

Key Points
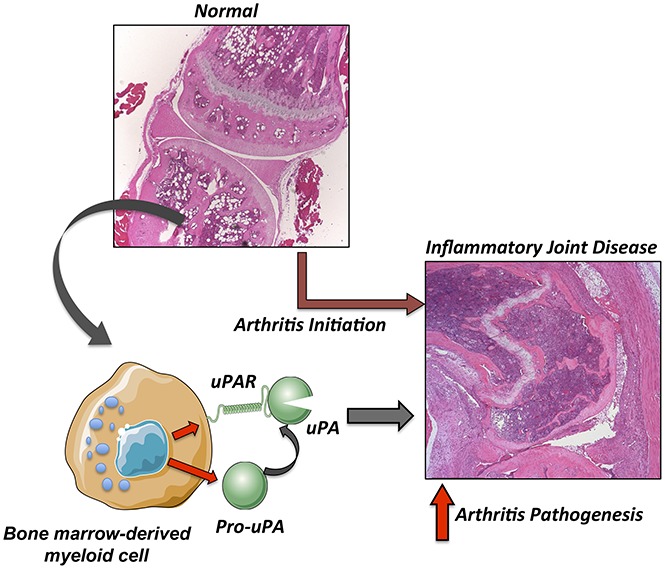
Deficiency in uPA or uPAR renders DBA/1 mice less susceptible to CIA.
Expression of uPAR in bone marrow–derived cells promotes arthritis pathogenesis.
Abstract
The plasminogen activation (PA) system has been implicated in driving inflammatory arthritis, but the precise contribution of PA system components to arthritis pathogenesis remains poorly defined. Here, the role of urokinase plasminogen activator (uPA) and its cognate receptor (uPAR) in the development and severity of inflammatory joint disease was determined using uPA- and uPAR-deficient mice inbred to the strain DBA/1J, a genetic background highly susceptible to collagen-induced arthritis (CIA). Mice deficient in uPA displayed a near-complete amelioration of macroscopic and histological inflammatory joint disease following CIA challenge. Similarly, CIA-challenged uPAR-deficient mice exhibited significant amelioration of arthritis incidence and severity. Reduced disease development in uPA-deficient and uPAR-deficient mice was not due to an altered adaptive immune response to the CIA challenge. Reciprocal bone marrow transplant studies indicated that uPAR-driven CIA was due to expression by hematopoietic-derived cells, as mice with uPAR-deficient bone marrow challenged with CIA developed significantly reduced macroscopic and histological joint disease as compared with mice with uPAR expression limited to non–hematopoietic-derived cells. These findings indicate a fundamental role for uPAR-expressing hematopoietic cells in driving arthritis incidence and progression. Thus, uPA/uPAR-mediated cell surface proteolysis and/or uPAR-mediated signaling events promote inflammatory joint disease, indicating that disruption of this key proteolytic/signaling system may provide a novel therapeutic strategy to limit clinical arthritis.
Visual Abstract
Introduction
Coagulation and fibrinolytic activity is observed in both experimental animals with inflammatory joint disease and patients with rheumatoid arthritis (RA). Analyses of hemostatic factors in RA disease models have indicated roles for fibrinogen, thrombin, plasminogen, and the plasminogen activators in promoting inflammatory arthritic disease.1-5 Components of the plasminogen activation (PA) system appear to be particularly powerful determinants of inflammatory arthritis.6-9 High levels of urokinase plasminogen activator (uPA) and tissue-type plasminogen activator activity have been documented in synovial tissue from RA patients.10 Similarly, increased levels of the physiological inhibitors of the PA system, plasminogen activator inhibitors 1 and 2, have been documented in RA tissue.11,12 Indeed, intriguing evidence from several studies has suggested that uPA can either exacerbate or limit the development of arthritis depending on the experimental model system.6,13-15 As with uPA, elevated expression levels of the uPA receptor (uPAR) have been reported in synovial tissue, as well as increased soluble uPAR protein in the circulation of RA patients and patients with primary Sjögrens syndrome.10,16 However, a potential functional contribution for uPAR in arthritis pathogenesis, be it in promoting or inhibiting disease, remains undefined.
