5.2, XIAP−/−→B6 Ly5.2) were generated as described previously.34-37 Briefly, B6 Ly5.2 animals received 11-Gy total-body irradiation (TBI; caesium-137 source) on day −1 and received transplants IV of 5 × 106 BM and 5 × 106 whole spleen cells from B6 wild-type (B6-WT) or cIAP1−/− or XIAP−/− donor animals on day 0. To generate B6 Ly5.2→B6, B6 Ly5.2→cIAP1−/−, and B6 Ly5.2→XIAP−/− chimeras, B6-WT or cIAP1−/− or XIAP−/− animals received 10-Gy TBI on day −1 and received transplants of 5 × 106 BM and 5 × 106 whole spleen cells from WT B6 Ly5.2 donor animals on day 0. Donor hematopoietic cell engraftment was confirmed using the CD45.2 or CD45.1 monoclonal antibody 3 to 4 months after BMT (donor type >95.0%).
BMT and treatment of AT-406
BMTs were performed as previously described.34,35,38 Briefly, splenic T cells from donors were purified, and the BM was depleted of T cells by autoMACS (Miltenyi Biotec, Bergisch Gladbach, Germany) utilizing CD90.2 microbeads (Miltenyi Biotec). We used well-established multiple BMT models. B6, BALB/c, B6D2F1, and C3H.sw animals were used as recipients and received either 8.5 (BALB/c), 10 (B6), 10.5 (C3H.sw), or 11 Gy (B6D2F1; caesium-137 source) on day −1 and 0.5 to 1.0 × 106 (B6→BALB/c), 2 × 106 (C3H.sw→B6), and 3 × 106 (B6→B6D2F1) CD90.2+ T cells along with 5 × 106 T cell–depleted BM (TCD-BM) cells from either syngeneic or allogeneic donors on day 0. For the treatment of AT-406, AT-406 (10 mg/kg) and its diluent control phosphate-buffered saline (PBS) were administered by subcutaneous injection at days −1, +1, +3, +5, and +7.
Systemic and histopathological analysis of GVHD
We monitored survival after allo-HCT daily and assessed the degree of clinical GVHD weekly, as described previously.39 Histopathological analysis of the liver, GI tract, and lung, which are the primary GVHD target organs, was performed as described40 utilizing a semiquantitative scoring system implemented by a single pathologist (C.L.).40 After scoring, the codes were broken, and the data were compiled.
DC cultures
To obtain dendritic cells (DCs), BM cells from B6-WT or BALB/c animals were cultured with murine recombinant granulocyte-macrophage colony-stimulating factor (20 ng/mL; PeproTech, Rocky Mill, NJ) for 7 days and harvested as described previously.37 DCs were isolated by using CD11c (N418) MicroBeads (Miltenyi Biotec) and the autoMACS (Miltenyi Biotec) and used as a stimulator for mixed lymphocyte reaction (MLR).
FACS analysis
Fluorescence-activated cell sorting (FACS) analyses were performed as previously described.37,41 Briefly, to analyze donor T-cell expansion and activation markers, splenocytes, hepatic lymphocytes,42 and intraepithelial lymphocytes43 from transplant-recipient mice were resuspended in FACS wash buffer (2% bovine serum albumin in PBS) and stained with conjugated monoclonal antibodies (mAbs): fluorescein isothiocyanate (FITC)–conjugated mAbs to mouse H2Kd (clone SF1-1.1) and H2Kb (clone AF6-88.5); PerCP-Cy5.5–conjugated mAbs to mouse CD4 (clone GK1.5) and CD8 (clone 53-6.7); phycoerythrin (PE)-conjugated mAbs to mouse CD69 (clone H1.2F3), CD183 (CXCR3; clone CXCR3-173), LPAM-1 (integrin α4β7; clone DATK32), and CD62L (clone MEL-14); and APC-conjugated mAbs to mouse CD44 (clone IM7; BioLegend, San Diego, CA). After staining, the cells were washed twice and fixed with 2% formaldehyde. The procedure was performed as described previously.37 Cells were analyzed using a BD Accuri C6 flow cytometer (BD Bioscience, San Diego, CA). For intracellular staining to detect cytokines, cells were permeabilized after fixation and stained with PE-conjugated interferon γ (IFN-γ; clone XMG1.2), interleukin-2 (IL-2; clone JES6-5H4), and Foxp3 (clone MF-14) and APC-conjugated mAbs to mouse TNF-α (clone MP6-XT22) and IL-17A (clone TC11-18H10.1; BioLegend) according to manufacturer protocol. For intracellular staining to detect cIAP1, XIAP, BCL-2, BIM, BAX, and LC3, cells were fixed with 2% formaldehyde and permeabilized with BDPhosFlow (BD Bioscience) and then stained with FITC-conjugated polyclonal Abs to mouse BIM (orb15184) and BAX (orb7603) from Biorbyt (Berkeley, CA) and PE-conjugated mAbs to mouse BCL-2 (clone BCL/10C4; BioLegend). In the case of nonconjugated Abs, firstly, gout polyclonal Ab, anti-cIAP1(D-19, sc-1869), and rabbit polyclonal anti-XIAP (H-202, sc-11426; Santa Cruz Biotechnology, Dallas, TX) and anti-LC3B/MAP1LC3B (NB100-2220; Novus Biotechnology, Littleton, CO) were stained, and appropriate secondary antibodies were used to detect these expressions.
MLR
Splenic T cells from B6-WT, cIAP1−/−, or XIAP−/− animals (magnetically separated by MACS using CD90.2 microbeads) were used as responders and BALB/c- versus B6-WT–derived BM-derived DCs (BMDCs) were used as stimulators in an MLR. 1 × 105 T cells and irradiated (20 Gy) 2.5 × 103 BMDCs were cocultured on 96-well U-bottom plates for 96 hours. The incorporation of 3H-thymidine (1 µCi per well) by proliferating T cells during the final 16 hours of coculture was measured by a Betaplate reader (Wallad, Turku, Finland). In some experiments, BMDCs were pretreated with AT-406 (1 µM) for 6 hours and then used as a stimulator, or AT-406 (1 µM) and its diluent control were added in culture wells during reaction.
Nonspecific T-cell receptor stimulation
Isolated T cells (1 × 105 per well) from B6-WT, cIAP1−/−, or XIAP−/− animals (magnetically separated by MACS using CD90.2 microbeads) were stimulated with anti-CD3 (2 µg/mL) and CD28 (1 µg/mL) antibodies in the presence or absence of AT-406 (1 µM) on 96-well U-bottom plates for 24 or 48 hours. The incorporation of 3H-thymidine (1 µCi per well) by proliferating T cells during the final 6 hours of culture was measured by a Betaplate reader (Wallad).
DC stimulation by pathogen- and damage-associated molecular patterns
BMDCs were stimulated with lipopolysaccharide (500 ng/mL), proteoglycan (5 µg/mL), Pam3CSK4 (300 ng/mL), and HMGB1 (10 µg/mL) for 16 hours, and then supernatants were collected and DCs harvested and stained with conjugated mAbs to test the expression of costimulatory molecules: FITC-conjugated mAbs to mouse CD11c (clone N418) and PE-conjugated mAbs to mouse CD80 (clone 16-10A1), CD86 (clone GL-1), CD40 (clone 3/23), B7H1 (PDL-1, clone 10F.9G2), and I-Ab (AF6-120.1; BioLegend). Cells were analyzed using a BD Accuri C6 flow cytometer (BD Bioscience).
Cytokine ELISA
Concentrations of IFN-γ, TNF-α, IL-17A, and IL-6 were measured in the serum or culture supernatants by enzyme-linked immunosorbent assay (ELISA) with specific anti-mouse mAbs for capture and detection utilizing BD OptEIA (IFN-γ, IL-6; BD Bioscience), ELISA MAX (IL-17A; BioLegend), or Mouse TNF-α Quantikine ELISA Kit (R&D Systems, Minneapolis, MN). Assays were performed according to manufacturer protocol and read at 450 nm using a microplate reader (Model 3550; Bio-Rad Labs, Hercules, CA). All samples and standards were run in duplicate.
Apoptosis analysis
Non–T-cell receptor–stimulated T cells were washed twice with PBS and stained with a PE-conjugated anti–annexin V mAb (BioLegend) and PerCP-Cy5.5–conjugated anti-90.2 mAb (BioLegend) in the dark for 15 minutes at room temperature in labeling buffer and analyzed using an Accuri C6 cytometer (Accuri, Ann Arbor, MI).
