

Key Points
A CD34-selected, T-cell–depleted alternative donor graft after a reduced conditioning regimen resulted in engraftment in patients with sickle cell.
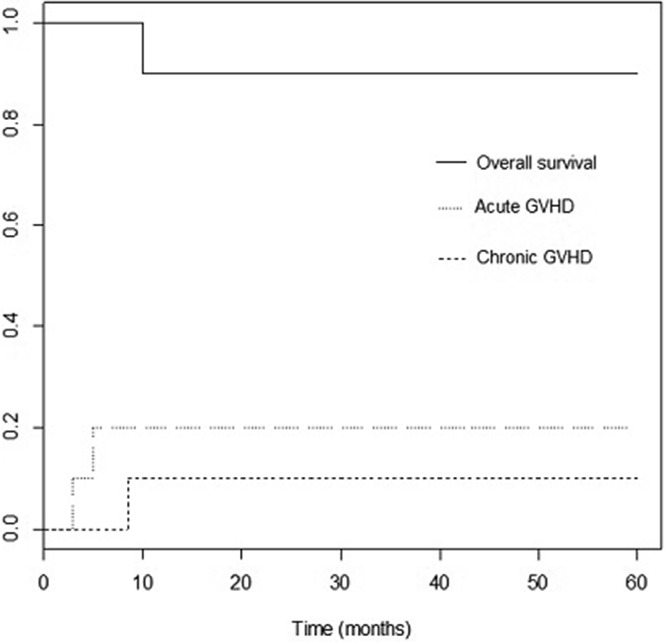
This approach was associated with a low incidence of acute and chronic graft-versus-host disease and very good survival.
Abstract
Most patients who could be cured of sickle cell disease (SCD) with stem cell transplantation do not have a matched sibling donor. Successful use of alternative donors, including mismatched family members, could provide a donor for almost all patients with SCD. The use of a reduced-intensity conditioning regimen may decrease late adverse effects. Ten patients with symptomatic SCD underwent CD34+ cell-selected, T-cell–depleted peripheral blood stem cell transplantation from a mismatched family member or unrelated donor. A reduced-intensity conditioning regimen including melphalan, thiotepa, fludarabine, and rabbit anti-thymocyte globulin was used. Patients were screened for a companion study for immune reconstitution that included a donor lymphocyte infusion given 30-42 days after transplant with intravenous methotrexate as graft-versus-host disease (GVHD) prophylaxis. Seven eligible patients were treated on the companion study. Nine of 10 patients are alive with a median follow-up of 49 months (range, 14-60 months). Surviving patients have stable donor hematopoietic engraftment (mean donor chimerism, 99.1% ± 0.7%). There were no sickle cell complications after transplant. Two patients had grade II-IV acute GVHD. One patient had chronic GVHD. Epstein-Barr virus–related posttransplant lymphoproliferative disorder (PTLD) occurred in 3 patients, and 1 patient died as a consequence of treatment of PTLD. Two-year overall survival was 90%, and event-free survival was 80%. A reduced-intensity conditioning regimen followed by CD34+ cell-selected, T-cell–depleted alternative donor peripheral blood stem cell transplantation achieved primary engraftment in all patients with a low incidence of GVHD, although PTLD was problematic. This trial was registered at clinicaltrials.gov as #NCT00968864.
Visual Abstract
Introduction
Sickle cell disease (SCD) is caused by an abnormal hemoglobin that results in sickling of red blood cells and obstruction of blood flow in small vessels. Vasoocclusion causes chronic organ damage, resulting in multisystem disease, and is associated with substantial morbidity and mortality.1,2 At this time, hematopoietic stem cell transplantation is the only curative treatment of SCD.3
Figure 1.

Lymphocyte recovery. Boxplots for lymphocyte subset for each point. The bottom and top of the box are the 25th (Q1) and 75th (Q3) percentiles of the data, and the band near the middle of the box is the median. The upper whisker is located at the smaller of the maximum value and Q3 + 1.5 IQR, where IQR = Q3 − Q1, the box length. The lower whisker is located at the larger of the smallest value, and Q1 − 1.5 IQR. Observations above Q3 + 1.5 IQR are shown by dots. (A) CD3+CD4+ cells/µL. (B) CD3+CD8+ cells/µL. (C) CD19+ cells/µL. T cells were isolated by negative selection using RosetteSep T-cell enrichment (StemCell Technologies; Vancouver, BC). T- and B-cell subsets were quantified using the TetraCXP system (antibodies recognizing CD45, CD3, CD4, CD8, and CD19) and a FC500 flow cytometer (Beckman Coulter, Indianapolis, IN).
Myeloablative HLA-matched sibling donor transplantation for pediatric SCD has been successful, with survival rates now approaching more than 95%.4-7 Most patients who could be cured of SCD by stem cell transplantation do not have a HLA-matched sibling donor or an 8/8 matched unrelated donor.8-10 Historically, alternative donor (unrelated donor or mismatched related) transplantation for SCD has been less successful than HLA-matched sibling transplantation as a result of graft failure or GVHD, resulting in failure to cure the SCD and/or poor survival.11-15 Engraftment has been a major challenge in the mismatched related donor setting, with graft failure rates of 38% to 43%.15,16 Long-term complications of myeloablative chemotherapy include progressive declines in renal, pulmonary, and cardiac function over time.16 Gonadal toxicity and fear of transplant-related complications including chronic GVHD have been cited as reasons to not transplant otherwise eligible patients with SCD.17,18 Reduced-intensity conditioning (RIC) has been used successfully for HLA-matched sibling donor transplantation for SCD and may reduce the long-term sequelae compared with myeloablative regimens.19-21
Successful use of alternative donors including mismatched family members, with reliable engraftment and a low incidence of GVHD, could provide a donor for almost all patients with SCD, as nearly all patients will have a related HLA-haploidentical donor. Matched unrelated and mismatched related donors were used in the study because both are associated with a significant risk for GVHD. T-cell depletion offers the possibility of a very low risk for GVHD and the avoidance of short- and long-term toxicities associated with calcineurin inhibitors. The use of RIC may limit long-term adverse effects. We report our experience using CD34+ cell-selected, T-cell–depleted peripheral blood stem cell (PBSC) alternative donor transplantation after a RIC regimen for pediatric and young adult patients with SCD.
