

Key Points
CLEC-1 is restricted to CD16− myeloid DCs in human blood and acts as an inhibitory receptor to restrain downstream Th17 activation.
CLEC-1–deficient rats highlight an in vivo function for CLEC-1 in preventing excessive T-cell priming and effector Th responses.
Abstract
Dendritic cells (DCs) represent essential antigen-presenting cells that are critical for linking innate and adaptive immunity, and influencing T-cell responses. Among pattern recognition receptors, DCs express C-type lectin receptors triggered by both exogenous and endogenous ligands, therefore dictating pathogen response, and also shaping T-cell immunity. We previously described in rat, the expression of the orphan C-type lectin-like receptor-1 (CLEC-1) by DCs and demonstrated in vitro its inhibitory role in downstream T helper 17 (Th17) activation. In this study, we examined the expression and functionality of CLEC-1 in human DCs, and show a cell-surface expression on the CD16− subpopulation of blood DCs and on monocyte-derived DCs (moDCs). CLEC-1 expression on moDCs is downregulated by inflammatory stimuli and enhanced by transforming growth factor β. Moreover, we demonstrate that CLEC-1 is a functional receptor on human moDCs and that although not modulating the spleen tyrosine kinase-dependent canonical nuclear factor-κB pathway, represses subsequent Th17 responses. Interestingly, a decreased expression of CLEC1A in human lung transplants is predictive of the development of chronic rejection and is associated with a higher level of interleukin 17A (IL17A). Importantly, using CLEC-1–deficient rats, we showed that disruption of CLEC-1 signaling led to an enhanced Il12p40 subunit expression in DCs, and to an exacerbation of downstream in vitro and in vivo CD4+ Th1 and Th17 responses. Collectively, our results establish a role for CLEC-1 as an inhibitory receptor in DCs able to dampen activation and downstream effector Th responses. As a cell-surface receptor, CLEC-1 may represent a useful therapeutic target for modulating T-cell immune responses in a clinical setting.
Visual Abstract
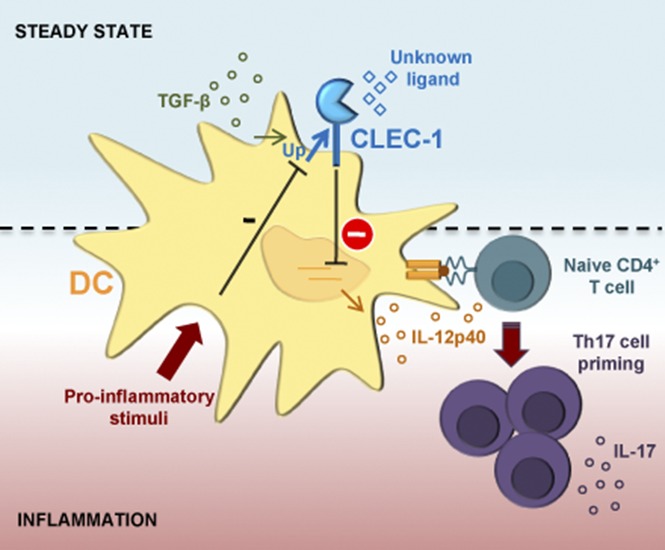
Introduction
Dendritic cells (DCs) are the sentinels of the immune system that are potentially activated to mediate efficient T-cell priming via a set of pattern-recognition receptors (PRRs). These receptors comprise the C-type lectin receptors (CLRs) that are able to recognize exogenous pathogen-associated molecular patterns, which are common to many types of bacteria, fungi, viruses, helminths, and also endogenous self-ligands of dying cells or glycans.1,2 The C-type lectin-like receptors (CTLRs) represent subtypes of these receptors, which lack the residues required for calcium-dependent carbohydrate binding and that by alternative mechanisms, recognize more diverse ligands such as proteins and lipids.3 Following triggering, most CLRs expressed on DCs modulate nuclear factor-κB (NF-κB) activation via the spleen tyrosine kinase (SYK) signaling pathway to enhance or suppress cellular activation, and fine-tune the magnitude and quality of downstream T-cell responses.3 We previously identified the CTLR, C-type lectin-like receptor-1 (CLEC-1), to be upregulated in a heart allograft model of tolerance in rats.4 We demonstrated that CLEC-1 is expressed by rat myeloid and endothelial cells (ECs), and is downregulated by pro-inflammatory stimuli and enhanced by transforming growth factor β (TGF-β). Moreover, our in vitro studies demonstrated that CLEC-1 inhibition in rat DCs via RNA interference enhanced subsequent DC-mediated CD4+ T helper 17 (Th17) activation.4 CLEC-1 belongs to the DC-associated C-type lectin-1 (DECTIN-1) cluster of CTLRs, and although identified a long time ago,5,6 corresponding exogenous and endogenous ligands are unknown and downstream signaling remains uncharacterized. CLEC-1 does not contain an immunoreceptor tyrosine-based activation or inhibitory motif in the cytoplasmic tail, but rather a tyrosine residue in a noncharacterized signaling sequence [YSST], in addition to a tri-acidic motif [DDD].3,7 In humans, CLEC-1 protein was reported to be expressed intracellularly in ECs.8 Nevertheless, its protein expression in human DCs as well as its biological effect remains uncharacterized. In this study, we have investigated CLEC-1 protein expression and regulation in human DCs, and its functional role on orchestration of T-cell responses. In addition, using CLEC-1–deficient rats and CLEC-1 fragment constant (Fc) fusion protein, we evaluated in vitro and in vivo, the consequence of CLEC-1 signaling disruption on DC function and downstream T-cell immunity.
Materials and methods
Patient and healthy donor material
Blood was obtained from healthy donors. Lung transplant biopsies from stable patients and from patients prior to chronic rejection (CR) were obtained from the multicentric longitudinal cohort “COhort in Lung Transplantation” (#NCT00980967). All material of healthy donors and patients was obtained after written informed consent, according to institutional guidelines.
Animals
Male 6- to 8-week-old LEW.1A (RT1a) and fully major histocompatibility complex–mismatched LEW.1W (RT1u) rats were purchased from the Centre d’Elevage Janvier (Genest, Saint-Isle, France), and experimental procedures were carried out in strict accordance with the protocols approved by the Committee on the Ethics of Animal Experiments of Pays de la Loire and authorized by the French Government’s Ministry of Higher Education and Research. Clec1a knockout rats were generated in the inbred LEW.1A background by the Transgenic Rats and Immunophenomics Platform facility (Structure Fédérative de Recherche [SFR]–Nantes) with the zinc finger nucleases technology (supplemental Figure 1). Absence of CLEC-1 at the expected size of 32 kDa was confirmed by western blot (supplemental Figure 2). For each experiment, 6- to 12-week-old sex-matched wild-type (WT) and CLEC-1–deficient (knockout) littermate rats were used.