Regarding epithelial cell isolation, small intestines and colons were digested once with 0.1-mM EDTA at 37°C for 45 minutes.
Western blot analysis
Epithelial cells of the small intestine were isolated as described above, and whole cells were lysed in RIPA lysis extraction buffer (Thermo Scientific, Rockford, IL). Equal amounts of protein lysates quantified with the BCA Protein Assay Kit (Thermo Scientific) were separated with 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis. Rabbit LC3 antibody (Novus Biologicals, Littelton, CO) was used according to manufacturer instructions. Secondary antibody conjugated to horseradish peroxidase (Jackson Immunoresearch) was used to detect the primary antibody. Densitometric analysis was performed using ImageJ software.
TUNEL staining
Histopathological samples of the small and large intestines were harvested at day 7 after BMT and fixed with paraffin. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed with the In Situ Cell Death Detection Kit, AP (Roche, Basel, Switzerland) according to manufacturer instructions. Briefly, tissues were dewaxed and rehydrated with 2 washes in xylene 100% followed by serial washes in decreasing concentrations of ethanol in water (100% to 95%-75% to 50%-10% to 0%). Proteinase K at a concentration of 20 µg/mL was incubated for 30 minutes at 37°C. After washes, the TUNEL reaction mix was incubated for 60 minutes at 37°C, followed by the AP converter for 30 minutes at 37°C. A negative control was incubated with the label solution of the reaction mix only. A positive control was pretreated with a DNase recombinant. A nucleus counterstain was then used. At the end of the procedure, the tissues were mounted on a 9:1 ratio of glycerol toPBS, and representative pictures were taken.
Statistical analysis
The Mann-Whitney U test was used for the statistical analysis of in vitro data, and the Wilcoxon rank test was used to analyze survival data. A P value < .05 was considered to be statistically significant.
Results
IAP inhibitor exacerbates GVHD responses
IAPs are broadly expressed in immune cells, such as T cells and APCs as well as parenchymal cells, and play an important role in regulating innate and adaptive immune responses.2,44 IAPs regulate TNF-α and NF-κB activation.45,46 Because the pan-IAP inhibitor AT-406, an SMAC mimetic, regulates TNFα,47 one of the most important cytokines mediating GVHD,48 and shows good bioavailability in vivo,30,49,50 we tested the hypothesis that administration of AT-406 would mitigate GVHD. On the basis of its half-life, we first tested various dosing regimens of AT-406 to determine the optimal dose for targeting IAPs after allo-BMT. In the lethally irradiated major histocompatibility complex (MHC)–mismatched B6-into-BALB/c model, the dose of 10 mg/kg from day −1 to day 7, every other day of AT-406 consistently targeted XIAP in donor T cells and host nonhematopoietic target cells (CD326+ intestinal epithelial cells [IECs]; Figure 1A-B). Similar results were also observed for cIAP (supplemental Figure 1).
Figure 1.

AT-406 decreases the expression of XIAP in donor T cells and host nonhematopoietic target cells after allo-BMT. (A-B) BALB/c animals received 8.5 Gy on day −1 and received transplants of 0.5 × 106 CD90.2+ splenic T cells along with 5 × 106 TCD-BM cells from either syngeneic BALB/c or allogeneic MHC-mismatched B6 donors. Recipient animals received either AT-406 (10 mg/kg) or its diluent subcutaneously on days −1, +1, +3, +5, and +7 after BMT. (A) Schema of experiment. (B) XIAP expression of donor CD3+ T cells in the spleen and CD326+ epithelial cells of the small intestine was evaluated by FACS staining on day 8. Representative figure (left) and pooled data (right) from 5 to 8 animals are shown. *P < .05, **P < .01. The bar shows the mean ± standard error of the mean (SEM).
Next, we used the above dose and schedule regimen to evaluate the impact of AT-406 on the regulation of allogeneic immune responses and the severity of GVHD in the same MHC-mismatched B6-into-BALB/c BMT model. Recipient BALB/c animals were irradiated with 8.5 Gy on day −1 and received transplants of 0.5 × 106 T cells along with 5 × 106 TCD-BM cells from either syngeneic BALB/c or allogeneic MHC-mismatched B6 donors. Both groups received either AT-406 or its diluent. As shown in Figure 2A, all the syngeneic mice that received AT-406 or diluent survived, thus ruling out nonspecific toxicity from AT-406. However, contrary to our hypothesis, allogeneic animals that received AT-406 showed significantly worse GVHD and died more rapidly than control B6-WT animals (P = .0012; Figure 2A).
Figure 2.

The IAP inhibitor AT-406, an SMAC mimetic, exacerbates GVHD. (A) Survival. BALB/c animals received 8.5 Gy on day −1 and received transplants of 0.5 × 106 CD90.2+ splenic T cells along with 5 × 106 TCD-BM cells from either syngeneic BALB/c or allogeneic MHC-mismatched B6 donors. Recipient animals received either AT-406 (10 mg/kg) or its diluent subcutaneously on days −1, +1, +3, +5, and +7 after BMT; n = 6-21 per group). Pooled data from 4 independent experiments are shown. **P < .01, when allogeneic treated and control animals are compared. (B) Survival. B6D2F1 animals received 11 Gy on day −1 and received transplants of 3 × 106 CD90.2+ splenic T cells along with 5 × 106 TCD-BM cells from either syngeneic B6D2F1 or allogeneic MHC-mismatched haploidentical B6 donors. Recipient animals received either AT-406 (10 mg/kg) or its diluent subcutaneously on days −1, +1, +3, +5, and +7 after BMT (n = 6-30 per group). Pooled data from 6 independent experiments are shown. *P < .05, when allogeneic treated and control animals are compared. (C-E) Expansion of donor T cells (H-2kb+CD4+CD8+) (C), CD44+CD62L− effector T cells (D), and IFN-γ–producing T cells (E) in the spleen on day 6 after allo-BMT (n = 4-5 per group, pooled from 2 experiments). The bar shows the mean ± SEM. (F-H) The representative figures of small (F) and large intestines (G) (original magnification ×200; hematoxylin and eosin stain) and GVHD histopathological score of GI tract (small and large intestines) (H) on day 6 after allo-BMT (n = 6-10 per group, pooled from 3 experiments). *P < .05. The bar shows the mean ± SEM.
To eliminate strain-dependent factors, we used another well-established MHC-mismatched haploidentical B6-into-B6D2F1 model of BMT. Recipient B6D2F1 animals received 11 Gy on day −1 and received transplants of 3 × 106 T cells along with 5 × 106 TCD-BM cells from either syngeneic B6D2F1 or allogeneic MHC-mismatched B6 donors. Both groups received the same treatment as above. Allogeneic B6D2F1 recipients treated with AT-406 showed significantly greater GVHD severity and mortality (P = .012; Figure 2B).
We next explored whether the enhanced GVHD in the allogeneic animals that were treated with AT-406 was associated enhanced donor T-cell expansion and cytokine secretion when compared with control-treated allogeneic animals on day 7 after BMT. There was no significantly greater expansion of total donor T cells or donor CD44+CD62L− effector T cells or proinflammatory cytokine secretion from splenic donor T cells (Figure 2C-E; supplemental Figure 2A-C). However, the GVHD-specific histopathological damage of the GI tract was substantially worse in AT-406–treated animals (Figure 2F-H). To clarify whether AT-406 exacerbates irradiation toxicity in the gut, we also tested the histopathological damage in the gut in syngeneic animals with AT-406 treatment and found that histopathological damage in these animals was comparable to that in WT syngeneic animals (Figure 2H). These data suggest that AT-406 exacerbated GI damage from GVHD in multiple BMT models.
IAPs in T cells are not required for enhancing GVHD severity
Donor T cells are indispensable to GVHD.51 Therefore, we next explored whether the augmentation of GVHD severity by the systemically administered AT-406 resulted from inhibition of IAPs on donor T cells. To dissect the cellular components, we took a genetic approach. To this end, we used 2 genetically deficient IAP animals, namely, XIAP and cIAP1 knockout animals. The role of IAPs in T-cell responses seems to be context dependent.52,53 We first examined the absolute numbers of CD4+ and CD8+ T cells and then determined detailed phenotypes of various T-cell subsets and the expression of the activation marker in naïve B6-WT, B6-cIAP1−/−, and B6-XIAP−/− animals. We found similar numbers of CD4+ and CD8+ T cells (supplemental Figure 3A) and an equivalent distribution of naïve (CD44−CD62L+), central memory (CD44+CD62L+), effector memory (CD44+CD62L−), and regulatory (CD4+CD25+Foxp3+) T cells in all 3 mouse strains (supplemental Figure 3B-D).