A primary role of uPA is the conversion of plasminogen to the active serine protease plasmin and subsequent fibrinolysis or degradation of fibrin matrices. Fibrin deposits are observed throughout hyperplastic synovial tissues and along affected articular surfaces as a hallmark of arthritis.1 In addition, fibrin is a major component of insoluble “rice bodies” found in the joint space.17 Previous studies highlighted that fibrin deposition within inflamed joint tissue can contribute to the incidence and severity of arthritis, as mice deficient in fibrinogen displayed decreased incidence and severity of disease.1 In addition to mediating fibrinolysis, uPA and uPAR have the capacity to contribute to arthritis pathogenesis by mediating local inflammation and tissue remodeling through cell-signaling mechanisms. For example, uPA has been linked to cell migration and proliferation.18 Interleukin 1β (IL-1β) and tumor necrosis factor α (TNF-α) elicit expression and secretion of uPA by several cells, including monocytes, endothelial cells, and neutrophils.19-21
The present study sought to determine the contribution of both uPA and uPAR to collagen-induced arthritis (CIA) in the highly susceptible DBA/1 strain of mice. Deficiencies in both uPA and uPAR in the context of the DBA/1 genetic background indicate a role for both uPA and its receptor, uPAR, in the pathogenesis of inflammatory arthritis. Furthermore, bone marrow transplant studies indicate that uPAR deficiency specific to the bone marrow cell compartment suppresses the incidence and severity of CIA.
Materials and methods
CIA
Cohorts of littermate 6- to 8-week-old male wild-type (WT), uPA-deficient (Plau−/−), and uPAR-deficient (Plaur−/−) mice that were backcrossed (8 generations) to the CIA-susceptible DBA/1J strain were injected intradermally at the base of the tail with 100 µg bovine collagen type II (CII) (Elastin Products) in complete Freund’s adjuvant on day 1 and once again on day 21 as previously described.1 Mice were evaluated for arthritis using an arthritic index macroscopic scoring system ranging from 0 to 4 (0 = no detectable arthritis, 1 = swelling and/or redness of paw or 1 digit, 2 = 2 joints involved, 3 = 3 joints involved, and 4 = severe arthritis of the entire paw and digit). Knee joints were fixed for 48 hours in 10% neutral-buffered formalin (Sigma) and then decalcified in TBD-2 (ThermoShandon) or 10% EDTA for 3 weeks. Four-micrometer sections of paraffin-embedded tissue were placed on Colorfrost Plus slides (Fisher Scientific) and processed for hematoxylin and eosin or fibrin(ogen) immunohistochemistry with a rabbit anti–mouse fibrinogen antisera as described previously.2 A semiquantitative histopathology analysis was performed for each knee joint based on the following scoring criteria for CIA: inflammation (0-3), synovial hyperplasia (0-3), edema (0-3), pannus (0-1), and bone/cartilage loss (0-3) for a total histopathology index (0-26) for knees of a given animal.1 The Cincinnati Children’s Hospital Medical Center Institutional Animal Care and Use Committee approved all experiments performed on mice.
Analysis of T-cell and B-cell responses
For CII-specific cell proliferation assays, spleen cells (5 × 105/well) were plated in triplicate in RPMI media containing 0.5% normal DBA/1 mouse serum at 37°C in 96-well flat-bottom microtiter plates that had been precoated with heat-denatured (56°C for 10 min) CII or antibody 145-2C11 (anti-CD3)–containing cell culture supernatant as a positive control. Cells were incubated for 72 hours, followed by addition of 1 µCi [3H]thymidine for an additional 18 hours, and then harvested. [3H]thymidine incorporation per well was measured and averaged for each triplicate. Background counts from unstimulated cells were subtracted from each group. Anti–CII antibody titers in plasma samples were determined by enzyme-linked immunosorbent assay, as described previously.22
RNA isolation and quantitative RT-PCR
Frozen (−80°C) hindpaws were homogenized in TRIzol reagent (Invitrogen) using a tissumizer (Tekmar) to extract total RNA and further purified using the RNeasy MicroKit (QIAGEN). Complementary DNA synthesis was performed using a High Capacity RNA-to-cDNA kit (Applied Biosystems), and quantitative reverse transcription polymerase chain reaction (RT-PCR) was performed with a StepOne Plus instrument (Applied Biosystems) using TaqMan probes for mouse IL-6 (Mm00446190_m1), IL-1β (Mm01336189_m1), and mouse TNF-α (Mm99999068_m1), in conjunction with TaqMan Gene Expression Master Mix (Applied Biosystems). Data were analyzed using the Pfaffl method in which β2-microglobulin (Mm00437762_m1) was used as the normalizing gene. Fold differences are depicted with levels in normal unchallenged WT mice set to 1.