For generation of chimeric rats, 50 million hematopoietic cells from WT or CLEC-1–deficient rats were IV injected into WT lethally irradiated rats (9 Gy, X-ray [SFR] day −1).
Antibodies
Anti-human CLEC-1 monoclonal antibody (anti-hCLEC-1 mAb, immunoglobulin G1 [IgG1]) was generated by Biotem (Apprieu, France) by lymphocyte somatic hybridization by immunization of Balb/c mice with a peptide encoding the extracellular domain of hCLEC-1 (CERRAGMVKPESLHVPPETLGEGD), screened by enzyme-linked immunosorbent assay (ELISA) (hCLEC-1 protein; R&D Systems, Minneapolis, MN) and purified by chromatography on protein A. Anti–hCLEC-1 mAb (IgG1-D6) was purchased from Santa Cruz (Dallas, TX). Purified anti-rat CD3 (G4.18), anti-rat T-cell receptor αβ-Alexa647 or -Alexa488 (R73), CD4-PECy7 (OX35), interleukin-17 (IL-17)–allophycocyanin (APC) (ebio17B7), interferon γ (IFN-γ)–phycoerythrin (PE), and anti-human phosphotyrosines (p-Tyr) (4G10), CD4-PE, CD3-APC or CD3-fluorescein isothiocyanate (FITC), CD45-PercP, CD16-PE or -FITC, CD209-PE, CD14-FITC, HLA–antigen D related (HLA-DR)–APC/Cy7 or –FITC, CD11c-PECy7, CD11b-FITC, CD80-FITC, CD86-FITC, CD83-FITC, and IgG1 isotype control were all purchased from BD Biosciences (Franklin Lakes, NJ). Phospho-IκBα (Ser32/36) (5A5) and IκBα mAbs were from Cell Signaling (Danvers, MA). Secondary mAbs were from Jackson Immunoresearch (West Grove, PA).
Flow cytometry and cell sorting
Before staining, cells were subjected to Fc block (BD Biosciences). For intracellular cytokine staining, cells were stimulated for 4 hours with phorbol 12-myristate 13-acetate (50 ng/mL), ionomycin (1 μg/mL) in the presence of GolgiStop, and subjected to fixation and permeabilization (all reagents from BD Biosciences). Fluorescent labeling of stained cells (2.5 µg/mL) was measured using a fluorescence-activated cell sorter LSR II (BD Biosciences) and analyzed with FloJo software (Tree Star Inc., Ashland, OR).
For cell sorting, total or CD4+ T cells were purified from the spleen of naïve LEW.1A rats by positive selection by T-cell receptor+ and CD4+ staining, and human neutrophils and monocytes by SSChighCD45+CD16+ and SSClowCD45+CD14+CD16+, respectively, using a fluorescence-activated cell sorter Aria flow cytometer (BD Biosciences). Purity was >99%.
Dead cells were excluded by gating on 4′,6-diamidino-2-phenylindole (DAPI)-negative cells.
In vivo models of immunization and in vitro secondary challenge
LEW.1W heart allografts were transplanted to WT and CLEC-1–deficient recipients as previously described9 and were rejected in 7 days.10 Spleens and grafts were recovered at day 5 after transplantation. Carboxyfluorescein diacetate succinimidyl ester (CFSE) (Thermo Fisher, Waltham, MA)-labeled (5 μM) splenic purified CD4+ T cells (1 × 105) from recipients were subjected in vitro to secondary challenge (mixed leukocyte reaction [MLR]) with T-cell–depleted splenocytes (1 × 105) from naïve LEW.1W rats for 3 days.
WT, CLEC-1–deficient, and chimeric rats were immunized subcutaneously in the footpad with keyhole limpet hemocyanin (KLH) protein (Sigma-Aldrich, St. Louis, MO) (100 μg) emulsified (volume-to-volume) in 100 μL of complete Freund adjuvant (CFA) (Difco, Lawrence, KS). Popliteal lymph nodes (LNs) were harvested 10 days after immunization. CFSE-labeled (5 μM) total cells or purified CD4+ T cells (1 × 105) plus T-cell–depleted splenocytes (1 × 105) from naïve LEW.1A rats were subjected to in vitro secondary challenge with KLH or ovalbumin protein as control (25 μg/mL) for 3 days.
Cell generation, in vitro stimulation, and MLR
Human monocytes were obtained by elutriation. Human monocyte-derived DCs (moDCs) were generated as previously described from elutriated monocytes cultured for 7 days in complete RPMI 1640 medium (10% endotoxin-free fetal calf serum [Thermo Fisher], 2 mM L-glutamine, 1 mM sodium pyruvate, 1 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid [all reagents from Sigma-Aldrich]), supplemented with IL-4 and granulocyte macrophage–colony-stimulating factor (1000 U/mL; AbCys, Paris, France).11 Cells were then stimulated for 24 hours (1 × 106/mL) with lipopolysaccharide (LPS) (0.5 μg/mL) (Sigma-Aldrich), Poly I:C (2 μg/mL) (Invivogen, San Diego, CA), R848 (2.5 μg/mL) (Invivogen), recombinant human TGF-β1 (20 ng/mL) (R&D Systems) alternatively in the presence of 10 μg/mL of coated anti–hCLEC-1 mAb or IgG1 isotype control. Cells were then subjected to flow cytometry or cultured (12.5 × 103) with 5 × 104 allogeneic human T cells (Pan T Cell Isolation Kit [Miltenyi, Bergisch Glabach, Germany]) for 5 days (MLR). Proliferation was measured by flow cytometry by CFSE profile in CD3+CD4+ T cells. All cytokines were assessed in supernatants of moDCs or MLR by ELISA (BD Biosciences).
Human embryonic kidney cells 293T (HEK293T) were obtained from American Type Culture Collection;
Human aortic ECs (HAECs) or human umbilical vein ECs (HUVECs) were isolated and cultured as previously described12; and
Bone marrow-derived DCs (BMDCs) from naïve, WT, or CLEC-1–deficient LEW.1A rats were obtained as previously described by culturing cells for 8 days in complete RPMI medium, supplemented with rat IL-4 (4 ng/mL) and murine granulocyte macrophage–colony-stimulating factor (1.5 ng/mL).13 BMDCs were then stimulated with LPS (1 μg/mL) or zymosan (20 μg/mL) (Invivogen, San Diego, CA) and cocultured (MLR) for 5 days with purified allogeneic CFSE-labeled LEW.1W CD4+ T cells. IL-17 and IFN-γ ELISA were from BD Biosciences. Rat CLEC-1 Fc or control human secreted alkaline phosphatase (hSEAP)-Fc fusion protein (supplemental Figure 3) (10 μg/mL) were added in MLR and in rat purified CD4+ T cell, stimulated with plate-bound (pb) anti-CD3 (G4.18 clone) (5 μg/mL). Stimulation was performed in the presence of endotoxin inhibitor polymyxine B (10 μg/mL) (Invivogen).