We next explored whether IAPs in donor T cells have a critical role in the aggravation of GVHD responses in vivo. We used the MHC completely mismatched B6-into-BALB/c model. B6-WT, XIAP−/−, or cIAP1−/− splenic T cells were used as sources of donor T cells, but the TCD-BM was obtained from B6-WT animals such that only the differences in the infused mature naïve T cells could be evaluated for their effect on GVHD. Recipient BALB/c animals were irradiated with 8 Gy on day −1 and received transplants of 0.5 × 106 T cells as above along with 5 × 106 TCD-BM cells from either syngeneic BALB/c or allogeneic MHC-mismatched B6 donors. We found that all of the allogeneic recipients showed similar GVHD severity and mortality regardless of whether they received T cells from WT or XIAP−/− or cIAP1−/− animals (Figure 3A-B). The role of XIAP and cIAP1 in donor T cells was next determined in a second, lethally irradiated MHC-mismatched haploidentical B6-into-B6D2F1 model. Recipient B6D2F1 animals received 11 Gy on day −1 and received transplants of 3 × 106 T cells along with 5 × 106 TCD-BM cells from either syngeneic B6D2F1 or allogeneic MHC-mismatched B6 donors. Donor WT, XIAP−/−, and cIAP1−/− T cells induced equivalent GVHD severity and mortality (Figure 3C). The similar GVH responses were confirmed by equivalent donor T-cell expansion (Figure 3D), activation (Figure 3E), and IFN-γ secretion (Figure 3F) in the spleen 14 days after BMT. The GVHD-specific histopathological damage of the GI tract and liver on day 14 was also equivalent among these groups (Figure 3G-H).
Figure 3.

IAPs are not required in donor T cells to modulate GVH responses. (A-B) BALB/c animals received 8.5 Gy on day −1 and received transplants of 0.5 × 106 CD90.2+ splenic T cells from either syngeneic BALB/c or allogeneic MHC-mismatched B6-WT, B6-cIAP1−/−, or B6-XIAP−/− animals along with 5 × 106 TCD-BM cells from either syngeneic BALB/c or allogeneic B6 donors. Survival (A) and GVHD clinical score (B) (n = 11-29 per group). Pooled data from 4 independent experiments are shown. (C) B6D2F1-recipient animals received 11 Gy on day −1 and received transplants of 3 × 106 CD90.2+ splenic T cells from either syngeneic B6D2F1 or haploidentical MHC-mismatched B6-WT, B6-cIAP1−/−, or B6-XIAP−/− animals along with 5 × 106 TCD-BM cells from either syngeneic B6D2F1 or allogenic B6 donors. Survival (n = 5-11 per group). Pooled data from 2 independent experiments are shown. (D-F) Expansion of donor T cells (H-2kb+CD4+CD8+) (D), CD69+ T cells (E), and IFN-γ–producing T cells (F) in the spleen on day 14 after allo-BMT (n = 3-4 per group, pooled from 2 experiments). The bar shows the mean ± SEM. (G-H) The histopathological GVHD score in the GI tract (small and large intestines) (G) and liver (H) on day 14 after allo-BMT (n = 3-4 per group, pooled from 2 experiments). The bar shows the mean ± SEM. (I) In vitro MLR. Isolated splenic CD90.2+ T cells from either BALB/c or B6-WT animals were cultured with BMDCs derived from animals in the presence or absence of AT-406 (1 µM) for 96 hours and analyzed for proliferation after 3H-thymidine incorporation during the last 16 hours of incubation. A representative figure from 3 independent experiments is shown. The bar shows the mean ± SEM. (J) In vitro MLR. Isolated splenic CD90.2+ T cells from either B6-WT, B6-cIAP1−/−, or B6-XIAP−/− animals were cultured with BMDCs derived from syngeneic B6 or allogeneic BALB/c animals for 72 hours and analyzed for proliferation after 3H-thymidine incorporation during the last 16 hours of incubation. The bar shows the mean ± SEM. CPM, counts per minute.
To further determine whether there were any T cell–intrinsic differences in response to allogeneic stimulus, we also determined in vitro whether XIAP−/− and cIAP1−/− T cells showed any differences when compared with WT T cells in the presence or absence of the SMAC mimetic AT-406. Stimulation of WT T cells with allogeneic BALB/c-derived BMDCs in an MLR showed no significant differences in T-cell proliferation regardless of presence of AT-406 (Figure 3I). Furthermore, the WT, XIAP−/−, and cIAP1−/− T cells showed a similar proliferation and secretion of IFN-γ and IL-2 after stimulation with allogeneic MLR in both the presence and absence of AT-406 (Figure 3J; supplemental Figure 4A-B). However, we found that AT-406 augmented nonspecific T-cell receptor responses when stimulated with anti-CD3/28 in vitro, as shown in a previous report (supplemental Figure 4C).53 Collectively, these results demonstrate that the inhibition of IAPs or the expression of XIAP1 or cIAP1 in donor T cells plays a dispensable role in allogeneic T-cell responses, especially GVHD.
IAPs in host animals mitigate GVHD
We next reasoned that the inhibition of IAPs in host tissues might be driving the exacerbation of GVHD after treatment with SMAC mimetics. To test this, we first examined whether the IAP levels in the IECs (CD326+) were altered after irradiation to eliminate radiation-dependent confounding factors. We found that IAP levels in CD326+ cells were transiently decreased at 24 hours after irradiation but spontaneously recovered in 48 hours (supplemental Figure 5A-B). We next explored whether IAP deficiency had an impact on the radiation toxicity in an intensity-dependent manner; B6-WT, XIAP−/−, cIAP1−/− animals received 9, 13, and 15 Gy on day −1 and received transplants of 5 × 106 BM cells and 3 × 106 splenic T cells from syngeneic B6 animals. We found that the overall survival of IAP-deficient animals was comparable to that of WT animals even with increased radiation dose (supplemental Figure 5C-E). In addition, histopathological damage in IAP-deficient animals was comparable to that in B6-WT animals after irradiation (supplemental Figure 5F). These data suggest that change in IAP expression in the CD326+ cells because of radiation toxicity may have had a negligible role in our BMT models. Next, we used XIAP−/− animals as recipients of allo-BMT. Recipient B6-WT and B6-XIAP−/− animals were lethally irradiated and received transplants on day 0 of 5 × 106 TCD-BM cells and 3 × 106 splenic CD90.2+ T cells from either syngeneic B6-WT or allogeneic MHC-mismatched BALB/c donors. As shown in Figure 4A, all the syngeneic B6-WT and B6 XIAP−/− animals survived the period of observation, ruling out any significant effect of conditioning-related damage on survival after BMT. By contrast, allogeneic B6-XIAP−/− animals showed significantly worse survival and clinical GVHD severity than B6-WT animals (P = .0003; Figure 4A; supplemental Figure 6A). However, the increased mortality was associated neither with an increased donor T-cell expansion in target organs on day 7 or 14 (Figure 4B; supplemental Figure 6B) nor with an elevation in serum levels of inflammatory cytokines, such as IFN-γ, TNF-α, IL-6, and IL-17A on day 7 (Figure 4C-D; supplemental Figure 6C-F). Nonetheless, consistent with survival data, the GVHD-specific histopathological score was higher only in the GI tract of allogeneic XIAP−/− animals on day 7 when compared with allogenic B6-WT animals (Figure 4E), but the scores of the other organs were similar (data not shown).
Figure 4.