Patients were also offered treatment on a companion study of donor lymphocyte infusion (DLI) and methotrexate (MTX) GVHD prophylaxis to accelerate immune recovery. MTX was used to prevent GVHD from the DLI by killing T cells responding to alloantigens in the recipient while sparing a sufficient number of viral-specific T cells to provide some immunity and protection from infection. MTX is an antiproliferative chemotherapy drug that has been used for the prevention of GVHD for many years.22
Methods
Patients were treated on a phase 2 study (IND 14045) to determine the efficacy of CD34+ cell selection for the prevention of severe acute GVHD in alternative donor stem cell transplantation. The study was approved by the Carolinas HealthCare System Institutional Review Board and was monitored by the Pediatric Blood and Marrow Transplant Consortium Data Safety Monitoring Committee. All patients (or a parent or guardian for minors) gave written informed consent in accordance with the Declaration of Helsinki. This report addresses only the consecutive sickle cell patients treated in this study.
An unrelated donor search was performed for all patients, and those without an 8/8 HLA-matched donor were offered transplant using a mismatched related donor. Donors were prioritized based on cytomegalovirus (CMV) status (CMV-positive donor required for a CMV-positive recipient after patient 2), lowest number of HLA mismatches, ABO compatibility, and younger age. Donors could only be used if the recipient did not have anti-HLA antibodies directed against donor antigens. Related donors could have sickle cell trait.
Inclusion criteria included age younger than 30 years, lack of a matched sibling donor, and the presence of debilitating SCD, as described previously,4 with minor modifications. Debilitating disease was defined as at least 1 of the following: recurrent painful events that could not be explained exclusively by other causes (at least 2 events per year in the 2 years before enrollment), acute chest syndrome with the development of a new infiltrate on chest radiograph and/or having a perfusion defect demonstrable on a lung radioisotope scan that required hospitalization and/or transfusion therapy (at least 2 acute chest syndrome episodes in the 2 years before enrollment), a history of any clinically significant neurologic event (stroke or hemorrhage), any combination of painful events and acute chest syndrome episodes that total 4 events within the 2 years before enrollment, an abnormal transcranial Doppler documented on 2 occasions, the presence of osteonecrosis, recurrent priapism that required hospitalization and/or transfusion therapy (at least 2 episodes in the past 2 years before enrollment), stage I or II sickle lung disease, sickle retinopathy and visual impairment in at least 1 eye, red cell alloimmunization during chronic transfusion therapy, chronic transfusion therapy for manifestations of SCD, sickle nephropathy (glomerular filtration rate 30%-50% of normal), and other unusual scenarios if there was agreement between the patient’s hematologist and transplant physician that the patient’s condition warranted transplant. All patients except patient 3, who had Kostmann’s neutropenia as a result of an ELA-2 mutation, failed hydroxyurea before transplant.
Exclusion criteria included adjusted diffusion capacity for carbon monoxide, forced expiratory volume in the first second, forced vital capacity of less than 50% of predicted, cardiac ejection fraction less than 40%, creatinine clearance below 40 mL/min/1.73 m2, serum alanine aminotransferase levels higher than 5 times or direct bilirubin levels higher than 2 times the upper limit of normal, performance score below 50, HIV or HTLV-I/II infection, active CMV or hepatitis infection, or serious infection within the 4 weeks preceding transplant.
Donors underwent 4 days of filgrastim mobilization (10-16 μg/kg per day) followed by large-volume leukapheresis, with a goal of collecting at least 20 × 106 CD34+ cells/kg recipient body weight for mismatched related donors and 10 × 106 CD34+ cells/kg body weight for matched unrelated donors. CD34+ cells were isolated using an automated cell selection device based on magnetic cell separation technology (CliniMACS; Miltenyi Biotec, Bergisch Gladbach, Germany). The cells were processed and cryopreserved at the University of California, San Francisco, and then shipped when needed.23 The conditioning regimen included melphalan 140 mg/m2 on day −7, thiotepa 5 mg/kg ×2 doses on day −6, fludarabine 40 mg/m2 daily on days −6 through −2, and rabbit anti-thymocyte globulin (Thymoglobulin; Genzyme, Quebec, Canada) 2.5 mg/kg daily on days −5 through −2. Because 2 of the first 3 patients had Epstein-Barr virus (EBV) reactivation, rituximab 375 mg/m2 was added on day −1 for EBV seropositive recipients (patients 4, 7, 8, and 9). Patient 5 received a reduced rituximab dose of 150 mg/m2 because at the time, CMV seropositive patients received a lower dose. All patients were screened for participation in a phase 1/2 companion study to hasten immune reconstitution (registered at clinicaltrials.gov as #NCT01027702). The study included a DLI of 3 to 5 × 104 CD3+ cells/kg 30 to 42 days after transplant, followed by intravenous MTX. MTX 10 mg/m2 was given 24 hours after DLI and then weekly starting 3 days after the DLI (ie, days +3, +10, +17, +24) for 1 patient. The duration of MTX was extended to 80 days after transplant for 6 patients because of late acute GVHD on the companion study. The dosing of the additional weekly MTX was 7.5 mg/m2 for 2 weeks and 5 mg/m2 for 6 weeks.