RNA extraction and quantitative real-time polymerase chain reaction (qRT-PCR)
RNAs from lung transplant biopsies were extracted with FastPrep Lysing Matrix (MP Biomedicals, Santa Ana, CA) and amplified with MessageAmp II aRNA Amplification Kit (Thermo Fisher).
Total RNAs from BMDCs or allografts were prepared using Trizol (Thermo Fisher). Retro-transcription and qRT-PCR was performed using the ViiA 7 Real-Time PCR System and SYBR Green PCR Master Mix (Thermo Fisher) as previously described.14 Oligonucleotides used in this study are described in Table 1. Human HPRT, IL17A, IFNG, and TGFB1 probes were from Life Technology. HPRT was used as an endogenous control gene for normalization. Relative expression was calculated using the 2−ΔΔCt method14,15 and expressed in arbitrary units (AUs).
Table 1.
Oligonucleotides (5′ to 3′) used for rat qRT-PCR
| Gene | Oligonucleotides (5′ to 3′) |
| r Hprt 5′ | CCTTGGTCAAGCAGTACAGCC |
| r Hprt 3′ | CTGTCTCACAAGGGAAGTGACAA |
| r Il6 5′ | ACAGCGATGATGCACTGTCA |
| r Il6 3′ | GGAACTCCAGAAGACCAGAGC |
| r Il12p35 5′ | TGATGATGACCCTGTGCCTT |
| r Il12p35 3′ | GCATGGAGCAGGATACAGAGC |
| r Il12p40 5′ | ATCATCAAACCGGACCCACC |
| r Il12p40 3′ | CAGGAGTCAGGGTACTCCCA |
| r Il23p19 5′ | GGACTCGGACATCTTCACAGG |
| r Il23p19 3′ | GGAACGGAGAAGAGAACGCT |
| r Il10 5′ | CCTCTGGATACAGCTGCGAC |
| r Il10 3′ | GTAGATGCCGGGTGGTTCAA |
| r Tgfb1 5′ | CTCAACACCTGCACAGCTCC |
| r Tgfb1 3′ | ACGATCATGTTGGACAACTGCT |
| r Il17a 5′ | TGCTGTTGCTGCTACTGAACC |
| r Il17a 3′ | AACTTCCCCTCAGCGTTGAC |
| r Ifng 5′ | AGTGTCATCGAATCGCACCTG |
| r Ifng 3′ | TTCTGGTGACAGCTGGTGAAT |
Hprt, hypoxanthine phosphoribosyltransferase; r, rat.
Immunoprecipitation and western blot
Human moDCs were plated on anti–hCLEC-1 (IgG1) or control IgG1 isotype (Invitrogen) mAb coated-plates (10 μg/mL) for 5 or 20 minutes with or without zymosan (20 μg/mL). Human moDCs, HAECs, HUVECs, and HEKs were lyzed in Nonidet P-40 1% lysis buffer with protease inhibitors cocktail (Sigma-Aldrich). CLEC-1 immunoprecipitation was performed with 4 μg of anti–hCLEC-1 mAb (D6) followed by incubation with protein G-sepharose beads. Proteins were then treated overnight with PNGase F (Sigma-Aldrich) and dissolved by boiling for 5 minutes in Laemmli sample buffer. Protein concentration was determined using the BC Assays Kit with bovine serum albumin (BSA) as standard (Interchim, San Pedro, CA). Polyvinylidene difluoride membranes were blocked with Tween-20–Tris-buffered saline, 5% BSA, and incubated with anti-phosphotyrosine (4G10), anti–hCLEC-1 (in house), anti–phospho-IκBα (Ser32/36), or anti-IκBα mAbs (0.5 µg/mL) followed by horseradish peroxidase-conjugated secondary antibodies. Detection by chemiluminescence was revealed using West Femto chemiluminescence substrate (Thermo Fisher) and monitored by Las 4000 (Fuji).
Immunohistochemistry (IHC)
Human HUVECs, moDCs, monocytes, and neutrophils were fixed in 4% paraformaldehyde (Electron Microscopy Science, Hatfield, PA) and alternatively permeabilized with Triton X-100 (0.1%). Cells were stained with anti–hCLEC-1 mAb (D6) or IgG1 isotype control (Invitrogen) (4 μg/mL) in phosphate-buffered saline 1% fetal calf serum, 1% BSA, and then with secondary Alexa-488 anti-mouse IgG1 antibodies for 1 hour. After 10 minutes in phosphate-buffered saline 1% DAPI, slides were mounted using Prolong Antifade Reagent (Invitrogen) and observed by confocal fluorescence microscopy (Nikon A1-RSi). Images were obtained (×60 Plan Apo N.A.: 1.4, zoom 2) with sequential mode, and analyzed by using ImageJ software. Quantitation of CLEC-1 was performed by Volocity software (PerkinElmer, Waltham, MA).
Statistical analysis
Statistical analyses were performed using GraphPad Prism software (La Jolla, CA) with 2-tailed unpaired nonparametric Student t test (Mann-Whitney). Results were considered significant if P values were < .05.
Results
Human myeloid DCs express CLEC-1 at the cell surface
So far, only limited information has been published on CLEC-1 expression in humans.4,5,8 qRT-PCR analysis in pooled organs demonstrates a strong expression of CLEC1A transcripts in placenta and lung, and a more moderate expression in the lymphoid organs including spleen, LNs, thymus, and tonsils (supplemental Figure 4i). In human cell subtypes, abundant CLEC1A transcripts were found in neutrophils, monocytes, moDCs, and HAECs (supplemental Figure 4ii). No transcripts were detected in T and B cells. We demonstrate by CLEC-1 immunoprecipitation followed by western blot, a high level of CLEC-1 protein in moDCs and ECs (Figure 1A). By generating a mAb directed against the extracellular domain of anti-human CLEC-1, we observed a low ectopic expression of CLEC-1 at the cell-surface of transfected HEKs (supplemental Figure 5A). These data are in accordance with previous studies5,6,16 and suggest that CLEC-1 requires other adaptor chains, other PRRs, or sufficient glycosylation for efficient expression, transport, and cell-surface stability as described for other CLRs.17,18 No improvement was observed by cotransfecting cells with plasmids encoding the adaptor proteins DAP12 and FcRγ (data not shown). Cell-surface CLEC-1 expression was confirmed by IHC in transfected cells with anti–hCLEC-1 mAb (D6 clone) (supplemental Figure 5B).
Figure 1.