Absence of IAPs in host exacerbates GVHD in allo-BMT. B6-WT and B6-XIAP−/− animals received 10 Gy on day −1 and received transplants of 3 × 106 CD90.2+ splenic T cells along with 5 × 106 TCD-BM cells from either syngeneic B6 or allogeneic MHC-mismatched BALB/c donors. (A) Survival (n = 5-13 per group). Pooled data from 3 independent experiments are shown. ***P < .001, when allogeneic WT control and allogeneic XIAP−/− animals are compared. (B) Donor T-cell (H-2kd+CD4+ or H-2kd+CD8+) expansion in spleen, liver, and intraepithelial cells (IECs) on day 7 after allo-BMT (n = 4-6 per group, pooled from 2 experiments). The bar shows the mean ± SEM. (C-D) Serum levels of IFN-γ (C) and TNF-α (D) on day 7 after allo-BMT (n = 4-6 per group, pooled from 2 experiments). *P < .05. The bar shows the mean ± SEM. (E) The histopathological GVHD score in GI tract (small and large intestines) on day 7 after allo-BMT (n = 4-6 per group, pooled from 2 experiments). **P < .01. The bar shows the mean ± SEM. (F) Survival. B6-WT and B6-cIAP1−/− animals received 10 Gy on day −1 and received transplants of 3 × 106 CD90.2+ splenic T cells along with 5 × 106 TCD-BM cells from either syngeneic B6 or allogeneic MHC-mismatched BALB/c donors (n = 3-16 per group). Pooled data from 3 independent experiments are shown. *P < .05, when allogeneic WT control and allogeneic cIAP1−/− animals are compared.
Similarly, enhanced GVHD mortality and clinical severity were observed in allogeneic cIAP1−/− animals when compared with WT animals after BALB/c-into-B6 BMT (P = .02; Figure 4F; supplemental Figure 6G). These data indicate that the absence of IAPs in hosts exacerbates GVHD and that use of the SMAC mimetic AT-406 may have similar adverse effects that are intrinsic to the host. They further suggest that XIAP and cIAP1 do not compensate for the absence of each other and that they have distinct effects on GVHD mortality.
IAPs in host hematopoietic-derived APCs are dispensable for exacerbation of GVHD
Host hematopoietic-derived APCs are central for initiating allogeneic reactive T-cell responses,28,29 and because expression of IAPs in the host is important for increasing GVHD severity, we next reasoned that IAP expression in host hematopoietic cells might be the critical cellular compartment mediating IAP-dependent severity in the recipients. Therefore, to evaluate the impact of IAP expression in hematopoietic APCs, we next explored in vitro the role of IAPs in DCs, the most potent APCs. We determined the expression of costimulatory molecules on splenic DCs from naïve B6-WT, cIAP1−/−, and XIAP−/− animals to determine their phenotypes at homeostasis. Although the numbers of CD86+CD11c+ and class II+CD11c+ cells in cIAP1−/− animals were significantly higher than those in B6-WT animals, the absolute numbers of CD11c+, CD80+CD11c+, PDL-1+CD11c+, and CD40+CD11c+ cells were equivalent among these animals (supplemental Figure 7A-F). BMDCs from cIAP1−/− or XIAP−/− animals showed a similar allogeneic T-cell stimulation capacity in an MLR when compared with B6-WT (Figure 5A). To further confirm whether AT-406 alters the allogeneic T-cell stimulation capacity of DCs, we pretreated B6-WT DCs with AT-406 before using them as stimulators in an MLR. We found that allogeneic T-cell stimulation was equivalent in both groups (Figure 5B). We next examined whether AT-406 altered the innate immune responses of DCs, such as TLR4-mediated responses after stimulation with lipopolysaccharide, but found similar responses in untreated and AT-406–treated DCs, as determined by their expression of costimulatory molecules as wells as secretion of TNF-α and IL-6 (Figure 5C-E). Because cIAP1 and XIAP are recognized as regulators of nucleotide-binding oligomerization domain-containing protein (NOD)-1– and NOD-2–mediated innate immune responses,44,47 we also tested the responses to other pathogen-associated molecular patterns utilizing peptidoglycan and Pam3CSK4 as well as damage-associated molecular patterns utilizing high-mobility group protein 1 (HMGB-1) in DCs in vitro. Again, AT-406–treated DCs showed no alteration in their response to these various stimuli (supplemental Figure 7G-I).
Figure 5.

The role of IAPs in host hematopoietic-derived APCs is dispensable for GVHD. (A-B) In vitro MLR. Isolated splenic CD90.2+ T cells from either syngeneic B6 or allogeneic BALB/c animals were cultured with BMDCs derived from B6-WT, B6-cIAP1−/−, or B6-XIAP−/− animals for 96 hours (A) or with AT406-pretreated (1 µM; 6 hours) BMDCs derived from B6-WT for 96 hours (B) and analyzed for proliferation after 3H-thymidine incorporation during the last 16 hours of incubation. Representative data of 3 independent experiments are shown. The bar shows the mean ± SEM. (C-E) Both nontreated and AT406-pretreated (1 µM) BMDCs were harvested and stimulated through TLR4 with lipopolysaccharide (LPS; 500 ng/ml) for 16 hours. (C) Bar graphs depicting the percentages of CD80, CD86, I-Ab (MHC class II), and B7H1 (PD-L1) expression on CD11c+ DCs are shown. The supernatants were analyzed for IL-6 (D) and TNF-α (E) by ELISA. The data are representative of 3 independent experiments and show the means ± SEMs. (F-G) To make BM chimeras, B6Ly5.2 animals were lethally irradiated with 11 Gy and infused with 5 × 106 BM cells and 5 × 106 splenocytes from syngeneic B6-WT, B6-cIAP1−/−, or B6-XIAP−/− donors. Three to 4 months later, B6→B6Ly5.2, cIAP1−/−→B6Ly5.2, and XIAP−/−→B6Ly5.2 animals were irradiated with 9 Gy and received 3 × 106 CD90+ T cells along with 5 × 106 TCD-BM cells from either syngeneic B6 or allogeneic MHC-mismatched BALB/c donors. Survival (F) and clinical GVHD score (G) (n = 4-10 per group). Pooled data from 2 independent experiments are shown.
To determine if the lack of in vitro differences was also observed in vivo in the context of GVHD, we next explored whether XIAP or cIAP1 expression in host hematopoietic-derived cells was critical for GVHD; we generated BM chimeras such that only the radiosensitive host hematopoietic cells either expressed or lacked IAPs. B6Ly5.2 animals were lethally irradiated with 11 Gy and infused with 5 × 106 BM cells and 5 × 106 splenocytes from syngeneic B6-WT, B6-cIAP1−/−, or B6-XIAP−/− donors. Chimerism analyses of the donor hematopoietic-derived cells showed complete donor types (>95%) 3 months after the first transplantation. The B6→B6Ly5.2, cIAP1−/−→B6Ly5.2, and XIAP1−/−→B6Ly5.2 animals were then used as recipients in a second allo-BMT 3 to 4 months after the primary BMT. In the second transplantation, all chimera animals received 9 Gy and received transplants of 3.0 × 106 CD90+ T cells along with 5 × 106 BM cells from either syngeneic B6 or MHC-mismatched allogeneic BALB/c donors. All chimeras that received syngeneic T cells and BM cells survived the duration of the observation period, with no signs of GVHD. In contrast, the allogeneic B6→B6Ly5.2, cIAP1−/−→B6Ly5.2, and XIAP1−/−→B6Ly5.2 animals showed similar GVHD mortality and severity (Figure 5F-G). These results suggest that the exacerbation of GVHD in the absence of IAPs in the host or after AT-406 treatment is independent of IAPs in host hematopoietic-derived APCs.
IAP expression in host nonhematopoietic cells plays an important role in GVHD
Because host hematopoietic cell expression of IAPs was not essential to GVHD severity, we next determined whether their expression by nonhematopoietic cells in the host was critical for modulation of GVHD severity.41,54 Therefore, we hypothesized that IAP deficiency in nonhematopoietic GVHD target tissues exacerbates GVHD. To this end, we made the reverse chimeras, namely, B6Ly5.2→B6, B6Ly5.2→XIAP−/−, and B6Ly5.2→cIAP1−/−, where IAPs were absent only in the nonhematopoietic host cells. These chimeras received 9 Gy and received transplants of 3.0 × 106 CD90+ T cells along with 5 × 106 BM cells from either syngeneic B6 or MHC-mismatched allogeneic BALB/c donors. The allogeneic B6Ly5.2→XIAP−/− animals lacking IAPs in nonhematopoietic tissues developed more severe GVHD and demonstrated significantly worse survival compared with B6Ly5.2→B6 recipients (P < .0006; Figure 6A). Similarly, the allogeneic B6Ly5.2→cIAP1−/− animals also showed significantly worse survival compared with B6Ly5.2→B6 recipients (P < .01; Figure 6B). These data demonstrate mutually exclusive roles of XIAP and cIAP1 expression in contributing toward GVHD. Collectively, they demonstrate that IAP expression in host nonhematopoietic target tissues plays an important role in regulating GVHD severity.
Figure 6.