Bone marrow transplantation
Recipient mice (6-8 weeks of age) were administered lethal irradiation (137Cs source; 700 cGy followed by 400 cGy spaced 4 hours apart) to eradicate all bone marrow cells. Irradiated mice were administered a tail vein injection of 2-3 × 106 harvested donor bone marrow cells, and marrow reconstitution was allowed to proceed for 8 weeks. At this time point, mice were bled and checked for 3 consecutive weeks for analysis of complete blood count and standard and quantitative polymerase chain reaction analysis of WT and mutant alleles, which indicated a complete reconstitution of the bone marrow compartment with donor derived cells. Following affirmation of bone marrow reconstitution, mice were challenged with CIA as described above.
Statistical analysis
Percentage of mice arthritis-free data were analyzed using the Kaplan-Meier log-rank test. Cytokine messenger RNA (mRNA) levels were subjected to Grubb’s test to identify outliers and analyzed by analysis of variance and Student-Newman-Keuls post hoc test. Differences in macroscopic and histological parameters of inflammatory joint disease were analyzed using the Mann-Whitney U test or the paired t test, as indicated.
Results
uPA and uPAR deficiency dramatically diminishes the incidence and severity of CIA
To determine the contribution of uPA and its receptor, uPAR, to inflammatory joint disease development and progression, mice with genetically imposed uPA or uPAR deficiencies (Plau−/− and Plaur−/−, respectively) were immunized with bovine type II collagen (CII). The response to CII immunization is dependent on multiple genetic factors, including major histocompatibility complex haplotype. Thus, to ensure a robust response to the immunization and minimize variability linked to response differences in genetic background, the previously characterized Plau-knockout23 and Plaur-knockout alleles24 were separately backcrossed for 8 generations (ie, >99% backcrossed) to the highly CIA-susceptible DBA/1 (major histocompatibility complex class II H-2q haplotype) strain.
Macroscopic evaluation of the fore- and hindpaws of control and Plau−/− mice shortly after secondary CII immunization revealed an obvious distinction in the development of arthritis as indicated by the percentage of Plau−/− mice that remained free of macroscopic arthritis (Figure 1A). Plau−/− mice developed appreciably less joint disease as indicated by a statistically significant diminution in macroscopic arthritis index scores at each time point beginning 27 days after primary CII injection (Figure 1B). Individual arthritis scores at day 42 following primary CII injection (Figure 1C) indicated a lack of arthritic disease in Plau−/− animals as compared with WT mice. Plau−/− animals also exhibited a statistically significant decrease in arthritis severity as evaluated using a score based on the degree of swelling for each paw (Figure 1D) from day 27 to the end of the evaluation period. As observed with the arthritis index, individual severity scores at day 42 following primary CII injection (Figure 1E) indicate a significant decrease in paw swelling in Plau−/− animals as compared with WT mice.
Figure 1.

Genetically imposed uPA deficiency is protective against the development of CIA in DBA/1 mice. (A) Kaplan-Meier log-rank analysis indicating the percentage of WT and Plau−/− mice with macroscopic arthritis in the paws is shown for the 3-week evaluation period after the second CII immunization. Note that ∼75% of DBA/1 Plau−/− mice remained arthritis-free based on visible inspection through the CIA study period. (B-C) The median arthritis index revealed a significant diminution in number of arthritic joints per mouse in the absence of uPA (B), with individual scores indicated for day 42 (C). (D-E) The median arthritis severity for the same cohort of mice revealed a significant reduction in paw swelling for CIA-challenged Plau−/− mice (n = 16 in the WT group and n = 11 in the Plau−/− group) (D), with individual scores indicated for day 42 (E). (F) Kaplan-Meier log-rank analysis indicating the percentage of WT and Plaur−/− mice with macroscopic arthritis in the paws is shown for the 3-week evaluation period after the second CII immunization. Note that ∼60% of DBA/1 Plaur−/− mice remained arthritis-free based on visible inspection through the CIA study period. (G and H) The median arthritis index revealed a significant diminution in number of arthritic joints per mouse in the absence of uPAR (G), with individual scores indicated for day 42 (H). (I-J) The median arthritis severity for the same cohort of mice revealed a significant reduction in paw swelling for CIA-challenged Plaur−/− mice (n = 13 in the WT group and n = 14 in the Plaur−/− group) (I), with individual scores indicated for day 42 (J). P values were determined by Mann-Whitney U test. *P < .05.