Supportive care
All patients had a hemoglobin S level of 35% or lower as a result of erythrocytapheresis within 1 week of admission for transplant or chronic red blood cell transfusions. All patients had platelet and red cell transfusions to maintain platelet counts greater than 50 000/μL and hemoglobin 9 to 11 g/dL. Infection prophylaxis included antiviral prophylaxis, Pneumocystis prophylaxis, antifungal prophylaxis, and intravenous gammaglobulin. Intravenous gammaglobulin was given for at least 6 months, until both the immunoglobulin G level was normal (when intravenous gammaglobulin was due) and the immunoglobulin M level was normal. Patients were monitored by real-time polymerase chain reaction testing for CMV, human herpesvirus-6 (HHV-6), EBV, and adenovirus, and preemptive therapy (foscarnet, ganciclovir, valganciclovir, cidofovir, or rituximab as appropriate) was initiated for any positive test until T-cell recovery. All patients received seizure prophylaxis.
Statistical analysis
Patients were to be followed for 5 years after transplant. Graft failure was defined as primary (lack of absolute neutrophil count >500 by day +28 after transplant) or late (secondary; initial evidence for marrow recovery and engraftment with subsequent pancytopenia without another cause [ie, infection and/or drug therapy] and decrease in donor chimerism by >50% from highest level achieved or decrease in donor chimerism to <10%). Acute and chronic GVHD were graded per standard criteria.24-26 A paired t test was used to compare pre- and posttransplant values. STATA v10 (StataCorp, College Station, TX) was used to generate analysis. Overall survival was defined as time from transplant to death from any cause, and event-free survival was defined as death from any cause or graft failure. Overall and event-free survival were calculated using the Kaplan-Meier method.27
Results
Characteristics of donors, recipients, and grafts
Ten patients were enrolled between August 2009 and August 2015. Eight alternative donors were mismatched related, and 2 were unrelated. The median donor age was 21 years (range, 13-49 years). Six of 8 mismatched related donors and neither of the 2 unrelated donors had sickle cell trait. Donors underwent a median of 2 days of apheresis collection for PBSCs (range, 1-3 days). Median recipient age was 14 years (range, 5-23 years). The CD34+ cell selection procedure resulted in a median 5.5 (range, 5.1-5.8) log reduction of CD3+ cells and median 63% (range, 52%-86%) recovery of CD34+ cells. The median total CD34+ cell dose infused was 18 × 106 CD34+ cells/kg (range, 9-25 × 106 CD34+ cells/kg), and all patients received <1 × 104 CD3+ cells/kg. Recipient and donor characteristics are summarized in Table 1. Seven patients were enrolled on the companion immune recovery study. Three patients were not eligible because of asymptomatic HHV-6 reactivation (subsequently removed as an exclusion criterion), decreasing donor chimerism, and EBV-PTLD. The companion study involved an escalation of the DLI dose. At a dose of 5 × 104 CD3+ cells/kg, which was determined to be the optimal dose per protocol, 6/9 evaluable patients had CD4 higher than 100/µL at 4 months after transplant, and there was an 11% incidence of acute GVHD.
Table 1.
Recipient and donor characteristics
| Patient ID | Age at HSCT (y) | Sex | Type of sickle Hgb | Coexisting conditions and indications for HSCT | Donor relationship (match and mismatched loci | Donor sickle cell trait (yes/no) | Donor blood type | Recipient blood type |
|---|---|---|---|---|---|---|---|---|
| 1 | 13 | F | SS | Recurrent VOC, acute chest syndrome, osteonecrosis | Father (7/10, HLA-A,B,C) | Yes | O+ | B+ |
| 2 | 15 | F | SS | Recurrent VOC, red cell alloimmunization | Half-brother (5/10, HLA-A,B,C,DR,DQ) | Yes | O+ | A+ |
| 3 | 13 | M | Sβ0thal | Recurrent VOC, Kostmann’s neutropenia | Unrelated donor (10/10) | No | O+ | AB+ |
| 4 | 8 | F | SS | Conditional TCD, nocturnal hypoxia, narrowing of CNS arteries on MRA | Sister (9/10, HLA-A) | Yes | A+ | B+ |
| 5 | 5 | F | SS | Stroke, narrowing of CNS arteries on MRA | Unrelated donor (9/10, HLA-DQ) | No | O+ | O+ |
| 6 | 17 | M | SS | Recurrent VOC, acute chest syndrome | Mother (5/10, HLA-A,B,C,DR,DQ) | Yes | O+ | B+ |
| 7 | 19 | M | SS | Stroke, narrowing of CNS arteries on MRA | Sister (6/10, HLA-A,B,C,DR) | Yes | O+ | O+ |
| 8 | 18 | M | SS | Recurrent VOC | Half-sister (6/10, HLA-A,B,C,DR) | No | A+ | B+ |
| 9 | 12 | M | SS | Acute chest syndrome | Uncle (5/10, HLA-A,B,C,DR,DQ) | No | AB+ | O+ |
| 10 | 23 | F | SS | Recurrent VOC, osteonecrosis | Brother (6/10 HLA-A,B,DR,DQ) | Yes | AB+ | AB+ |
CNS, central nervous system; F female; Hgb, hemoglobin; HLA, human leukocyte antigen; M, male; MRA, magnetic resonance angiogram; Sβ0thal, hemoglobin S-β0 thalassemia; SS, homozygous hemoglobin S; TCD, transcranial Doppler ultrasound; VOC, vaso-occlusive crisis.