Absence of IAPs in host target tissues plays an important role in aggravating GVHD. To make BM chimeras, B6-WT, B6-cIAP1−/−, and B6-XIAP−/− animals were lethally irradiated with 10 Gy and infused with 5 × 106 BM cells and 5 × 106 splenocytes from syngeneic B6Ly5.2 donors. Three to 4 months later, B6 Ly5.2→B6, B6Ly5.2→cIAP1−/−, and B6Ly5.2→XIAP−/− animals were irradiated with 9 Gy and received 3 × 106 CD90+ T cells along with 5 × 106 TCD-BM cells from either syngeneic B6 or allogeneic MHC-mismatched BALB/c donors. (A-B) Survival. ***P < .001 or **P < .01, when allogeneic B6Ly5.2→B6 vs B6Ly5.2→XIAP−/− animals or allogeneic B6Ly5.2→B6 vs B6Ly5.2→cIAP−/− animals were compared (n = 9-27 per group, pooled from 3 to 4 independent experiments). The bar shows the mean ± SEM.
IAP expression alters apoptosis of GI epithelial cells and mitigates GVHD
GVHD is characterized by target-cell apoptosis.25 Because IAPs regulate apoptosis1-3 and their role in nonhematopoietic target cells is critical for GI GVHD severity,25 we next tested the effect of IAPs on expression of anti- or proapoptotic proteins in CD326+ IECs, which are critical for maintaining mucosal integrity in the gut after allo-BMT.55,56 We first confirmed whether allogeneic XIAP−/− animals showed increased apoptosis in the GI tract. We performed TUNEL staining in the intestine on day 7 after allo-BMT. Consistent with GVHD severity, allogeneic XIAP−/− animals showed increased numbers of IECs stained with TUNEL compared with allogeneic WT animals (Figure 7A). To determine the potential mechanisms for enhanced apoptosis in target tissue, we next tested the expression of antiapoptotic (B-cell lymphoma-2 [BCL-2]) and proapoptotic (BCL-2–like protein 11 [BIM] and BCL-2–associated X protein [BAX]) protein expression in the CD326+ IECs from B6-WT and XIAP−/− animals harvested on day 7 after allo-BMT. Both allogeneic XIAP−/− and cIAP1−/− animals showed significantly reduced expression of antiapoptotic protein BCL-2 but equivalent expression of proapoptotic proteins (Figure 7B-D). In addition, the expression ratio of BIM or BAX to BCL-2 in allogeneic XIAP−/− animals was significantly increased (Figure 7E-F). These data suggest that enhanced apoptosis in the target tissues in the absence of IAPs contributes to greater GVHD severity.
Figure 7.

IAPs on host target tissues coordinately regulate gut homeostasis by distinct mechanisms in GVHD. B6-WT and B6-XIAP−/− or cIAP1−/− animals received 10 Gy on day −1 and received transplants of 3 × 106 CD90.2+ splenic T cells along with 5 × 106 TCD-BM cells from either syngeneic B6 or allogeneic MHC-mismatched BALB/c donors. TUNEL staining and anti- or proapoptotic protein expression in intestinal epithelial cells were evaluated on day 7 after allo-BMT. (A) The representative figures of TUNEL staining of small intestine on day 7 after allo-BMT are shown. Original magnification ×40; TUNEL staining (nuclei purple) with a counterstain of Nuclear Fast Red (nuclei pink). (B-F) The expression of anti- or proapoptotic proteins in CD326+ intestinal epithelial cells on day 7 after allo-BMT: BCL-2 (B), BIM (C), BAX (D), and the ratios of BIM/BCL-2 (E) and BAX/BCL-2 (F). (G) LC3 expression in CD326+ intestinal epithelial cells from animals receiving transplants on day 7 after BMT was analyzed by FACS (n = 4-15, pooled from 3 independent experiments). *P < .05, **P < .01, ****P < .0001. The data are the means ± SEMs. (H) ρ-A expression in CD326+ intestinal epithelial cells from animals receiving transplants on day 7 after BMT was analyzed by FACS (n = 4-15, pooled from 3 independent experiments). **P < .01, when compared between allogeneic B6-WT and cIAP−/− animals. The data are the means ± SEMs.
Because apoptosis and autophagy coordinate to regulate cell survival and maintain homeostasis of the gut mucosa,57,58 we also examined whether IAP deficiency affected autophagy. We did not find any differences in the LC3 I to II conversion rate in the epithelial cells of the small intestine between B6-WT and XIAP−/− animals on day 7 or 14 after allo-BMT. However, there was a significant decrease in the expression of LC3 in both small and large intestines of allogeneic XIAP−/− animals at day 7, indicating decreased autophagy (Figure 7G; supplemental Figure 8A). These data suggest that the enhanced apoptosis in both XIAP−/− and cIAP1−/− animals and the reduced autophagy in the target tissues in the absence of XIAP may be potential mechanisms of enhanced GI GVHD. By contrast, allogeneic cIAP1−/− but not XIAP−/− animals showed reduced level of Ras homolog gene family member A (ρ-A) expression, which is known to control many cellular functions,59-61 cellular development,62 and gut homeostasis63 and is regulated by IAPs, in the small intestine at day 7 after allo-BMT (Figure 7H). These data indicate that the absence of IAPs likely exacerbates GVHD damage by multiple, distinct cellular mechanisms.
Discussion
Our study demonstrates a novel role of IAPs in the regulation of GVHD and suggests that the effects of XIAP and cIAP1 expression and SMAC mimetic treatment after BMT are independent of effects on donor T cells and host hematopoietic cells. Our data suggest that IAP expression in host nonhematopoietic target cells is important for regulation of GI GVHD damage.
The current understanding of the role of IAP regulation of innate immune responses seems to be dependent on the context. For example, IAPs have been suggested to regulate innate responses through the pattern recognition receptors24,44,47,64 by inhibiting cell-death signaling,16,17,44 controlling MAPK and NF-κB signaling pathways,45,65 and modifying inflammasomes or in the context of candida infections.20,66 In contrast, loss of cIAP or XIAP in the myeloid lineage caused overproduction of many inflammatory cytokines and severe sterile inflammation.21 We, however, found that the absence of IAPs in hematopoietic-derived APCs was not associated with increased GVHD severity or mortality. Rather, the absence of IAPs in host nonhematopoietic target tissues exacerbated GVHD in second BMT animals utilizing BM chimeras. Our data are consistent with and extend those in a recent report showing that cIAP1 protects TNF-α–mediated destruction of IECs in a colitis model in part.67 We found that both XIAP and cIAP1 reduced GI GVHD. A remarkable observation of this study is that neither the absence of IAPs nor treatment with the SMAC mimetic AT-406 exacerbated conditioning-related GI damage, as demonstrated by the absence of increased damage in syngeneic recipients (Figure 1H; supplemental Figure 5C-F). Interestingly, IAP expression in IECs was temporally reduced at 24 hours after irradiation but spontaneously recovered at 48 hours (supplemental Figure 5A). This quick alteration may have an indispensable role in the cell-intrinsic self-tissue repair mechanism to maintain gut homeostasis after injury such as radiation toxicity. Therefore, our observations suggest that IAPs protect tissue damage in IECs only in the allogeneic immunity context. The reasons for this remain unknown. Whether IAPs mitigate damage mediated by donor T cells and are dispensable for nonspecific sterile inflammation or whether they also protect tissue damage from non-TBI conditioning will need to be further studied. In addition, whether IAPs regulate the gut microbiome, which is linked with gut GVHD,68 will need to further investigated.
Consistent with our observations, SMAC mimetics induce increased enterocyte shedding and loss of barrier function in piglet ileal mucosa after oral infection with Cryptosporidium parvum.69 Furthermore, a clinical study demonstrated that some patients with advanced solid tumors who received LCL16, which induces the degradation of cIAP1, experienced grade 3 to 4 cytokine release syndrome.9 Altogether, these data suggest that inflammatory responses after the inhibition of IAPs may differ and depend on the context of inflammation and the target tissues.