Evaluation of the macroscopic disease in Plaur−/− mice as compared with WT littermates indicated that uPAR deficiency also results in a significant reduction in the number of animals displaying any clinical arthritis (Figure 1F). Similarly, Plaur−/− mice exhibited less severe disease overall than their WT littermates, with significant differences observed from day 27 to the end of the evaluation period (Figure 1G). Individual macroscopic scores at day 42 (Figure 1H) depict the significantly reduced arthritis in Plaur−/− as compared with WT mice. Plaur−/− animals also showed a significant decrease in arthritis severity (Figure 1I), and individual severity scores at day 42 following primary CII injection (Figure 1J) indicate a significant decrease in paw swelling in Plaur−/− animals as compared with WT mice.
uPA or uPAR deficiency results in reduced CIA-mediated joint destruction
To ascertain if the genotype-dependent differences in macroscopic evidence of arthritis were associated with differential degenerative microscopic disease, histological analyses were performed on midline sagittal knee joint sections from CIA-challenged mice of each genotype. Representative sections of knee joints from control and mutant mice (Figure 2A) highlight the substantial qualitative benefit of eliminating uPA or uPAR to reduce local inflammatory joint disease. The knee joints of CIA-challenged WT animals were typically characterized by significant inflammatory infiltrates, invading granulation tissue (pannus), and loss of intact cartilage and bone (Figure 2A), whereas the overall architecture of knee joints from CIA-challenged Plau−/− or Plaur−/− mice was typically unperturbed. Analysis of fibrin deposition by immunohistochemistry revealed an absence of fibrin deposits in unchallenged mice regardless of genotype (Figure 2B). CIA-challenged WT mice had significant fibrin deposits along articular surfaces and throughout hyperplastic synovial tissue (Figure 2B). In contrast, CIA-challenged Plau−/− and Plaur−/− mice displayed very little fibrin deposition within knee joint tissue, consistent with a lack of inflammatory disease development (Figure 2B). A quantitative histological comparison was performed on knee joint sections from cohorts of both Plau−/− and WT littermates as well as on Plaur−/− and WT littermates 42 days after primary CII immunization. Two independent investigators unaware of animal genotype evaluated and scored knee joint sections with regard to inflammation, synovial hyperplasia, edema, pannus formation, and cartilage/bone destruction as previously described.1 Each individual disease parameter scored for histology was significantly diminished in Plau−/− mice as compared with their littermate uPA-sufficient control mice (Figure 2B), consistent with the view that the genetic elimination of uPA broadly limits the pathological process. The composite histopathology index (Figure 2C) in Plau−/− mice showed a statistically significant diminution relative to that for uPA-sufficient mice. A similar pattern of significantly reduced histological disease was observed for knee joint sections from CIA-challenged Plaur−/− mice. Again, individual histology parameters as well as the total composite histopathology index was significantly diminished compared with WT mice (Figure 2D-E), but to a lesser degree than Plau−/− mice.
Figure 2.

Elimination of uPA or uPAR in DBA/1 mice results in diminished CIA knee joint pathology. (A) Representative hematoxylin and eosin–stained knee joint sections from unchallenged WT and CIA-challenged WT, uPA−/−, uPAR WT, and uPAR−/− cohorts. Upon CIA challenge, significant inflammation, synovial hyperplasia, and erosive pannus are apparent in WT mice, whereas knee joints of uPA−/− and uPAR−/− mice display markedly attenuated pathological features. Scale bar represents 200 μm. (B) Representative knee joint sections of unchallenged and CIA-challenged WT, uPA−/−, and uPAR−/− mice stained by immunohistochemistry for fibrin. Note the fibrin staining along articular surfaces (arrows) and within hyperplastic synovial tissue (asterisk) as CIA-challenged WT mice. Scale bar represents 200 μm. (C) Semiquantitative microscopic analysis of knee joint pathological features from WT (n = 29, red bars) and Plau−/− (n = 17, white, blue outline bars) mice. Student t test. Scatter plot of composite histopathology index analysis (see Materials and methods) of hematoxylin and eosin–stained knee joint sections. (D) Semiquantitative microscopic analysis of knee joint pathological features from WT (n = 8, red bars) and Plaur−/− (n = 16, white, green outline bars) mice. Student t test. Scatter plot of composite histopathology index analysis (see Materials and methods) of hematoxylin and eosin–stained knee joint sections. Each symbol represents the composite score for individual knees and bars denote median values for each genotype. P values were determined by Mann-Whitney U test. #P < .001 and *P < .05.
uPA and uPAR deficiency results in decreased cytokine expression in paw joints
To determine whether the loss of uPA and uPAR resulted in changes in local proinflammatory cytokine production, mRNA levels of TNF-α, IL-1β, and IL-6 were assessed in hindpaw tissue at day 42 of disease. These cytokines were specifically selected for analysis, as each has been linked to arthritis pathogenesis. As shown in Figure 3, mRNA levels of TNF-α, IL-1β, and IL-6 in paws from unchallenged mice were each significantly lower than that of WT CIA-challenged mice, which was consistent with the development of inflammatory joint disease. Plau−/− CIA-challenged mice showed significantly reduced mRNA levels of TNF-α and IL-6 mRNA as compared with WT CII-challenged mice, whereas levels of Il-1β trended lower in Plau−/− mice (Figure 3A-C). CIA-challenged Plaur−/− mice similarly demonstrated significantly decreased mRNA levels of IL-1β and IL-6 mRNA in paws from CII-challenged WT mice (Figure 3D-F). Thus, expression of key inflammatory cytokines mediating arthritis disease progression is reduced by the elimination of uPA or uPAR.