Outcomes
Table 2 provides detailed transplant outcomes. All recipients tolerated the conditioning regimen and engrafted. Neutrophil recovery (first of 3 days of absolute neutrophil count >500/μL) occurred at a median of 14 days (range, 10-17 days). Platelet recovery to more than 50 000/μL (without a platelet transfusion in the preceding 7 days) occurred at a median of 19 days (range, 16-31 days). At last follow-up, all recipients have overall donor chimerism of 94% or more. T-cell chimerism was not checked routinely because in the first few months, patients had very few T cells, and later, most patients were 95% or more donor overall.
Table 2.
Interventions and outcome after CD34-selected, T-cell–depleted alternative donor stem cell transplant
| Patient ID | DLI CD3+ cell dose (×104/kg) (prophylactic/therapeutic) | Day after HSCT DLI given | MTX duration (days) | 4 wk after HSCT donor chimerism | 3 mo after HSCT donor chimerism | 6 mo after HSCT donor chimerism | 12 mo after HSCT donor chimerism | aGVHD (day after HSCT) | cGVHD | Major complications | Follow-up (months) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 (T) | +65 | N/A | 99 | 97 | 100 | 100 | None | None | Septic arthritis of prosthetic hip | 60+ |
| 2 | 2.5 (T), 5 (T) | +44, +70 | N/A | 94 | 85 | 100 | N/A | Grade I after HSCT; grade IV after DLI #2 (+10, +86) | None | PTLD, TMA, PRES, renal failure, seizures, Aspergillosis | 10 |
| 3 | 3 (P) | +35 | 24 | 98 | 99 | 100 | 97 | None | None | None | 60+ |
| 4 | 4 (P) | +33 | 80 | 94 | 98 | 96 | 95 | None | None | PRES, seizure | 51+ |
| 5 | 0.1 (T)* | +35 | N/A | 100 | 77 | 87 | 92 | None | None | PTLD | 50+ |
| 6 | 4 (P) | +33 | 80 | 98 | 51 | N/A | 97† | None | None | PRES, acquired aplastic anemia, second HSCT† | 49+ |
| 7 | 5 (P) | +35 | 80 | 100 | 100 | 100 | 100 | None | None | None | 28+ |
| 8 | 5 (P) | +34 | 80 | 100 | 100 | 100 | 100 | None | None | PTLD | 15+ |
| 9 | 4 (P) | +35 | 80 | 100 | 100 | 100 | 100 | None | None | None | 15+ |
| 10 | 5 (P) | +40 | 80 | 100 | 100 | 100 | 100 | Grade I (+127), grade II (+149) | Extensive, skin | Hemorrhagic cystitis caused by BKV | 14+ |
aGVHD, acute graft-versus-host disease; BKV, BK virus; cGVHD, chronic graft-versus-host disease; DLI, donor lymphocyte infusion; HSCT, hematopoietic stem cell transplant; MTX, methotrexate; N/A not applicable; PRES, posterior reversible leukoencephalopathy; PTLD, posttransplant lymphoproliferative disorder; TMA, thrombotic microangiopathy. Chimerism is on whole blood. The last 3 patients in the table received rituximab 200 mg/m2 on the day of DLI because of autoimmunity seen in patients transplanted with the same approach for diseases other than SCD. For follow-up, a + means that the patient is surviving.
One patient received a very low dose DLI and EBV-specific third-party cytotoxic T lymphocytes.
Second transplant from sickle trait negative half-brother (HLA 5/10) with CD34+ cell dose of 20.3 × 106/kg and 0.3 × 104 CD3+/kg on day +210 after first transplant. Transplant conditioning regimen was pentostatin 1.5 mg/m2 × 3 d, rabbit antithymocyte globulin 2 mg/kg × 4 d, and total lymphoid irradiation 400 cGy. Twelve months after HSCT donor chimerism refers to the second donor.
Patient 2 had decreasing donor chimerism and was treated with therapeutic DLI at day +44 after transplant. She continued to have falling chimerism and developed PTLD. She was treated with a second DLI at day +77. She developed GVHD and had a shift of chimerism to 100% donor. Patient 4 had T-cell chimerism checked starting at 4 months after transplant because overall chimerism fell from 98% to 94%. Her T-cell chimerism at 4 months was 72%, gradually rising to 91% at 12 months without intervention and without GVHD. Patient 5 had falling overall donor chimerism that was probably a result of expansion of recipient antiviral T cells in response to CMV and RSV. Donor T-cell chimerism fell as low as 13% at 4 months, but rose without intervention and without GVHD (23% at 5 months, 49% at 6 months, and 96% at 9 months). Her myeloid chimerism was always higher than 94%. Late pancytopenia occurred on day +63 after transplant in 1 patient (patient 6) after antiviral therapy for HHV-6 reactivation. Despite not qualifying as late graft failure per protocol because of alternative causes (viral infection and myelosuppressive drug therapy) and greater than 50% donor chimerism, the patient was considered to have graft rejection/graft failure because he required a second transplant using a second mismatched related donor (Table 2). He did not have anti-HLA antibodies directed against the original donor antigens. He engrafted after second transplant and remains 100% donor. Nine patients are alive with median follow-up of 49 months (range, 14-60 months). Estimated 2-year overall survival was 90%, and event-free survival was 80%.