Human DCs express cell-surface CLEC-1. (A) Western blot analysis of CLEC-1 expression in human moDCs, HAECs, HUVECs, and HEKs. Cell extracts were immunoprecipitated with anti-human CLEC-1 mAb (D6 clone) and then analyzed by western blot with a second in-house anti-human CLEC-1 mAb (IgG1). Arrows indicate human CLEC-1 and IgG HC and LC at the expected size of 32, 50, and 25 kDa, respectively. M line represents molecular-weight size markers. (B) Representative dot plots and histograms of IgG1 isotype or CLEC-1 (IgG1) staining in non-permeabilized (non-perm) and permeabilized (perm) conditions, evaluated by flow cytometry for human blood: (i) CD16+ and CD16− subpopulation of CD45+CD14−CD11c+HLA-DRhigh DCs; (ii) CD45+CD14+CD16+ monocytes; (iii) SSChighCD16+ neutrophils; and (iv) HAECs. Histograms represent the overlay image of CLEC-1 staining (gray filled histogram) matching the isotype control IgG1 staining (open histogram). (C) Representative dot plots or histograms of IgG1 isotype or CLEC-1 (IgG1) staining vs DC-SIGN or CD16 staining for human moDCs in non-perm or perm conditions and evaluated by flow cytometry (i-ii). Histograms represent the overlay image of CLEC-1 staining (gray filled histogram) matching the isotype control IgG1 staining (open histogram). (iii) Cell-surface expression of CLEC-1 vs HLA-DR on unstimulated (US), TLR 4-L (LPS), TLR 3-L (Poly I:C), TLR 7-L (R848), and TGF-β–stimulated moDCs. (iv) Histogram represents MFI ± standard error of the mean (SEM) of CLEC-1 staining of 6 independent experiments. Statistical analysis of CLEC-1 MFI staining was performed between US and each stimuli. Panel Di shows representative confocal microscopy images, and (ii) quantitation of CLEC-1 protein in non-perm and perm conditions for human HUVECs, moDCs, CD16+ monocytes and neutrophils. Panels exhibiting DAPI (blue) and CLEC-1 (green) staining revealed by anti-human CLEC-1 mAb (D6 clone) followed by secondary anti-mouse Alexa-488 antibody. Original magnification ×600. Images are representative of 4 independent experiments. CLEC-1 protein quantitation was performed by velocity software and expressed as histogram of mean ± SEM of numbers of fluorescent spots per cell (n ≥7). *P < .05; **P < .01; ***P < .001. IgG HC, IgG heavy chain; IgG LC, IgG light chain; MFI, mean fluorescence intensity; mono, monocytes; neutro, neutrophils.
With this generated mAb, we investigated CLEC-1 protein expression in human cell subtypes. Interestingly, we observed in blood that cell-surface CLEC-1 expression is restricted to the CD16− subpopulation of myeloid DCs and to CD14+CD16+ monocytes (Figure 1Bi-ii). Low CLEC-1 expression was observed at the cell-surface of neutrophils and HAECs where expression is mostly intracellular, as previously reported6,8 (Figure 1Biii-iv). No expression of CLEC-1 was observed at the cell-surface or intracellularly in blood BDCA3+ myeloid or CD123+ plasmacytoid DCs (supplemental Figure 6). Almost all cultured moDCs express cell-surface CLEC-1, which is largely coexpressed with the other CLR DC-specific ICAM-3–grabbing nonintegrin (DC-SIGN); however, in contrast to blood, CLEC-1 does not appear to be restricted to the CD16− subpopulation (Figure 1Ci-ii). Importantly, as we previously described in rats,4 CLEC-1 expression is decreased on human moDCs by inflammatory stimuli such as toll-like receptor (TLR) ligands, and is upregulated by TGF-β (Figure 1Ciii-iv). By IHC, we confirmed that CLEC-1 expression is mostly intracellular for ECs, and at the cell-surface for moDCs and monocytes (Figure 1Di-ii). These results demonstrate that CLEC-1, as for the CLR DC-SIGN,19 is restricted to a particular cell subtype of DCs in blood while being largely expressed by moDCs, suggesting that CLEC-1 is enhanced in particular conditions.
In vitro CLEC-1 triggering on human moDCs suppresses downstream allogeneic Th17 activation
Because CLEC-1 natural ligands have not yet been identified, we used anti-human CLEC-1 mAb to mimic the ligand and crosslink CLEC-1 at the cell-surface of moDCs. By CLEC-1 immunoprecipitation in low stringent conditions, we observed no tyrosine phosphorylation at the expected size of CLEC-1 (32 kDa) following CLEC-1 ligation, suggesting that tyrosine motif in the cytoplasmic tail is not phosphorylated (Figure 2A). Nevertheless, we observed several changes in the tyrosine phosphorylation pattern with enhanced or decreased phosphorylation of several bands around 40 to 50 kDa in size, strongly suggesting that CLEC-1 is a functional receptor that signals via binding partners that remain to be identified.
Figure 2.

CLEC-1 triggering on human moDCs prevents downstream Th17 activation. (A) Human moDCs were stimulated with pb anti–CLEC-1 or IgG1 isotype control mAb for 5 minutes. CLEC-1 and binding partners were immunoprecipitated in low-stringent conditions (D6 clone) and were revealed by western blot using anti-phosphotyrosine mAb (4G10). Representative image of western blot with arrows indicating bands with changes in phosphorylation intensity between isotype control and anti–hCLEC-1 mAb stimulation, and IgG HC and LC chains of immunoprecipitating antibody (at the expected size of 50 and 25 kDa, respectively). M line represents molecular-weight size markers. Data are representative of 3 independent experiments. (B) Human moDCs were incubated with or without (−) pb anti–hCLEC-1 or IgG1 isotype control mAbs, and were alternatively stimulated simultaneously with TLR 4-L (LPS) or zymosan for 24 hours, and CD80, CD86, CD83, and HLA-DR were evaluated by flow cytometry (overlays are representative of 8 independent experiments). (C) Tumor necrosis factor-α, IL-12p70, IL-6, IL-23, and IL-10 were assessed by ELISA in supernatants (histograms represent mean ± SEM of 8 independent experiments). (D) Human moDCs were stimulated with pb anti–hCLEC-1 or IgG1 Iso control mAbs for 20 minutes and with or without zymosan. Representative images of western blot revealing phosphorylation of IκBα (PSer32/36) or the degradation of total IκBα at the expected size of 40 and 39 kDa, respectively. Data are representative of 3 independent experiments. M line represents molecular-weight size markers. (E) Following 24 hours of CLEC-1 triggering, human moDCs were extensively washed and subjected to MLR with allogeneic T cells for 5 days. (i) T-cell proliferation was assessed (CFSE dilution) by flow cytometry in allogeneic T cells, and (ii) IL-17 and IFN-γ production was evaluated by ELISA in supernatants. Data were expressed in histograms as mean ± SEM of 8 independent experiments. **P < .01; ***P < .001. IgG HC, IgG heavy chain; IgG LC, IgG light chain; Iso, isotype; UT, untreated.