Figure 3.

Reduced inflammatory cytokine expression in CIA-challenged uPA- and uPAR-deficient mice. (A-C) Quantitative RT-PCR analysis of messenger RNA levels for the inflammatory cytokines TNF-α (A), IL-1β (B), and IL-6 (C) in the hindpaws obtained from unchallenged WT and Plau−/− mice (n = 5 and 4, respectively) and day 42 of CIA WT and Plau−/− mice (n = 8 per genotype). (D-F) Quantitative RT-PCR analysis of mRNA levels for the inflammatory cytokines TNF-α (D), IL-1β (E), and IL-6 (F) in the hindpaws obtained from unchallenged wild-type and Plaur−/− mice (n = 6 per genotype) and day 42 of CIA WT and Plaur−/− mice (n = 6 per genotype). Data are expressed as average fold change over unchallenged WT group with error bars denoting standard error of the mean. Data were analyzed by 2-way analysis of variance followed by Student-Newman-Keuls post hoc test. #P < .05 for comparing unchallenged and CIA-challenged of the same genotype, *P < .05 for comparing WT to Plau−/− or Plaur−/− at day 42 of CIA, and **P < .01 for comparing WT to Plau−/− or Plaur−/− at day 42 of CIA.
uPA and uPAR deficiencies have little effect on the adaptive immune response to CII
To determine whether the significantly reduced inflammatory joint disease observed in Plau−/− and Plaur−/− mice was linked to an ameliorated response to the immunization protocol, CII-specific T- and B-cell adaptive immune responses were measured. Plasma was collected from control and Plau−/− or Plaur−/− animals on day 42 following primary CII immunization and analyzed for anti–CII antibody titers. As shown in Figure 4A-C, anti–CII antibody titers of total immunoglobulin G (IgG) and the IgG1 and IgG2a subclasses were similar between control animals and Plau−/− or Plaur−/− animals, with the exception of the IgG2a subclass being significantly lower in Plau−/− animals. In parallel, the development of cellular immunity was not impaired in Plau−/− or Plaur−/− mice. Splenocytes prepared from animals 42 days following primary CII immunization were analyzed for their ability to proliferate in response to CII. Spleen cells from CIA-challenged Plau−/− mice and their WT controls, as well as from Plaur−/− mice and their controls, responded similarly to stimulation with CII (Figure 4D). As a positive control, splenocyte preparations were also stimulated with an activating T-cell receptor antibody, 145-2C11, and a similar robust proliferative response was observed across genotypes (Figure 4E). Together, these data indicate that Plau−/− or Plaur−/− mice are able to mount an adaptive immune response similar to that of control animals following CII immunization.
Figure 4.

The loss of uPA or uPAR does not alter CII-specific B- and T-cell responses. (A-C) CII-specific antibody titers were analyzed in the plasma of untreated (no CII) DBA/1J mice and CIA-challenged uPA WT and Plau−/− or uPAR WT and Plaur−/− mice (n = 5 or more mice per genotype). (D-E) Proliferation of splenocytes harvested from CIA-challenged uPA WT, Plau−/−, uPAR WT, and Plaur−/− mice following stimulation with either (D) heat-inactivated CII (100 μg/mL) or (E) direct T-cell receptor stimulation using anti–CD3 antibody as evaluated by [3H] thymidine incorporation. Note that for all T-cell analyses, n = 4 mice per genotype. All data are presented as mean ± standard error of the mean. P values were determined by Student t test.
The bone marrow compartment is the source of uPAR-expressing cells promoting the incidence and severity of CIA
Previous studies identified a requirement of uPA expression from bone marrow–derived cells for the full development of CIA.14 To similarly determine whether the cells within the bone marrow compartment or cells local to the joint expressing uPAR are driving arthritis, WT and Plaur−/− bone marrow cells were transferred into WT irradiated recipients. Recipients of Plaur−/− bone marrow developed appreciably less joint disease than recipients of WT bone marrow (Figure 5A) accompanied by a significant diminution in clinical arthritis scores (Figure 5B) and severity index (Figure 5D) from day 33 to the end of the evaluation period at day 42. Individual arthritis (Figure 5C) scores at day 42 of disease indicated a lack of arthritis observed in WT mice reconstituted with Plaur−/− bone marrow as compared with WT bone marrow recipients.