Our data demonstrate that XIAP, cIAP1, or SMAC inhibition do not significantly alter T-cell responses to an allogeneic stimulus, either in vitro or in vivo. The differential effect on nonspecific antibody-mediated in vitro stimulation from an allogeneic stimulus may be a consequence of the strength of stimulation and the absence of physiological engagement of the full panoply of costimulatory and coinhibitory receptors on an APC in the former context. IAPs are closely linked to inflammasome activation, as shown by studies utilizing XIAP−/− or XIAP−/−cIAP2−/−cIAP1LysMcre/LysMcre animals as well as SMAC mimetics.22,64 Inflammasome activation, such as NLRP3 in APCs or myeloid-derived suppressor cells, are involved in the development and aggravation of GVHD.70-72 However, we found that the serum levels of IL-1β and IL-18 in XIAP−/− or cIAP1−/− animals were equivalent to those in WT animals after allo-BMT (data not shown). Thus, the contribution of IAPs to inflammasome activation is unlikely to be a critical component for its role in mitigating GVHD severity, but this would need to be formally tested.
Our observations suggest that the role of IAPs in gut GVHD is limited to nonhematopoietic target tissues, such as IECs. With regard to the function of IAPs in tissue homeostasis, increased IAP expression in IECs after IL-11 treatment seems to promote wound healing and regeneration in IBD, and the disruption of IAP signaling impairs regenerative process in the intestine and worsens IBD.73 In addition, inhibition of IAPs with SMAC mimetics increased enterocyte shedding and loss of barrier function in piglet ileal mucosa after oral infection with Cryptosporidium parvum, a significant diarrhea-inducing pathogen.69 These data are in part consistent with our results and indicate that a delicate balance may exist in the function of IAPs. Our data might also explain clinical observations in XIAP patients. For example, these data are consistent with the clinical observation that patients with X-linked lymphoproliferative disease 2 who have an XIAP-inactivating mutation develop severe intestinal inflammation.74 Furthermore, a recent clinical study demonstrated that the outcome of XIAP-deficient recipients who underwent allo-HCT after myeloablative conditioning was poor.75 XIAP-deficient patients show increased apoptosis of their lymphocytes after various stimulations and a decreased number of NK T cells.6 Up to 20% of patients with XIAP mutations develop IBD and systemic inflammation.74,76,77 Our data show how IAPs contribute to GI GVHD and, when taken in light of earlier observations in IBD and XIAP patients, point to the contribution of tissue tolerance in the maintenance of gut homeostasis in the context of allogeneic immunity. Interestingly, recent clinical observations suggests that XIAP or cIAP1 polymorphisms are presumably associated with hereditary periodic fever syndromes78 and asthma susceptibility.79 Whether specific human polymorphisms of IAPs are involved in the severity of human host susceptibility to GI GVHD would certainly be of great interest, but would represent a risk factor rather than a biomarker. We found that antiapoptotic protein BCL-2 was decreased in the IECs of cIAP1−/− and XIAP−/− animals and was associated with a significant increase in the ratio of proapoptotic to antiapoptotic proteins. XIAP inhibits the mitochondrial apoptosis pathway by activating caspases 3, 7, and 9.80 Therefore, inhibiting XIAP may increase this signaling and promote apoptosis of epithelial cells. We also found that IECs in XIAP−/− animals showed low LC3 expression after allo-BMT. Because autophagy plays an important role in maintaining GI homeostasis and its absence has a negative impact on gut inflammation,57,58,81,82 it may be a potential mechanism of increased gut damage in XIAP−/− animals. How XIAP regulates autophagy in IECs will be great interest, and additional detailed studies will be required. To explore potential mechanisms of increased gut GVHD in IAP-deficient animals, we also tested IL-22, which is a critical regulator of gut homeostasis,83 and lgr5 expression, which is an intestinal stem-cell marker, deletion of which is involved in the severity of gut GVHD83,84; however, both IL-22 and lgr5 expression in IAP-deficient animals were comparable to those in WT animals (supplemental Figure 8B-C). Interestingly, cIAP1−/− animals showed reduced levels of ρ-A expression. IAPs directly regulate ρ GTPases, which are known to control many cellular functions,59-61 cellular development,62 and gut homeostasis.63 Recent studies have suggested that ρ-A inactivation63 and Ras-related C3 botulium toxin substrate (Rac1) activation85 are associated with IBD63,86 and that IAPs moderate these signaling pathways.87 Therefore, the ρ-A signaling pathway, in addition to increasing apoptosis, may be a potential mechanism of enhancing GVHD in cIAP1−/− animals. Because both autophagy and ρ-A signaling play important roles in maintaining GI homeostasis and their absence has a negative impact on gut inflammation,63,88,89 enhancing autophagy or ρ-A signaling might protect against GI damage and ameliorate GVHD after allo-BMT. Although these will be explored in future studies, whether they are uniquely relevant for cIAP- or also important for XIAP-induced mechanisms remains unknown. A notable aspect of our data is that the significant impact of IAP expression was observed only with regard to the severity of GI GVHD, but not with skin or liver GVHD. The mechanisms for GI-specific reduction of GVHD will be explored in future studies. However, these data point to the possibility that tissue-intrinsic mechanisms that protect them from immune-mediated damage might be distinct among various GVHD target organs. Collectively, these results suggest that the expression of IAPs in host target tissues might regulate a complex signaling network that protects the GI tract from severe damage in settings of intense inflammatory injury, such as GVHD.
In conclusion, our data demonstrate a novel role for IAPs in the regulation of GVHD within target organs and independent of their role in donor T cells or host hematopoietic cells. Our data suggest that altering IAP-dependent target tissue sensitivity to apoptosis may represent a novel strategy to reduce GVHD severity.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
This work was supported by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (HL090775 and HL128046) and National Cancer Institute (CA173878 and CA203542), an American Society of Blood and Marrow Transplantation New Investigator Award (T.T.), and a Dragon Bleu Foundation fellowship from Switzerland and Kurt and Senta Herrmann-Stiftung fellowship from Lichtenstein (C.R.).
Authorship
Contribution: T.T. designed and performed experiments, analyzed the data, and wrote the paper; C.R. performed experiments, analyzed the data, and wrote the paper; K.O.-W. performed experiments; C.Z. performed experiments; C.L. performed experiments and histopathological analysis; S.-R.J.W., Y.S., H.F., H.T., D.P., I.H., M.R., and S.B. performed experiments; C.S.D. contributed reagents and the XIAP/cIAP1-deficient animals and contributed intellectually; S.W. contributed intellectually and contributed the reagents and SMAC mimetic; and P.R. designed experiments, analyzed the data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Pavan Reddy, Department of Internal Medicine, Division of Hematology and Oncology, Blood and Marrow Transplantation Program, University of Michigan Comprehensive Cancer Center, 3312 CCGC, 1500 E. Medical Center Dr, Ann Arbor, MI 48105-1942; e-mail: reddypr@med.umich.edu.