We next investigated whether CLEC-1 triggering potentiates or suppresses PRR-induced moDCs maturation, NF-κB pathway activation, or downstream T-cell polarization as previously described for other activating or inhibitory CLRs.3 We observed that CLEC-1 triggering on moDCs does not induce by itself nor does it modulate the expression of the activation markers CD80, CD86, CD83, and HLA-DR, or the production of tumor necrosis factor-α, IL-12p70, IL-6, IL-23, and IL-10 induced by TLR 4 ligand (TLR 4-L), zymosan (agonist of both DECTIN-1 and TLR 2) (Figure 2B-C), TLR 3, and TLR 7 ligands (data not shown). Moreover, CLEC-1 triggering does not induce by itself nor does it modulate the zymosan-induced activation of the SYK-dependent canonical NF-κB pathway, evaluated by the phosphorylation of the NF-κB inhibitor IκBα (Ser32/36) and by its degradation (Figure 2D). Nevertheless, because CLEC-1 activation was achieved by antibody crosslinking, it will be important to determine whether true ligands, once identified, elicit similar responses. These results suggest that CLEC-1 may engage an alternative pathway of NF-κB activation that requires further investigation.
Interestingly, we observed that CLEC-1 triggering on moDCs alone (Figure 2Ei-ii) or in combination with TLR L or zymosan (data not shown), although not modulating the subsequent proliferation of allogeneic T cells, reduced Th17 activation and skewed the response toward a Th1 polarization.
These data demonstrate that CLEC-1 triggering on human moDCs inhibits downstream Th17 response.
Downregulation of CLEC1A expression in human lung transplants is predictive of CR
Given the strong expression of CLEC1A in lung, we investigated its modulation in lung biopsies from stable patients or from patients prior to the diagnostic of CR. At that time, biopsies do not exhibit histopathology signs of rejection (supplemental Figure 7). Interestingly, we observed a lower expression of CLEC1A transcripts in biopsies from patients prior to CR, and this was associated with a higher level of IL17A transcripts (Figure 3). No difference was observed for IFNG or TGFB1 expression. These data suggest that the strong expression of CLEC-1 in lung, as a CLR potentially involves in the first-line of defense against pathogens, may play also a crucial role in adaptive immune response to locally dampen Th17 responses.
Figure 3.

Decreased CLEC1A expression in lung transplants is predictive of CR. Lung transplants from stable patients or from patients prior to the development of CR were subjected to qRT-PCR for HPRT, CLEC1A, IL17A, IFNG, and TGFB1. Results were expressed in histograms as mean ± SEM of 7 samples in each group and were expressed in AU of specific cytokine/HPRT ratio. *P < .05; **P < .01. mRNA, messenger RNA.
Disruption of CLEC-1 signaling in rat BMDCs enhances in vitro T-cell responses
To gain insight into the function of CLEC-1, we generated CLEC-1–deficient rats that are viable, healthy, and born from heterozygote breeding with the expected Mendelian frequency. At steady-state, CLEC-1–deficient rats exhibited regular myeloid and lymphoid immune cell compartments in blood and peripheral lymphoid organs (data not shown).
We previously showed that rat BMDCs express CLEC-1.4 Therefore, we compared these cells from WT and CLEC-1–deficient rats for their phenotype and function. We observed no difference in their ability to maturate in response to TLR4 L or zymosan, according to the maturation markers CD80, CD86, and Class-I and -II major histocompatibility complex (Figure 4A). However, we found that CLEC-1–deficient BMDCs expressed a higher level of Il12p40 subunit than WT BMDCs in US condition, and following LPS and zymosan activation (Figure 4B). An increase in Il12p35 was also observed following zymosan stimulation. No difference was observed for Il23p19, Il6, Il10, and Tgfb1 expression. Furthermore, CLEC-1–deficient BMDCs induced an enhanced proliferation of allogeneic CD4+ T cells in MLR that was associated with an increased number of IL-17+ CD4+ T cells (Figure 4Ci-ii, respectively).
Figure 4.

Rat CLEC-1–deficient BMDCs enhance Th17-cell activation. (A) BMDCs from WT and CLEC-1–deficient rats were stimulated with TLR 4-L or zymosan for 24 hours, and CD80, CD86, and Class I and II major histocompatibility complex (MHC) were assessed by flow cytometry. Data were expressed in histograms as mean ± SEM of 6 independent experiments. (B) BMDCs were stimulated with TLR4-L or zymosan for 8 hours, and Il12p40, Il12p35, Il23p19, Il6, Il10, and Tgfb1 were assessed by qRT-PCR. Results were expressed in histograms as mean ± SEM of 6 independent experiments and were expressed in AU of specific cytokine/Hprt ratio. (C) BMDCs were incubated for 4 days in MLR with allogeneic purified CD4+ T cells. (i) Histogram and representative staining of proliferation (CFSE dilution) assessed in CD4+ T cells by flow cytometry, and (ii) histogram and representative dot plots of percentage of IL-17+ and IFN-γ+ cells among gated CD4+ T cells assessed by flow cytometry. Data were expressed in histograms as mean ± SEM of 6 independent experiments. *P < .05; **P < .01. KO, knockout; MFI, mean fluorescence intensity; mRNA, messenger RNA; NS, nonstimulated.
To confirm these data, we generated rat CLEC-1 Fc fusion protein (supplemental Figure 3) that should block CLEC-1 interaction on BMDCs with its putative ligand(s), and thus mimic CLEC-1 deficiency. Similarly, we observed in the presence of CLEC-1 Fc fusion protein in MLR, a more prominent proliferation of allogeneic T cells and more IL-17 production (Figure 5Ai-ii, respectively). As control, no direct effect of CLEC-1 Fc was observed on proliferation or on IL-17+ cells number, and IL-17 production of anti-CD3 polyclonally activated CD4+ T cells (Figure 5Bi-ii, respectively). This demonstrates that the increase of Th17 activation in MLR was specific to CLEC-1 signaling disruption in BMDCs, and not due to ligation of CLEC-1 Fc and possible agonist effect on a putative ligand on T cells.
Figure 5.

Blocking CLEC-1 Fc fusion protein enhances rat BMDC-mediated Th17-cell activation. (A) BMDCs from naïve rats were incubated for 4 days in MLR with allogeneic purified CD4+ T cells, together with CLEC-1 Fc or irrelevant hSEAP-Fc fusion proteins (produced and purified under the same conditions) (10 μg/mL). (i) Histogram of proliferation (CFSE dilution) of CD4+ T cells assessed by flow cytometry, and (ii) IL-17 and IFN-γ cytokine production assessed in supernatants of MLR by ELISA. Data were expressed in histograms as mean ± SEM of 4 independent experiments. (B) Purified CD4+ T cells from naïve rats were stimulated with pb anti-CD3 (5 μg/mL) in combination with CLEC-1–Fc or irrelevant hSEAP-Fc fusion proteins (10 μg/mL) for 4 days. (i) Proliferation (CFSE dilution) was assessed by flow cytometry, and (ii) IL-17 cytokine production was assessed in supernatants by ELISA and in CD4+ T cells by flow cytometry as FSC vs isotype or IL-17 staining. Data were expressed as histograms as mean ± SEM of 4 independent experiments. **P < .01; ***P < .001. FSC, forward scatter.