Figure 5.

Expression of uPAR by hematopoietic cells promotes susceptibility to CIA. (A-D) Bone marrow from either uPAR WT or Plaur−/− mice was transplanted into WT recipients. (A) The percentage of mice free from macroscopic arthritis in the paws is shown for the 3-week evaluation period following the second CII immunization. Note that ∼60% of Plaur−/− transplanted mice remained arthritis-free based on visible inspection through the CIA study period (P < .05, Kaplan-Meier log-rank analysis). (B-C) The median arthritis index revealed a significant diminution in number of arthritic joints with transplant of Plaur−/− bone marrow cells (B), with individual scores indicated for day 42 (C). The median arthritis severity for the same cohort of mice revealed a reduction in paw swelling for CIA-challenged mice with transplant of Plaur−/− bone marrow cells (D). (E-H) Bone marrow from WT or Plaur−/− mice was transplanted into Plaur−/− recipients. (E) The percentage of mice free from macroscopic arthritis in the paws is shown for the 3-week evaluation period following the second CII immunization, with ∼65% of Plaur−/− bone marrow recipients remaining arthritis-free. (F-G) The median arthritis index revealed a significant diminution in number of arthritic joints with transplant of Plaur−/− bone marrow cells (F), with individual scores indicated for day 42 (G). The median arthritis severity for the same cohort of mice revealed a reduction in paw swelling for CIA-challenged mice with transplant of uPAR−/− bone marrow cells (H). For WT recipient experiments, n = 11 uPAR WT mice and n = 12 Plaur−/− mice. For uPAR-deficient recipients, n = 11 uPAR WT mice and n = 12 Plaur−/− Mice. P values were determined by Mann-Whitney U test.
A reciprocal study design was used to determine if WT bone marrow could restore inflammatory joint disease in Plaur−/− mice. Bone marrow from both WT littermates and Plaur−/− mice was transferred into uPAR-deficient recipients. Plaur−/− recipients with WT bone marrow developed joint disease at a rate that trended higher than that of uPAR-deficient recipients of Plaur−/− bone marrow (Figure 5E). Macroscopic disease was significantly exacerbated in WT bone marrow recipients from day 32 of CIA to the end point of the study (Figure 5F). Individual arthritis index scores (Figure 5G) were also significantly higher in Plaur−/− recipients of WT bone marrow at day 42 of disease course. The severity of disease trended higher in Plaur−/− recipients of WT bone marrow (Figure 5H), but the differences were not statistically significant.
Discussion
The present study indicates that mice deficient in uPA and uPAR on the highly susceptible DBA/1 background show a significant reduction in inflammatory joint disease incidence and ultimately develop less severe inflammatory arthritis, further supporting key roles for these 2 hemostatic factors in the pathogenesis of arthritis. Macroscopic, histological, and molecular examination of joint tissue indicated a reduction in clinically evident arthritis, a reduction in expression of key inflammatory cytokines that contribute to arthritis, and a reduction in pathological joint inflammation and destruction in the absence of uPA and uPAR. However, adaptive immune responses did not differ between uPA- and uPAR-sufficient and deficient animals. Furthermore, our studies utilizing bone marrow transplantation indicated that uPAR-expressing cells from the bone marrow compartment promote the pathogenesis of inflammatory arthritis.
Previous studies utilizing the CIA model or direct antibody transfer models (ie, K/BxN or collagen monoclonal antibody–induced arthritis) implicated uPA in the pathogenesis of arthritis. However, there are notable distinctions between these previous reports and the current work. Similar to results reported here, uPA-deficient CIA-challenged mice on the less susceptible C57Bl/6 (H-2b) mouse strain exhibited decreased disease,6 indicating that uPA drives pathogenesis of inflammatory joint disease. Notably, in that study, 62% of C57Bl/6 uPA-deficient mice developed some signs of macroscopic CIA, which was not significantly different from C57Bl/6 control mice (73% incidence). In addition, in C57Bl/6 mice deficient in uPA and backcrossed to DBA/1 for 1 generation, 40% of uPA-deficient mice exhibit arthritis as compared with 70% to 80% of WT mice.4 In contrast, the current study indicated only ∼25% of DBA/1 uPA-deficient mice developed any evidence of disease, which was a statistically significant difference in comparison with the 100% of control DBA/1 mice that developed disease. Both our study in DBA/1 mice and the prior study using C57Bl/6 mice indicated that among the mice that do develop arthritis, the overall severity is significantly reduced in uPA-deficient mice.