References
- 1.Beug ST, Cheung HH, LaCasse EC, Korneluk RG. Modulation of immune signalling by inhibitors of apoptosis. Trends Immunol. 2012;33(11):535-545. [DOI] [PubMed] [Google Scholar]
- 2.Pedersen J, LaCasse EC, Seidelin JB, Coskun M, Nielsen OH. Inhibitors of apoptosis (IAPs) regulate intestinal immunity and inflammatory bowel disease (IBD) inflammation. Trends Mol Med. 2014;20(11):652-665. [DOI] [PubMed] [Google Scholar]
- 3.Vandenabeele P, Bertrand MJ. The role of the IAP E3 ubiquitin ligases in regulating pattern-recognition receptor signalling. Nat Rev Immunol. 2012;12(12):833-844. [DOI] [PubMed] [Google Scholar]
- 4.Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388(6639):300-304. [DOI] [PubMed] [Google Scholar]
- 5.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132(3):344-362. [DOI] [PubMed] [Google Scholar]
- 6.Rigaud S, Fondanèche MC, Lambert N, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature. 2006;444(7115):110-114. [DOI] [PubMed] [Google Scholar]
- 7.Marsh RA, Madden L, Kitchen BJ, et al. XIAP deficiency: a unique primary immunodeficiency best classified as X-linked familial hemophagocytic lymphohistiocytosis and not as X-linked lymphoproliferative disease. Blood. 2010;116(7):1079-1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Speckmann C, Ehl S. XIAP deficiency is a mendelian cause of late-onset IBD. Gut. 2014;63(6):1031-1032. [DOI] [PubMed] [Google Scholar]
- 9.Infante JR, Dees EC, Olszanski AJ, et al. Phase I dose-escalation study of LCL161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2014;32(28):3103-3110. [DOI] [PubMed] [Google Scholar]
- 10.Schimmer AD, Estey EH, Borthakur G, et al. Phase I/II trial of AEG35156 X-linked inhibitor of apoptosis protein antisense oligonucleotide combined with idarubicin and cytarabine in patients with relapsed or primary refractory acute myeloid leukemia. J Clin Oncol. 2009;27(28):4741-4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov. 2012;11(2):109-124. [DOI] [PubMed] [Google Scholar]
- 12.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102(1):33-42. [DOI] [PubMed] [Google Scholar]
- 13.Yang Y, Fang S, Jensen JP, Weissman AM, Ashwell JD. Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science. 2000;288(5467):874-877. [DOI] [PubMed] [Google Scholar]
- 14.Hay BA, Wassarman DA, Rubin GM. Drosophila homologs of baculovirus inhibitor of apoptosis proteins function to block cell death. Cell. 1995;83(7):1253-1262. [DOI] [PubMed] [Google Scholar]
- 15.Harlin H, Reffey SB, Duckett CS, Lindsten T, Thompson CB. Characterization of XIAP-deficient mice. Mol Cell Biol. 2001;21(10):3604-3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Varfolomeev E, Blankenship JW, Wayson SM, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell. 2007;131(4):669-681. [DOI] [PubMed] [Google Scholar]
- 17.Vince JE, Wong WW, Khan N, et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell. 2007;131(4):682-693. [DOI] [PubMed] [Google Scholar]
- 18.Bai L, Smith DC, Wang S. Small-molecule SMAC mimetics as new cancer therapeutics. Pharmacol Ther. 2014;144(1):82-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gaither A, Porter D, Yao Y, et al. A Smac mimetic rescue screen reveals roles for inhibitor of apoptosis proteins in tumor necrosis factor-alpha signaling. Cancer Res. 2007;67(24):11493-11498. [DOI] [PubMed] [Google Scholar]
- 20.Vince JE, Wong WW, Gentle I, et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity. 2012;36(2):215-227. [DOI] [PubMed] [Google Scholar]
- 21.Wong WW, Vince JE, Lalaoui N, et al. cIAPs and XIAP regulate myelopoiesis through cytokine production in an RIPK1- and RIPK3-dependent manner. Blood. 2014;123(16):2562-2572. [DOI] [PubMed] [Google Scholar]
- 22.Yabal M, Müller N, Adler H, et al. XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Reports. 2014;7(6):1796-1808. [DOI] [PubMed] [Google Scholar]
- 23.Mayer BA, Rehberg M, Erhardt A, et al. Inhibitor of apoptosis proteins as novel targets in inflammatory processes. Arterioscler Thromb Vasc Biol. 2011;31(10):2240-2250. [DOI] [PubMed] [Google Scholar]
- 24.Tseng PH, Matsuzawa A, Zhang W, Mino T, Vignali DA, Karin M. Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat Immunol. 2010;11(1):70-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet. 2009;373(9674):1550-1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783-801. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Chen GY, Zheng P. CD24-Siglec G/10 discriminates danger- from pathogen-associated molecular patterns. Trends Immunol. 2009;30(12):557-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shlomchik WD, Couzens MS, Tang CB, McNiff J, Robert ME, Liu J, et al. Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science. 1999;285(5426):412-415. [DOI] [PubMed] [Google Scholar]
- 29.Teshima T, Ordemann R, Reddy P, Gagin S, Liu C, Cooke KR, et al. Acute graft-versus-host disease does not require alloantigen expression on host epithelium. Nat Med. 2002;8(6):575-581. [DOI] [PubMed] [Google Scholar]
- 30.Cai Q, Sun H, Peng Y, et al. A potent and orally active antagonist (SM-406/AT-406) of multiple inhibitor of apoptosis proteins (IAPs) in clinical development for cancer treatment. J Med Chem. 2011;54(8):2714-2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun H, Lu J, Liu L, Yang CY, Wang S. Potent and selective small-molecule inhibitors of cIAP1/2 proteins reveal that the binding of Smac mimetics to XIAP BIR3 is not required for their effective induction of cell death in tumor cells. ACS Chem Biol. 2014;9(4):994-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hurwitz HI, Smith DC, Pitot HC, et al. Safety, pharmacokinetics, and pharmacodynamic properties of oral DEBIO1143 (AT-406) in patients with advanced cancer: results of a first-in-man study. Cancer Chemother Pharmacol. 2015;75(4):851-859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DiPersio JF, Erba HP, Larson RA, et al. Oral Debio1143 (AT406), an antagonist of inhibitor of apoptosis proteins, combined with daunorubicin and cytarabine in patients with poor-risk acute myeloid leukemia—results of a phase I dose-escalation study. Clin Lymphoma Myeloma Leuk. 2015;15(7):443-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reddy P, Maeda Y, Liu C, Krijanovski OI, Korngold R, Ferrara JL. A crucial role for antigen-presenting cells and alloantigen expression in graft-versus-leukemia responses. Nat Med. 2005;11(11):1244-1249. [DOI] [PubMed] [Google Scholar]
- 35.Toubai T, Hou G, Mathewson N, et al. Siglec-G-CD24 axis controls the severity of graft-versus-host disease in mice. Blood. 2014;123(22):3512-3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toubai T, Sun Y, Luker G, Liu J, Luker KE, Tawara I, et al. Host-derived CD8+ dendritic cells are required for induction of optimal graft-versus-tumor responses after experimental allogeneic bone marrow transplantation. Blood. 2013;121(20):4231-4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Toubai T, Sun Y, Tawara I, et al. Ikaros-Notch axis in host hematopoietic cells regulates experimental graft-versus-host disease. Blood. 2011;118(1):192-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reddy P, Sun Y, Toubai T, et al. Histone deacetylase inhibition modulates indoleamine 2,3-dioxygenase-dependent DC functions and regulates experimental graft-versus-host disease in mice. J Clin Invest. 2008;118(7):2562-2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cooke KR, Hill GR, Crawford JM, et al. Tumor necrosis factor- alpha production to lipopolysaccharide stimulation by donor cells predicts the severity of experimental acute graft-versus-host disease. J Clin Invest. 1998;102(10):1882-1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hill GR, Cooke KR, Teshima T, et al. Interleukin-11 promotes T cell polarization and prevents acute graft-versus-host disease after allogeneic bone marrow transplantation. J Clin Invest. 1998;102(1):115-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Toubai T, Tawara I, Sun Y, Liu C, Nieves E, Evers R, et al. Induction of acute GVHD by sex-mismatched H-Y antigens in the absence of functional radiosensitive host hematopoietic-derived antigen-presenting cells. Blood. 2012;119(16):3844-3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y, Shlomchik WD, Joe G, et al. APCs in the liver and spleen recruit activated allogeneic CD8+ T cells to elicit hepatic graft-versus-host disease. J Immunol. 2002;169(12):7111-7118. [DOI] [PubMed] [Google Scholar]
- 43.Montufar-Solis D, Klein JR. An improved method for isolating intraepithelial lymphocytes (IELs) from the murine small intestine with consistently high purity. J Immunol Methods. 2006;308(1-2):251-254. [DOI] [PubMed] [Google Scholar]
- 44.Bertrand MJ, Doiron K, Labbé K, Korneluk RG, Barker PA, Saleh M. Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity. 2009;30(6):789-801. [DOI] [PubMed] [Google Scholar]
- 45.Mahoney DJ, Cheung HH, Mrad RL, et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci USA. 2008;105(33):11778-11783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Varfolomeev E, Goncharov T, Maecker H, et al. Cellular inhibitors of apoptosis are global regulators of NF-κB and MAPK activation by members of the TNF family of receptors. Sci Signal. 2012;5(216):ra22. [DOI] [PubMed] [Google Scholar]