Taken collectively, these data demonstrate in vitro that the absence of CLEC-1 signaling in rat BMDCs enhanced their Il12p40 subunit expression and their ability to induce allogeneic Th17 cell activation.
CLEC-1 deficiency enhances in vivo DC-mediated Th1 and Th17 responses
We previously described in rat that conventional DCs (cDCs) from secondary lymphoid organs express CLEC-1.4 Therefore, we investigated the potential function of CLEC-1 in DC-mediated antigen-presentation and Th responses following in vivo immunization.
Following subcutaneous injection of KLH and CFA, we observed after in vitro secondary challenge of total draining LNs or purified CD4+ T cells from CLEC-1–deficient rats, an increased proliferation of KLH-specific CD4+ T cells associated with an increased number of IL-17+, IL-17+ IFN-γ+, and IFN-γ+ CD4+ T cells (Figure 6Ai-ii, respectively). Importantly, similar results were obtained in chimeric rats (WT rats fully reconstituted with BM from CLEC-1–deficient rats), suggesting that this enhanced in vivo priming was due to the absence of CLEC-1 in myeloid cell compartment and not to the one in ECs (Figure 6Aiii).
Figure 6.

CLEC-1–deficient rats exhibit an exacerbation of in vivo DC-mediated CD4+Th1/Th17 responses. (A) WT, CLEC-1–deficient rats (i-ii) and chimeric rats reconstituted with BM from WT or CLEC-1–deficient rats (iii) were immunized subcutaneously in the footpad with CFA plus KLH protein (100 μg/mL). At day 10 after immunization, popliteal LNs were harvested and total LN cells or purified CD4+ T cells were re-stimulated in vitro with KLH or control ovalbumin (25 μg/mL) for 3 days. Histograms and representative plots of proliferation (CFSE dilution) and percentage of IL-17+, IL-17+ IFN-γ+, and IFN-γ+ cells in gated CD4+ T cells assessed by flow cytometry. Data were expressed as histograms as mean ± SEM of 4 independent experiments. Staining of isotypes was indicated as control. (B) WT and CLEC-1–deficient rats were transplanted with cardiac allografts. (i) At day 5 after transplantation, purified CD4+ T cells from spleen were re-stimulated in vitro with donor T-cell–depleted splenocytes (MLR) for 3 days. Histograms of proliferation (CFSE dilution) in gated CD4+ T cells assessed by flow cytometry and expressed as mean ± SEM of 4 independent experiments. (ii) Il17a and Ifng were assessed by qRT-PCR in cardiac allografts harvested at day 5 after transplantation. Results were expressed in histograms as mean ± SEM of 4 independent experiments and were expressed in AU of specific cytokine/Hprt ratio. *P < .05; **P < .01. mRNA, messenger RNA.
Similarly, following immunization with cardiac allografts, we observed after in vitro secondary challenge of splenic purified T cells from CLEC-1–deficient rats, an increased proliferation of allogeneic T cells (Figure 6Bi). In addition, although allografts were rejected with a similar kinetic at day 7 after transplantation (data not shown), we observed an increased expression of Il17a transcripts in allografts from CLEC-1–deficient recipients, suggesting more Th17 cell activation (Figure 6Bii).
These data demonstrate that the in vivo deficiency of CLEC-1 signaling in cDCs exacerbates priming, and downstream Th17 and Th1 responses in both innate and adaptive immunity.
Discussion
In this study, we demonstrate that CLEC-1 is a functional cell-surface inhibitory receptor on human DCs that restrains downstream Th17 activation. Furthermore, the use of CLEC-1–deficient animals brings to light an in vivo function for CLEC-1 in the prevention of excessive DC-mediated CD4+ T-cell priming, and Th17 and Th1 polarization. Interestingly, we found in human blood, a cell-surface CLEC-1 expression restricted to myeloid CD16− DCs and to CD14+CD16+ monocytes. Both cell subtypes are known to exhibit strong pro-inflammatory properties and to be potent inducers of Th17 cell expansion.20-23 Moreover, cell-surface CLEC-1 expression was observed on human moDCs, and as in rat,4 is decreased by inflammatory stimuli and is upregulated by TGF-β. In contrast, CLEC-1 appears to be mostly expressed intracellularly in ECs and neutrophils. Therefore, as for other CLRs such as LOX-1 or DC-SIGN, pattern, localization, and regulation of CLEC-1 expression may depend on cell subtypes and microenvironment.24-26 Cell-surface CLEC-1 may be enhanced in particular conditions such as an environment rich in TGF-β to locally impede excessive inflammatory response. Indeed, we originally identified CLEC-1 as upregulated in a TGF-β–dependent model of rat allograft tolerance, and demonstrated that regulatory CD4+CD25+ T cells enhance CLEC-1 expression and are necessary for DC-mediated Th17 inhibition.4 Importantly, we found a decreased expression of CLEC1A in human lung as predictive of the Th17-associated development of CR.27 Therefore, CLEC-1 may represent a new therapeutic target in a clinical setting to limit Th17 activation and notably lung tissue injury. This profile of expression in DCs with a decrease following inflammatory stimulation represents a classic response observed for other inhibitory receptors such as myeloid C-type lectin-like receptor28 or DC immunoreceptor,29 which have also been shown to suppress in vivo T-cell responses and inflammation.30,31 However, in contrast to these inhibitory CLRs,28,29 CLEC-1 does not hamper total DC maturation or pro-inflammatory cytokine production, and we only observed a decrease of Il12p40 subunit expression. Interestingly, the activating receptor DECTIN-1, has in contrast been shown to enhance Il12p40 subunit expression in DCs to promote bioactive IL-23 and IL-12p70 production, and Th17 and Th1 polarization.32-34 DECTIN-1 ligation on DCs was shown to trigger the SYK-dependent activation of both the canonical and noncanonical NF-κB pathways, with the latter repressing the Il12p40 expression. However, according to the ligand, DECTIN-1 can also signal through a SYK-independent Raf-1 proto-oncogene, serine/threonine kinase (RAF-1) pathway to prevent Il12p40 repression.33 For example, in response to Curdlan and Candida albicans challenge, DECTIN-1 leads in mice to both Th1 and Th17 polarization,33 whereas in response to Aspergillus fumigatus, it potentiates Th17 differentiation by inhibiting Th1 polarization.35 We observed in DCs that CLEC-1 signaling alone or in combination with TLR L or zymosan suppress particularly the subsequent Th17 activation in vitro, but both Th1 and Th17 responses following immunization with CFA in vivo. Because CFA is known to trigger numerous PRRs such as CLRs,36 this suggests that CLEC-1 may differently suppress the Th1 or Th17 responses according to the ligands and PRR coengagement. Alternatively, CLEC-1 in DCs may also shape the Th17/Th1 balance by mechanisms others than the only expression of polarizing cytokines. For example, DECTIN-1 signaling in DCs has been shown to influence T-cell polarization fate by modulating also the expression of the costimulatory molecules OX40 ligand.37
We found that CLEC-1 ligation does not induce by itself nor does it modulate the zymosan-induced activation of the SYK-dependent canonical NF-κB pathway. Nevertheless, we observed changes in the phosphorylation pattern of CLEC-1–binding partners. Therefore, CLEC-1 by itself or through partners could suppress Il12p40 expression by acting via the noncanonical NF-κB pathway. Alternatively, CLEC-1 could antagonize the SYK-independent RAF-1 pathway. Interestingly, CLEC-1 contains in its cytoplasmic tail a tri-acidic motif [DDD],3 which has been shown for other CLRs to promote RAF-1 signaling and to modulate PRRs pathways.38-40 However, if this tri-acidic motif is functional for CLEC-1 is currently unknown.