The precise cellular or molecular basis for the difference in uPA-dependent CIA disease development based on genetic background remains unknown. However, it is notable that analyses of CIA with C57Bl/6 mice revealed reduced T-cell proliferation and interferon γ production following secondary CII stimulation of cells harvested from CII-immunized uPA-deficient mice relative to uPA-sufficient mice.6 Previous studies suggested that C57Bl/6 uPA-deficient mice mount significantly diminished Th1 and Th2 responses following infection.25,26 We observed no differences in T-cell responses using DBA/1 uPA-deficient mice. Rather, a modest but statistically significant reduction in subclass IgG2a anti–CII antibody titers in uPA-deficient mice was observed. The IgG2a subtype has previously been shown to have a critical role in promoting arthritis pathogenesis,27,28 and thus, a reduction in this subtype of antibody would be consistent with diminished local disease. Analyses of CIA in C57Bl/6 uPA-deficient mice revealed no differences in anti–CII antibody titers.6 Whether the modest differences in cellular and humoral immune responses in the 2 genetic backgrounds make a biologically meaningful contribution to the robust reduction in CIA for uPA-deficient mice remains to be determined.
Hematopoietic cells have previously been identified as a dominant source of uPA promoting CIA.6 Here, we have performed reciprocal bone marrow transplant studies with Plaur−/− mice. Like uPA, cells expressing uPAR derived from the hematopoietic compartment are critical for full development of CIA in mice. In the bone marrow transplant experiments performed here, an overall reduction in CIA severity in Plaur−/− mice receiving WT bone marrow relative to nontransplanted WT mice was observed. It is possible that Plaur−/− mice developed an immune response to uPAR expressed by the WT hematopoietic cells. However, if such a host response did occur, it was not sufficient to prohibit arthritis development in Plaur−/− hosts. It is possible that the slightly diminished response observed in the bone marrow transplant experiments was simply due to an overall weakened immune system secondary to whole-body irradiation and recovery of the transplant. Regardless, our findings are consistent with the conclusion that it is expression by a hematopoietic cell that mediates uPAR-driven CIA disease progression.
The specific identity of the uPAR- (and uPA)-expressing cell or cells from the hematopoietic compartment driving CIA remains to be determined. One prominent hematopoietic-derived candidate cell type is the monocyte/macrophage. Macrophages are well characterized as expressing both uPA and uPAR, and the expression of both in macrophages increases following exposure to proinflammatory stimuli. In RA, macrophages accumulate throughout inflamed synovial tissue.29 Macrophages also secrete several inflammatory cytokines (eg, IL-1, TNF-α, and IL-6) that play a seminal role in RA pathogenesis. Additionally macrophages produce a spectrum of proteases (in addition to uPA) such as matrix metalloproteinases that drive tissue destruction in RA.30-32 In CIA, macrophages play a preeminent role in driving disease. Targeting molecules that promote macrophage proliferation, accumulation or activation (eg, CSF-1, granulocyte-macrophage colony-stimulating factor, TNF-α, IL-1β, and IL-6)33-40 suppresses CIA in mice. As has been reported using antibody-mediated macrophage depletion,41 we found that macrophage depletion by systemic clodronate administration resulted in amelioration of CIA to an extent analogous to what was observed with uPA-deficient mice (data not shown). Local macrophage depletion by intra-articular clodronate injections has shown some benefit in rheumatoid arthritis,42 and macrophage depletion is still explored as a therapeutic option within inflammatory diseases.43 In addition to macrophages, neutrophils and natural killer cells could be linked to the uPA/uPAR-mediated inflammatory arthritis. Each of these cell types has been shown to express both uPA and uPAR,19,44-47 and each has been linked to arthritis pathogenesis in both RA and CIA.20,33,48-51 Whether uPA/uPAR-dependent mechanisms of arthritis pathogenesis occur through multiple hematopoietic cell types and whether these mechanisms require uPA production by one cell type and uPAR expression by another remain to be determined.