Hemoglobin levels gradually increased after transplant. Hemolysis declined after transplant, as reflected by mean total bilirubin before transplantation of 2.1 mg/dL compared with a level 100 days after transplant of 0.5 mg/dL (P < .001). Hemoglobin S levels ranged from 0% to 39.5% among those with more than 1 year of posttransplant follow-up and were elevated in 4 patients despite 95% or more donor chimerism resulting from the use of donors with sickle cell trait. Immune recovery as assessed by CD4+ cells greater than 100/μL occurred at a median of 134 days (range, 59-342 days) after transplant and to CD4+ cells greater than 200/μL at a median of 163 days (range, 59-452 days) (Figure 1). The peak tricuspid regurgitant jet velocity before transplant was greater than 2.5 m/s in 3 of 10 patients. Two of the patients with elevated tricuspid regurgitant jet velocity jets had a decrease to less than 2.5 m/s at 1 year after transplant (2.73 to 2.25 and 2.55 to 2.36). The third patient died before this time. There were no significant declines in shortening (P = .24) or ejection fraction (P = .31) after 1 year of follow-up (data not shown). All patients had baseline pulmonary function testing completed before transplantation. There were no significant differences in the mean percentage predicted forced vital capacity, forced expiratory volume in the first second, forced expiratory flow (25%-75%), or adjusted diffusion capacity for carbon monoxide between baseline studies and at 1 year posttransplant (Table 3). There was no progression of cerebrovascular disease after transplantation in patients with abnormalities before transplant. No patient had a painful crisis or other sickle cell complication after transplant. All survivors are in school or employed. The patient with chronic GVHD is home schooled as a result of pain from osteonecrosis and severe lymphopenia.
Table 3.
Pulmonary function testing before and at 1 year after hematopoietic cell transplant
| Patient ID | FVC | Pre-HSCT | DLCOadj | FVC | 1 y post-HSCT | DLCOadj | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| FEV1 | FEF 25-75% | FEV1 | FEF 25-75% | |||||||
| 1 | 105 | 99 | 97 | 86 | 114 | 113 | 120 | 100 | ||
| 2 | 104 | 94 | 75 | 74 | N/A | N/A | N/A | N/A | ||
| 3 | 79 | 65 | 45 | 62 | 80 | 67 | 50 | 78 | ||
| 4 | 103 | 86 | 58 | 89 | 95 | 91 | 71 | 75 | ||
| 5 | 114 | 92 | 87 | N/A | 92 | 89 | 85 | N/A | ||
| 6 | 78 | 61 | 36 | 70 | 88 | 74 | 46 | 64 | ||
| 7 | 81 | 83 | 89 | 98 | 82 | 85 | 88 | 86 | ||
| 8 | 115 | 102 | 77 | 101 | 109 | 105 | 87 | 66 | ||
| 9 | 97 | 89 | 96 | 74 | 93 | 97 | 103 | 82 | ||
| 10 | 120 | 120 | 95 | 77 | 102 | 92 | 52 | 82 | ||
| Mean difference | 4.1 | 1.7 | 2.6 | 2.8 | ||||||
| Standard error | 3.6 | 4.1 | 6.2 | 6.0 | ||||||
| P value | .14 | .66 | .66 | .32 | ||||||
DLCOadj, diffusion capacity carbon monoxide adjusted for hemoglobin; FEF 25-75%, forced expiratory flow; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; N/A, not available.
Adverse events
Engraftment syndrome occurred in 2 patients. Both were treated with hydrocortisone 2 mg/kg/d on days +13 and +7, respectively, with complete resolution, but developed posterior reversible encephalopathy while receiving hydrocortisone. Both patients had clinical recovery and radiologic improvement.
Grade II-IV acute GVHD occurred in 1 of 10 patients after transplant and prophylactic DLI and was grade II in this patient. Another patient developed grade IV acute GVHD after a therapeutic DLI for PTLD. Acute GVHD involved skin and gastrointestinal tract in both patients. One patient developed extensive chronic GVHD with lichenoid skin involvement; this was the patient with a history of grade II acute GVHD.
EBV reactivation occurred in 4 patients, and 3 patients developed PTLD. PTLD occurred in 2 of 8 patients with a mismatched related donor and 1 of 2 with an unrelated donor. PTLD resolved in all patients clinically, by positron emission tomography scan, and by EBV viral load. Patient 2, who accounted for most of the severe adverse events on study, presented 2 months after transplant with high fevers and liver and bone lesions and had rapidly progressive plasmacytic PTLD. She had no response to rituximab and chemotherapy, so she was given a DLI (dose 5 × 104 CD3+ cells/kg). The PTLD resolved, but she developed grade IV gastrointestinal GVHD. She succumbed months later as a result of thrombotic microangiopathy, renal failure, and fatal disseminated aspergillosis. Patient 5 presented with fever and adenopathy and had PTLD in her nasopharynx, cervical nodes, and a retroperitoneal lymph node at 1 month after transplant. She was treated with low-dose radiation therapy, rituximab, and a very low dose DLI. She subsequently was treated with third-party EBV-specific cytotoxic T lymphocytes for persistent disease, and her PTLD resolved. Patient 8 developed EBV viremia that was not treated with rituximab per protocol because he was having T-cell recovery. Soon after, he had a mononucleosis presentation (fever, sore throat, adenopathy) at 5 months after transplant and had polymorphic PTLD. The PTLD resolved with 4 weekly doses of rituximab. This patient had a negative EBV viral load in blood for 6 months, but subsequently had EBV detected in blood intermittently between 9 and 12 months after transplant, despite immune recovery. He did not have clinical PTLD and was not treated with rituximab.