We have not been able to detect the cells expressing the endogenous ligands with a CLEC-1 Fc fusion protein. Nevertheless, our in vitro data suggest that CLEC-1 ligands may be expressed by hematopoietic cells themselves, or released “naturally” or during cell damage. For examples, DC immunoreceptor-2 and its endogenous ligand have been described to be both expressed at the cell-surface of cDCs in mice.22 DECTIN-2 was shown to recognize as “danger sensor” molecules released into DC culture and a putative ligand on regulatory T cells to suppress immune responses.41,42 An endogenous ligand for DECTIN-1 has also been reported on T cells that, in contrast to CLEC-1, acts as a costimulatory molecule enhancing T-cell proliferation.43 Therefore, the identification of CLEC-1 ligand(s) is urgently needed to better decipher cell signaling and function.
In conclusion, these findings establish in both human and rodent, the relevance of CLEC-1 in DCs in the tight control of downstream Th17 responses. CLEC-1 as an inhibitory cell-surface receptor may represent a therapeutic tool to manipulate the degree and quality of T-cell responses, and a new treatment paradigm in transplantation, autoimmunity, cancer vaccination, and infectious diseases.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the COhort in Lung Transplantation consortium (Coordinator: Antoine Magnan, Centre Hospitalier Universitaire of Nantes) for lung biopsies from lung-transplant recipients, Jerome Martin (INSERM UMR 1064, Nantes) for human neutrophil complementary DNAs, and Philippe Hulin (SFR, Nantes) for confocal microscopy.
This work was supported by the “CENTAURE” foundation, “Agence de la Biomédecine 2015-2016” and realized in the context of the Institut Hospitalo-Universitaire-Cesti project, and the LabEx Immunotherapy Graft-Oncology program, which received French government financial support managed by the National Research Agency via the investment of the future program (ANR-10-IBHU-005 and ANR-11-LABX-0016-01).
Authorship
Contribution: M.D.L.R., A.P., V.H., L.L.T., S.R., C. Braudeau, and L.D. performed experiments; M.D.L.R., A.P., V.H., and E.C. analyzed results and made the figures; A. Moreau, C.L., C. Brosseau, P-.J.R., A. Magnan, F.H., R.J., M.-C.C., I.A., and E.C. designed the research and critically revised the article; and E.C. designed the research and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Elise Chiffoleau, INSERM, UMR 1064, Centre Hospitalier Universitaire, University of Nantes, Hotel Dieu, 30 Bd Jean Monnet, 44093 Nantes, France; e-mail: elise.chiffoleau@univ-nantes.fr.
References
- 1.Geijtenbeek TB, Gringhuis SI. Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol. 2009;9(7):465-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dambuza IM, Brown GD. C-type lectins in immunity: recent developments. Curr Opin Immunol. 2015;32:21-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Plato A, Willment JA, Brown GD. C-type lectin-like receptors of the dectin-1 cluster: ligands and signaling pathways. Int Rev Immunol. 2013;32(2):134-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thebault P, Lhermite N, Tilly G, et al. . The C-type lectin-like receptor CLEC-1, expressed by myeloid cells and endothelial cells, is up-regulated by immunoregulatory mediators and moderates T cell activation. J Immunol. 2009;183(5):3099-3108. [DOI] [PubMed] [Google Scholar]
- 5.Colonna M, Samaridis J, Angman L. Molecular characterization of two novel C-type lectin-like receptors, one of which is selectively expressed in human dendritic cells. Eur J Immunol. 2000;30(2):697-704. [DOI] [PubMed] [Google Scholar]
- 6.Sobanov Y, Bernreiter A, Derdak S, et al. . A novel cluster of lectin-like receptor genes expressed in monocytic, dendritic and endothelial cells maps close to the NK receptor genes in the human NK gene complex. Eur J Immunol. 2001;31(12):3493-3503. [DOI] [PubMed] [Google Scholar]
- 7.Flornes LM, Nylenna Ø, Saether PC, Daws MR, Dissen E, Fossum S. The complete inventory of receptors encoded by the rat natural killer cell gene complex. Immunogenetics. 2010;62(8):521-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sattler S, Reiche D, Sturtzel C, et al. . The human C-type lectin-like receptor CLEC-1 is upregulated by TGF-β and primarily localized in the endoplasmic membrane compartment. Scand J Immunol. 2012;75(3):282-292. [DOI] [PubMed] [Google Scholar]
- 9.Ono K, Lindsey ES. Improved technique of heart transplantation in rats. J Thorac Cardiovasc Surg. 1969;57(2):225-229. [PubMed] [Google Scholar]
- 10.Chiffoleau E, Bériou G, Dutartre P, Usal C, Soulillou JP, Cuturi MC. Induction of donor-specific allograft tolerance by short-term treatment with LF15-0195 after transplantation. Evidence for a direct effect on T-cell differentiation. Am J Transplant. 2002;2(8):745-757. [DOI] [PubMed] [Google Scholar]
- 11.Spisek R, Bretaudeau L, Barbieux I, Meflah K, Gregoire M. Standardized generation of fully mature p70 IL-12 secreting monocyte-derived dendritic cells for clinical use. Cancer Immunol Immunother. 2001;50(8):417-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le Bas-Bernardet S, Hourmant M, Coupel S, Bignon JD, Soulillou JP, Charreau B. Non-HLA-type endothelial cell reactive alloantibodies in pre-transplant sera of kidney recipients trigger apoptosis. Am J Transplant. 2003;3(2):167-177. [DOI] [PubMed] [Google Scholar]
- 13.Pêche H, Heslan M, Usal C, Amigorena S, Cuturi MC. Presentation of donor major histocompatibility complex antigens by bone marrow dendritic cell-derived exosomes modulates allograft rejection. Transplantation. 2003;76(10):1503-1510. [DOI] [PubMed] [Google Scholar]
- 14.Louvet C, Heslan JM, Merieau E, Soulillou JP, Cuturi MC, Chiffoleau E. Induction of Fractalkine and CX3CR1 mediated by host CD8+ T cells in allograft tolerance induced by donor specific blood transfusion. Transplantation. 2004;78(9):1259-1266. [DOI] [PubMed] [Google Scholar]
- 15.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402-408. [DOI] [PubMed] [Google Scholar]
- 16.Kataoka H, Kume N, Miyamoto S, et al. . Biosynthesis and post-translational processing of lectin-like oxidized low density lipoprotein receptor-1 (LOX-1). N-linked glycosylation affects cell-surface expression and ligand binding. J Biol Chem. 2000;275(9):6573-6579. [DOI] [PubMed] [Google Scholar]