- 47.Krieg A, Correa RG, Garrison JB, et al. XIAP mediates NOD signaling via interaction with RIP2. Proc Natl Acad Sci USA. 2009;106(34):14524-14529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Piguet PF, Grau GE, Allet B, Vassalli P. Tumor necrosis factor/cachectin is an effector of skin and gut lesions of the acute phase of graft-vs.-host disease. J Exp Med. 1987;166(5):1280-1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang T, Li Y, Zou P, et al. Physiologically based pharmacokinetic and pharmacodynamic modeling of an antagonist (SM-406/AT-406) of multiple inhibitor of apoptosis proteins (IAPs) in a mouse xenograft model of human breast cancer. Biopharm Drug Dispos. 2013;34(6):348-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brunckhorst MK, Lerner D, Wang S, Yu Q. AT-406, an orally active antagonist of multiple inhibitor of apoptosis proteins, inhibits progression of human ovarian cancer. Cancer Biol Ther. 2012;13(9):804-811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Korngold R, Sprent J. Lethal graft-versus-host disease after bone marrow transplantation across minor histocompatibility barriers in mice. Prevention by removing mature T cells from marrow. J Exp Med. 1978;148(6):1687-1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gentle IE, Moelter I, Lechler N, et al. Inhibitors of apoptosis proteins (IAPs) are required for effective T-cell expansion/survival during antiviral immunity in mice. Blood. 2014;123(5):659-668. [DOI] [PubMed] [Google Scholar]
- 53.Dougan M, Dougan S, Slisz J, et al. IAP inhibitors enhance co-stimulation to promote tumor immunity. J Exp Med. 2010;207(10):2195-2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koyama M, Kuns RD, Olver SD, Raffelt NC, Wilson YA, Don AL, et al. Recipient nonhematopoietic antigen-presenting cells are sufficient to induce lethal acute graft-versus-host disease. Nat Med. 2012;18(1):135-142. [DOI] [PubMed] [Google Scholar]
- 55.Hawkins CJ, Uren AG, Häcker G, Medcalf RL, Vaux DL. Inhibition of interleukin 1 beta-converting enzyme-mediated apoptosis of mammalian cells by baculovirus IAP. Proc Natl Acad Sci USA. 1996;93(24):13786-13790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Uren AG, Pakusch M, Hawkins CJ, Puls KL, Vaux DL. Cloning and expression of apoptosis inhibitory protein homologs that function to inhibit apoptosis and/or bind tumor necrosis factor receptor-associated factors. Proc Natl Acad Sci USA. 1996;93(10):4974-4978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Groulx JF, Khalfaoui T, Benoit YD, et al. Autophagy is active in normal colon mucosa. Autophagy. 2012;8(6):893-902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mukhopadhyay S, Panda PK, Sinha N, Das DN, Bhutia SK. Autophagy and apoptosis: where do they meet? Apoptosis. 2014;19(4):555-566. [DOI] [PubMed] [Google Scholar]
- 59.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420(6916):629-635. [DOI] [PubMed] [Google Scholar]
- 60.Geisbrecht ER, Montell DJ. A role for Drosophila IAP1-mediated caspase inhibition in Rac-dependent cell migration. Cell. 2004;118(1):111-125. [DOI] [PubMed] [Google Scholar]
- 61.Oberoi-Khanuja TK, Murali A, Rajalingam K. IAPs on the move: role of inhibitors of apoptosis proteins in cell migration. Cell Death Dis. 2013;4:e784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kenneth NS, Duckett CS. IAP proteins: regulators of cell migration and development. Curr Opin Cell Biol. 2012;24(6):871-875. [DOI] [PubMed] [Google Scholar]
- 63.López-Posadas R, Becker C, Günther C, et al. Rho-A prenylation and signaling link epithelial homeostasis to intestinal inflammation. J Clin Invest. 2016;126(2):611-626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Damgaard RB, Nachbur U, Yabal M, et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell. 2012;46(6):746-758. [DOI] [PubMed] [Google Scholar]
- 65.Moulin M, Anderton H, Voss AK, et al. IAPs limit activation of RIP kinases by TNF receptor 1 during development. EMBO J. 2012;31(7):1679-1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Labbé K, McIntire CR, Doiron K, Leblanc PM, Saleh M. Cellular inhibitors of apoptosis proteins cIAP1 and cIAP2 are required for efficient caspase-1 activation by the inflammasome. Immunity. 2011;35(6):897-907. [DOI] [PubMed] [Google Scholar]
- 67.Grabinger T, Bode KJ, Demgenski J, et al. Inhibitor of apoptosis protein-1 regulates tumor necrosis factor-mediated destruction of intestinal epithelial cells. Gastroenterology. 2017;152(4):867-879. [DOI] [PubMed] [Google Scholar]
- 68.Jenq RR, Ubeda C, Taur Y, et al. Regulation of intestinal inflammation by microbiota following allogeneic bone marrow transplantation. J Exp Med. 2012;209(5):903-911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Foster DM, Stauffer SH, Stone MR, Gookin JL. Proteasome inhibition of pathologic shedding of enterocytes to defend barrier function requires X-linked inhibitor of apoptosis protein and nuclear factor kappaB. Gastroenterology. 2012;143(1):133-144.e4. [DOI] [PubMed] [Google Scholar]
- 70.Jankovic D, Ganesan J, Bscheider M, et al. The Nlrp3 inflammasome regulates acute graft-versus-host disease. J Exp Med. 2013;210(10):1899-1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen S, Smith BA, Iype J, et al. MicroRNA-155-deficient dendritic cells cause less severe GVHD through reduced migration and defective inflammasome activation. Blood. 2015;126(1):103-112. [DOI] [PubMed] [Google Scholar]
- 72.Koehn BH, Apostolova P, Haverkamp JM, et al. GVHD-associated, inflammasome-mediated loss of function in adoptively transferred myeloid-derived suppressor cells. Blood. 2015;126(13):1621-1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Naugler KM, Baer KA, Ropeleski MJ. Interleukin-11 antagonizes Fas ligand-mediated apoptosis in IEC-18 intestinal epithelial crypt cells: role of MEK and Akt-dependent signaling. Am J Physiol Gastrointest Liver Physiol. 2008;294(3):G728-G737. [DOI] [PubMed] [Google Scholar]
- 74.Zeissig Y, Petersen BS, Milutinovic S, et al. XIAP variants in male Crohn’s disease. Gut. 2015;64(1):66-76. [DOI] [PubMed] [Google Scholar]
- 75.Marsh RA, Rao K, Satwani P, et al. Allogeneic hematopoietic cell transplantation for XIAP deficiency: an international survey reveals poor outcomes. Blood. 2013;121(6):877-883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aguilar C, Latour S. X-linked inhibitor of apoptosis protein deficiency: more than an X-linked lymphoproliferative syndrome. J Clin Immunol. 2015;35(4):331-338. [DOI] [PubMed] [Google Scholar]
- 77.Pachlopnik Schmid J, Canioni D, Moshous D, et al. Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency). Blood. 2011;117(5):1522-1529. [DOI] [PubMed] [Google Scholar]
- 78.Ferretti M, Gattorno M, Chiocchetti A, et al. The 423Q polymorphism of the X-linked inhibitor of apoptosis gene influences monocyte function and is associated with periodic fever. Arthritis Rheum. 2009;60(11):3476-3484. [DOI] [PubMed] [Google Scholar]
- 79.Roscioli E, Hamon R, Ruffin RE, et al. BIRC3 single nucleotide polymorphism associate with asthma susceptibility and the abundance of eosinophils and neutrophils. J Asthma. 2017;54(2):116-124. [DOI] [PubMed] [Google Scholar]
- 80.Schimmer AD. Inhibitor of apoptosis proteins: translating basic knowledge into clinical practice. Cancer Res. 2004;64(20):7183-7190. [DOI] [PubMed] [Google Scholar]
- 81.Wu S, Zhang YG, Lu R, et al. Intestinal epithelial vitamin D receptor deletion leads to defective autophagy in colitis. Gut. 2015;64(7):1082-1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Whelan KA, Merves JF, Giroux V, et al. Autophagy mediates epithelial cytoprotection in eosinophilic oesophagitis. Gut. 2017;66(7):1197-1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hanash AM, Dudakov JA, Hua G, et al. Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity. 2012;37(2):339-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lindemans CA, Calafiore M, Mertelsmann AM, et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature. 2015;528(7583):560-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Winge MC, Ohyama B, Dey CN, et al. RAC1 activation drives pathologic interactions between the epidermis and immune cells. J Clin Invest. 2016;126(7):2661-2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Parikh K, Zhou L, Somasundaram R, et al. Suppression of p21Rac signaling and increased innate immunity mediate remission in Crohn’s disease. Sci Transl Med. 2014;6(233):233ra53. [DOI] [PubMed] [Google Scholar]
- 87.Oberoi TK, Dogan T, Hocking JC, et al. IAPs regulate the plasticity of cell migration by directly targeting Rac1 for degradation. EMBO J. 2012;31(1):14-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cadwell K, Liu JY, Brown SL, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456(7219):259-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gutierrez MG, Saka HA, Chinen I, et al. Protective role of autophagy against Vibrio cholerae cytolysin, a pore-forming toxin from V. cholerae. Proc Natl Acad Sci USA. 2007;104(6):1829-1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.