Viral reactivation (Table 4) was common, but disease was uncommon. Patient 2 had a CMV seronegative donor and developed CMV enteritis after intensive immunosuppression for acute GVHD. Six patients had HHV-6 reactivation at median day +22 (range, 8-59 days), and the only possible disease was pancytopenia after HHV-6 reactivation in 1 patient. No patients developed adenovirus reactivation.
Table 4.
Summary of virus reactivation and immune recovery after hematopoietic stem cell transplant
| Patient ID | CMV (D,R) | EBV (D,R) | CD4 >100 recovery | CD4 >200 recovery | HHV-6 reactivation day of onset | HHV-6 max level | Last day of HHV-6 reactivation | CMV reactivation and day of onset | CMV max level | Last day of CMV reactivation | EBV reactivation day of onset | EBV max level | Last day of EBV reactivation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | −/− | +/− | 132 | 176 | 8 | 7 300 | 400 | None | — | — | None | — | — |
| 2 | −/+ | +/+ | 92 | 96 | 33 | 400 | 41 | 68 | 46 500 | * | 26 | 50 018 | 98 |
| 3 | −/− | +/+ | 148 | 169 | 14 | 1 400 | 27 | None | — | — | 17 | 5 573 | 31 |
| 4 | +/− | +/+ | 148 | 149 | None | — | — | None | — | — | None | — | — |
| 5 | +/+ | +/+ | 59 | 59 | 59 | 522 | 140 | 10 | 85 500 | 192 | 24 | 89 250 | 59 |
| 6 | +/− | +/− | 342 | 452 | 17 | 31 800 | 153 | None | — | — | None | — | — |
| 7 | −/− | +/+ | 124 | 163 | 26 | 287 | 34 | None | — | — | None | — | — |
| 8 | +/− | +/+ | 153 | 163 | 65 | 76 | 90 | None | — | — | 139 | 1 095 | 365 |
| 9 | +/+ | +/+ | 136 | 167 | 24 | 98 | 97 | 35 | <137 | 115 | None | — | — |
| 10 | −/− | −/− | 131 | 146 | 32 | 54 075 | † | None | — | — | None | — | — |
HHV-6, CMV, and EBV maximum levels are in maximum copy number (copies/microliter) by real-time polymerase chain reaction in blood.
—, Not applicable; (D,R), viral serology of donor (D) and recipient (R).
Patient had reactivation at the time of death.
Patient has reactivation at time of last follow-up.
Discussion
The goal of allogeneic transplantation for SCD is to achieve adequate donor engraftment to reverse the SCD phenotype while minimizing GVHD and late therapy-related effects.3 Although matched sibling donor transplant has met this goal in SCD,20,28 less than 20% of patients with symptomatic SCD will have an unaffected HLA-matched sibling. In our study of 10 pediatric and young adult patients with symptomatic SCD, all patients had primary engraftment (ie, engrafted after transplant), and there was a low incidence of GVHD after reduced-intensity conditioning and alternative donor stem cell transplant. There was 1 case of possible late graft failure, which did not meet the protocol definition but could be considered as such because of the need for a second transplant. One patient had reversal of falling donor chimerism after therapeutic DLI and GVHD.
Acute GVHD grade II followed by chronic GVHD occurred in 1 patient. She initially had grade I skin GVHD. When her rash worsened and she developed diarrhea, she had calicivirus in her stool that may have contributed to the progression. Another patient had grade IV acute GVHD after DLI for refractory PTLD. DLI in the HLA-mismatched setting carries a significant risk for acute GVHD.29,30
The major complications of transplant were viral reactivation and PTLD, with 1 late transplant-related mortality. PTLD occurred more frequently than would be expected from a T replete allogeneic transplant31 and from a CD34+ cell-selected graft.32 Because 2 of the first 3 patients had EBV reactivation and 1 had PTLD in recipient cells, we reasoned that the reduced-intensity conditioning was not eradicating EBV-containing host B cells that could transform to PTLD.33 We added rituximab to the conditioning regimen on day −1 for patients who were EBV seropositive before transplant. Of the 7 seropositive recipients, 1 of 4 receiving rituximab 375 mg/m2 had EBV reactivation and PTLD compared with 3 of 3 patients not receiving rituximab (n = 2) or receiving a reduced dose of 150 mg/m2 (n = 1) having reactivation and 2 having PTLD. Lower doses of rituximab may not be adequate to prevent recipient-derived PTLD in patients who have not received chemotherapy previously.
Most patients were heavily transfused in the 3 years before transplant (median, 16 transfusions; range, 4-29 transfusions), but tolerated the conditioning regimen well. No patient had a painful crisis or other sickle cell complication after transplant. Long-term outcomes have been good. Elevated tricuspid regurgitant jet velocity is a surrogate marker for pulmonary hypertension and is associated with low hemoglobin, elevated reticulocyte count, and cerebral vasculopathy.34 Three patients had this early sign of pulmonary hypertension before transplant, with normalization in 2 after transplant. Three patients had narrowing of central nervous system arteries on magnetic resonance imaging before transplant. One had early Moya-Moya, with resolution and normal magnetic resonance imaging/magnetic resonance angiography 1 year after transplant. The other 2 had stable cerebrovascular disease.