- 17.Kerscher B, Dambuza IM, Christofi M, et al. . Signalling through MyD88 drives surface expression of the mycobacterial receptors MCL (Clecsf8, Clec4d) and Mincle (Clec4e) following microbial stimulation. Microbes Infect. 2016;18(7-8):505-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pöhlmann S, Zhang J, Baribaud F, et al. . Hepatitis C virus glycoproteins interact with DC-SIGN and DC-SIGNR. J Virol. 2003;77(7):4070-4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Engering A, Van Vliet SJ, Geijtenbeek TB, Van Kooyk Y. Subset of DC-SIGN(+) dendritic cells in human blood transmits HIV-1 to T lymphocytes. Blood. 2002;100(5):1780-1786. [DOI] [PubMed] [Google Scholar]
- 20.Passlick B, Flieger D, Ziegler-Heitbrock HW. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood. 1989;74(7):2527-2534. [PubMed] [Google Scholar]
- 21.Rossol M, Kraus S, Pierer M, Baerwald C, Wagner U. The CD14(bright) CD16+ monocyte subset is expanded in rheumatoid arthritis and promotes expansion of the Th17 cell population. Arthritis Rheum. 2012;64(3):671-677. [DOI] [PubMed] [Google Scholar]
- 22.Piccioli D, Tavarini S, Borgogni E, et al. . Functional specialization of human circulating CD16 and CD1c myeloid dendritic-cell subsets. Blood. 2007;109(12):5371-5379. [DOI] [PubMed] [Google Scholar]
- 23.Lundberg K, Rydnert F, Greiff L, Lindstedt M. Human blood dendritic cell subsets exhibit discriminative pattern recognition receptor profiles. Immunology. 2014;142(2):279-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alanne-Kinnunen M, Lappalainen J, Öörni K, Kovanen PT. Activated human mast cells induce LOX-1-specific scavenger receptor expression in human monocyte-derived macrophages. PLoS One. 2014;9(9):e108352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minami M, Kume N, Kataoka H, et al. . Transforming growth factor-beta(1) increases the expression of lectin-like oxidized low-density lipoprotein receptor-1. Biochem Biophys Res Commun. 2000;272(2):357-361. [DOI] [PubMed] [Google Scholar]
- 26.Draude G, Lorenz RL. TGF-beta1 downregulates CD36 and scavenger receptor A but upregulates LOX-1 in human macrophages. Am J Physiol Heart Circ Physiol. 2000;278(4):H1042-H1048. [DOI] [PubMed] [Google Scholar]
- 27.Vanaudenaerde BM, De Vleeschauwer SI, Vos R, et al. . The role of the IL23/IL17 axis in bronchiolitis obliterans syndrome after lung transplantation. Am J Transplant. 2008;8(9):1911-1920. [DOI] [PubMed] [Google Scholar]
- 28.Marshall AS, Willment JA, Pyz E, et al. . Human MICL (CLEC12A) is differentially glycosylated and is down-regulated following cellular activation. Eur J Immunol. 2006;36(8):2159-2169. [DOI] [PubMed] [Google Scholar]
- 29.Bates EE, Fournier N, Garcia E, et al. . APCs express DCIR, a novel C-type lectin surface receptor containing an immunoreceptor tyrosine-based inhibitory motif. J Immunol. 1999;163(4):1973-1983. [PubMed] [Google Scholar]
- 30.Redelinghuys P, Whitehead L, Augello A, et al. . MICL controls inflammation in rheumatoid arthritis. Ann Rheum Dis. 2016:75(7):1386-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Uto T, Fukaya T, Takagi H, et al. . Clec4A4 is a regulatory receptor for dendritic cells that impairs inflammation and T-cell immunity. Nat Commun. 2016;7:11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.LeibundGut-Landmann S, Gross O, Robinson MJ, et al. . Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007;8(6):630-638. [DOI] [PubMed] [Google Scholar]
- 33.Gringhuis SI, den Dunnen J, Litjens M, et al. . Dectin-1 directs T helper cell differentiation by controlling noncanonical NF-kappaB activation through Raf-1 and Syk. Nat Immunol. 2009;10(2):203-213. [DOI] [PubMed] [Google Scholar]
- 34.Lee EJ, Brown BR, Vance EE, et al. . Mincle activation and the Syk/Card9 signaling axis are central to the development of autoimmune disease of the eye. J Immunol. 2016;196(7):3148-3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rivera A, Hohl TM, Collins N, et al. . Dectin-1 diversifies Aspergillus fumigatus-specific T cell responses by inhibiting T helper type 1 CD4 T cell differentiation. J Exp Med. 2011;208(2):369-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stoppelkamp S, Reid DM, Yeoh J, et al. . Murine pattern recognition receptor dectin-1 is essential in the development of experimental autoimmune uveoretinitis. Mol Immunol. 2015;67(2 pt B):398-406. [DOI] [PubMed] [Google Scholar]
- 37.Joo H, Upchurch K, Zhang W, et al. . Opposing roles of dectin-1 expressed on human plasmacytoid dendritic cells and myeloid dendritic cells in Th2 polarization. J Immunol. 2015;195(4):1723-1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mahnke K, Guo M, Lee S, et al. . The dendritic cell receptor for endocytosis, DEC-205, can recycle and enhance antigen presentation via major histocompatibility complex class II-positive lysosomal compartments. J Cell Biol. 2000;151(3):673-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Underhill DM, Rossnagle E, Lowell CA, Simmons RM. Dectin-1 activates Syk tyrosine kinase in a dynamic subset of macrophages for reactive oxygen production. Blood. 2005;106(7):2543-2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Svajger U, Anderluh M, Jeras M, Obermajer N. C-type lectin DC-SIGN: an adhesion, signalling and antigen-uptake molecule that guides dendritic cells in immunity. Cell Signal. 2010;22(10):1397-1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mori D, Shibata K, Yamasaki S. C-type lectin receptor dectin-2 binds to an endogenous protein β-glucuronidase on dendritic cells. PLoS One. 2017;12(1):e0169562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aragane Y, Maeda A, Schwarz A, Tezuka T, Ariizumi K, Schwarz T. Involvement of dectin-2 in ultraviolet radiation-induced tolerance. J Immunol. 2003;171(7):3801-3807. [DOI] [PubMed] [Google Scholar]
- 43.Ariizumi K, Shen GL, Shikano S, et al. . Identification of a novel, dendritic cell-associated molecule, dectin-1, by subtractive cDNA cloning. J Biol Chem. 2000;275(26):20157-20167. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.