Outcomes in our study of alternative donors were similar to trials for matched sibling donors with myeloablative,4-7 RIC,20 and nonmyeloablative28 conditioning regimens. These trials have reported approximate overall survival of 90% to 100%, graft failure 10%, acute GVHD 0% to 10%, and chronic GVHD 0% to 20%.35 A recent study of unrelated donor bone marrow transplant with RIC for SCD had 2-year overall survival of 79%; grade III-IV (severe) acute GVHD, 17%; and chronic GVHD, 62%.11 Transplantation using unrelated cord blood on the same study was stopped because of the high incidence of graft rejection.12 Reduced-intensity transplant using posttransplant cyclophosphamide has been used with haploidentical donors and is associated with high survival, but also a high incidence (43%) of graft failure and many patients requiring prolonged or indefinite immunosuppression.15 One advantage of a T-cell depletion approach is that long-term use of oral calcineurin inhibitors is not needed. This may be of particular benefit to children with SCD, as high rates of medical noncompliance with outpatient medications are often observed in this population.36
In summary, our approach of CD34+ cell-selected PBSCs with a RIC regimen for pediatric and young adult patients with SCD was associated with rapid engraftment and a low incidence of GVHD. There appears to be stable, usually complete, donor chimerism despite the use of RIC. Although the initial engraftment was achieved before DLI, the persistence of engraftment may be in part a result of the DLI and methotrexate that was used in 7 of 9 surviving patients. EBV-related PTLD was a problem, but this improved with the addition of rituximab into the conditioning regimen for EBV seropositive patients. PTLD and the profound immunosuppression early after transplant are concerns with this approach, and additional study is needed to further evaluate these risks. Although there is relatively long follow-up, additional follow-up is required to assess important outcomes such as fertility. Our approach offers the potential to increase the number of people with SCD who can benefit from transplant, as most will have a matched unrelated donor or a mismatched related family donor available. The treatment of additional patients will be needed to confirm our results. Our approach including DLI with methotrexate for 80 days and rituximab with SCT conditioning may be considered a therapeutic option in patients with SCD who are considering an alternative donor transplant.
Acknowledgments
The authors thank Andrew Bensky for review of echocardiograms and Emily Buzzerio and Jessica Keim-Malpass for assistance with preparation of the manuscript.
Authorship
Contribution: A.L.G. conceived the study; A.L.G., C.B., S.E., and A.I. designed the study; T.F., M.H., K.S., and D.G. acquired the data; A.L.G., M.J.E., E.C., and M.C. analyzed and interpreted the data; M.J.E. and A.I. performed the statistical analysis; A.L.G. and M.J.E. wrote the manuscript; all other authors critically reviewed the manuscript; and all authors approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Andrew L. Gilman, PRA Health Sciences, 4130 Park Lake Ave, Raleigh, NC 22612; e-mail: docg756@gmail.com.
References
- 1.Lanzkron S, Carroll CP, Haywood C Jr. Mortality rates and age at death from sickle cell disease: U.S., 1979-2005. Public Health Rep. 2013;128(2):110-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quinn CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children and adolescents with sickle cell disease. Blood. 2010;115(17):3447-3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shenoy S. Hematopoietic stem-cell transplantation for sickle cell disease: current evidence and opinions. Ther Adv Hematol. 2013;4(5):335-344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walters MC, Patience M, Leisenring W, et al. Bone marrow transplantation for sickle cell disease. N Engl J Med. 1996;335(6):369-376. [DOI] [PubMed] [Google Scholar]
- 5.Bernaudin F, Socie G, Kuentz M, et al. ; SFGM-TC. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood. 2007;110(7):2749-2756. [DOI] [PubMed] [Google Scholar]
- 6.Panepinto JA, Walters MC, Carreras J, et al. ; Non-Malignant Marrow Disorders Working Committee, Center for International Blood and Marrow Transplant Research. Matched-related donor transplantation for sickle cell disease: report from the Center for International Blood and Transplant Research. Br J Haematol. 2007;137(5):479-485. [DOI] [PubMed] [Google Scholar]
- 7.Dedeken L, Lê PQ, Azzi N, et al. Haematopoietic stem cell transplantation for severe sickle cell disease in childhood: a single centre experience of 50 patients. Br J Haematol. 2014;165(3):402-408. [DOI] [PubMed] [Google Scholar]
- 8.Krishnamurti L, Abel S, Maiers M, Flesch S. Availability of unrelated donors for hematopoietic stem cell transplantation for hemoglobinopathies. Bone Marrow Transplant. 2003;31(7):547-550. [DOI] [PubMed] [Google Scholar]
- 9.Mentzer WC, Heller S, Pearle PR, Hackney E, Vichinsky E. Availability of related donors for bone marrow transplantation in sickle cell anemia. Am J Pediatr Hematol Oncol. 1994;16(1):27-29. [PubMed] [Google Scholar]
- 10.Justus D, Perez-Albuerne E, Dioguardi J, Jacobsohn D, Abraham A. Allogeneic donor availability for hematopoietic stem cell transplantation in children with sickle cell disease. Pediatr Blood Cancer. 2015;62(7):1285-1287. [DOI] [PubMed] [Google Scholar]
- 11.Shenoy S, Eapen M, Panepinto JA, et al. A BMT CTN phase II trial of unrelated donor marrow transplantation for children with severe sickle cell disease. Blood. 2016;128(21):2561-2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamani NR, Walters MC, Carter S, et al. Unrelated donor cord blood transplantation for children with severe sickle cell disease: results of one cohort from the phase II study from the Blood and Marrow Transplant Clinical Trials Network (BMT CTN). Biol Blood Marrow Transplant. 2012;18(8):1265-1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kharbanda S, Smith AR, Hutchinson SK, et al. Unrelated donor allogeneic hematopoietic stem cell transplantation for patients with hemoglobinopathies using a reduced-intensity conditioning regimen and third-party mesenchymal stromal cells. Biol Blood Marrow Transplant. 2014;20(4):581-586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Radhakrishnan K, Bhatia M, Geyer MB, et al. Busulfan, fludarabine, and alemtuzumab conditioning and unrelated cord blood transplantation in children with sickle cell disease. Biol Blood Marrow Transplant. 2013;19(4):676-677. [DOI] [PubMed] [Google Scholar]
- 15.Bolaños-Meade J, Fuchs EJ, Luznik L, et al. HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood. 2012;120(22):4285-4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dallas MH, Triplett B, Shook DR, et al. Long-term outcome and evaluation of organ function in pediatric patients undergoing haploidentical and matched related hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2013;19(5):820-830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hansbury EN, Schultz WH, Ware RE, Aygun B. Bone marrow transplant options and preferences in a sickle cell anemia cohort on chronic transfusions. Pediatr Blood Cancer. 2012;58(4):611-615. [DOI] [PubMed] [Google Scholar]
- 18.Kodish E, Lantos J, Stocking C, Singer PA, Siegler M, Johnson FL. Bone marrow transplantation for sickle cell disease. A study of parents’ decisions. N Engl J Med. 1991;325(19):1349-1353. [DOI] [PubMed] [Google Scholar]
- 19.Matthes-Martin S, Lawitschka A, Fritsch G, et al. Stem cell transplantation after reduced-intensity conditioning for sickle cell disease. Eur J Haematol. 2013;90(4):308-312. [DOI] [PubMed] [Google Scholar]
- 20.King AA, Kamani N, Bunin N, et al. Successful matched sibling donor marrow transplantation following reduced intensity conditioning in children with hemoglobinopathies. Am J Hematol. 2015;90(12):1093-1098. [DOI] [PubMed] [Google Scholar]
- 21.Madden LM, Hayashi RJ, Chan KW, et al. Long-term follow-up after reduced-intensity conditioning and stem cell transplantation for childhood nonmalignant disorders. Biol Blood Marrow Transplant. 2016;22(8):1467-1472. [DOI] [PubMed] [Google Scholar]
- 22.Ramsay NK, Kersey JH, Robison LL, et al. A randomized study of the prevention of acute graft-versus-host disease. N Engl J Med. 1982;306(7):392-397. [DOI] [PubMed] [Google Scholar]
- 23.Eckrich MJ, Barnhart C, Oon C-Y, et al. Successful use of cryopreservation and shipping of CD34-selected mismatched related donor grafts for treatment of children. Biol Blood Marrow Transplant. 2015;21(2):S153-S154. [Google Scholar]
- 24.Glucksberg H, Storb R, Fefer A, et al. Clinical manifestations of graft-versus-host disease in human recipients of marrow from HL-A-matched sibling donors. Transplantation. 1974;18(4):295-304. [DOI] [PubMed] [Google Scholar]
- 25.Lee SJ, Vogelsang G, Flowers ME. Chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2003;9(4):215-233. [DOI] [PubMed] [Google Scholar]
- 26.Filipovich AH, Weisdorf D, Pavletic S, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant. 2005;11(12):945-956. [DOI] [PubMed] [Google Scholar]
- 27.Kaplan EM, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457-481. [Google Scholar]
- 28.Hsieh MM, Fitzhugh CD, Weitzel RP, et al. Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA. 2014;312(1):48-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Handgretinger R, Klingebiel T, Lang P, et al. Megadose transplantation of purified peripheral blood CD34(+) progenitor cells from HLA-mismatched parental donors in children. Bone Marrow Transplant. 2001;27(8):777-783. [DOI] [PubMed] [Google Scholar]
- 30.Lang P, Klingebiel T, Bader P, et al. Transplantation of highly purified peripheral-blood CD34+ progenitor cells from related and unrelated donors in children with nonmalignant diseases. Bone Marrow Transplant. 2004;33(1):25-32. [DOI] [PubMed] [Google Scholar]
- 31.Uhlin M, Wikell H, Sundin M, et al. Risk factors for Epstein-Barr virus-related post-transplant lymphoproliferative disease after allogeneic hematopoietic stem cell transplantation. Haematologica. 2014;99(2):346-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oevermann L, Handgretinger R. New strategies for haploidentical transplantation. Pediatr Res. 2012;71(4 Pt 2):418-426. [DOI] [PubMed] [Google Scholar]
- 33.Lynch BA, Vasef MA, Comito M, et al. Effect of in vivo lymphocyte-depleting strategies on development of lymphoproliferative disorders in children post allogeneic bone marrow transplantation. Bone Marrow Transplant. 2003; 32(5):527-533. [DOI] [PubMed] [Google Scholar]
- 34.Ambrusko SJ, Gunawardena S, Sakara A, et al. Elevation of tricuspid regurgitant jet velocity, a marker for pulmonary hypertension in children with sickle cell disease. Pediatr Blood Cancer. 2006;47(7):907-913. [DOI] [PubMed] [Google Scholar]
- 35.Talano JA, Cairo MS. Hematopoietic stem cell transplantation for sickle cell disease: state of the science. Eur J Haematol. 2015;94(5):391-399. [DOI] [PubMed] [Google Scholar]
- 36.Drotar D, Bonner MS. Influences on adherence to pediatric asthma treatment: a review of correlates and predictors. J Dev Behav Pediatr. 2009;30(6):574-582. [DOI] [PubMed] [Google Scholar]