A primary function of uPA is PA leading to fibrinolysis. Fibrin deposits within the inflamed joint are a feature of CIA and inflammatory arthritis in general.1,17 PA by uPA and subsequent fibrin clearance is unlikely to be the mechanism of action by which uPA elimination reduces disease in CIA given that the genetic elimination of fibrinogen itself significantly reduces disease in this arthritis model.1 Further, despite the loss of uPA, we observed little fibrin deposition within joint tissue of Plau−/− or Plaur−/− unchallenged mice, suggesting that tissue-type plasminogen activator was sufficient to mediate baseline fibrin surveillance and clearance. Fibrin deposits were also minimal in CIA-challenged Plau−/− and Plaur−/− mice consistent with the protection from inflammatory joint disease development found in these animals. In contrast, in the model of antigen-induced arthritis, uPA-deficient mice exhibited increased disease pathology.8,13 The basis for this distinct phenotype was linked to the wound field generated by the intra-articular injection. In immune complex–driven arthritis, where uPA deficiency ameliorates disease, the elimination of uPA results in increased disease if a joint is simultaneously subjected to an intra-articular injection of saline.8 It was proposed that the local wound field resulted in exuberant fibrin deposition, due to loss of uPA/plasmin-mediated fibrinolysis that in turn promoted local inflammation driving arthritis severity. Thus, in the absence of a local secondary insult, the role of uPA is as a disease driver rather than a protective factor in models of systemic arthritis.
There are multiple potential mechanisms of action for uPA/uPAR in CIA other than strictly plasmin-mediated fibrinolysis. uPA/uPAR may be driving arthritis by supporting inflammatory cell migration to affected joints. Our analysis of cytokine expression in the joints of CII-challenged mice supports the presence of inflammatory cells producing cytokines in arthritic joints. uPA has been shown to support macrophage migration/chemotaxis in vitro as well as in animal models of atherosclerosis, skeletal muscle regeneration, and cancer.52-54 uPA/uPAR may modulate proinflammatory CIA effector cell functions in concert with integrins or other cell-surface receptors.55-57 It has also been proposed that uPA may be driving arthritis pathogenesis by supporting immune complex–mediated complement factor C5a activation and inflammation. Local disease in the CIA model is driven by C5a generation.58,59 uPA was shown to be required for immune complex–mediated neutrophilia in a peritonitis model.14 uPA/uPAR signaling crosstalk in macrophages was shown to alter the C5a-driven signaling through PKC-δ in a model of sepsis.60 uPA has been shown to regulate osteoclastogenesis and bone resorption through activation of a protease-activated receptor 1/adenosine monophosphate–activated protein kinase pathway.61 uPAR was also shown to influence osteoclastogenesis at multiple levels, including by driving production and release of macrophage colony-stimulating factor by osteoblasts as well as by directly mediating formation, differentiation, and functional properties of macrophage-derived osteoclasts.62 Notably, the impact of uPA or uPAR deficiency on arthritis pathogenesis may not be limited to a single pathway or event but may rather be representative of a multifactorial effect, a concept consistent with the near-curative effect of complete uPA or uPAR deficiency in the CIA model.
These studies further highlight the potent contribution of the PA system to arthritis pathogenesis. In the highly susceptible strain DBA/1, uPA deficiency resulted in the near-complete amelioration of disease development despite a robust adaptive immune response to the immunization challenge. Furthermore, these findings document a similarly potent role for uPAR derived from the hematopoietic compartment in driving susceptibility and severity to systemic arthritis. Although significant work remains to establish the precise mechanisms of action for uPA and uPAR in arthritis, the findings presented here provide the proof of principle that targeting uPA or uPAR could be effective in treating inflammatory arthritis.
Acknowledgments
The authors thank Alice Jone for technical assistance, Jeff Bailey and the Cincinnati Children’s Hospital Medical Center Comprehensive Mouse and Cancer Core, and Jonathan G. Schoenecker for providing a clinical perspective in considering the data.
This research was supported by National Institutes of Health, National Institute of Arthritis and Musculoskeletal and Skin Diseases, grant AR056990 (M.J.F.) and Cincinnati Rheumatic Diseases Center Animal Models of Inflammatory Diseases Core grant P30 AR47363 (M.J.F.). A portion of this research project was supported by Novo Nordisk.
Authorship
Contribution: S.T., H.R., and M.J.F. performed the mouse model CIA experiments; C.C. and M.D.F. generated mice, prepared histological sections, and assisted with T- and B-cell assays; J.S.P., E.S.M., K.A., and P.A.U. provided expert assistance and guidance in data analysis and interpretation; S.T. and M.J.F. drafted the manuscript; all authors participated in the study design and reviewed and revised the manuscript critically; and all authors read and approved the final manuscript.
Conflict-of-interest disclosure: P.A.U. and K.A. are employees of Novo Nordisk. The remaining authors declare no competing financial interests.
Correspondence: Matthew J. Flick, Division of Experimental Hematology and Cancer Biology–ML7015, Children’s Hospital Research Foundation, 3333 Burnet Ave, Cincinnati, OH 45229-3039; e-mail: matthew.flick@cchmc.org.
References
- 1.Flick MJ, LaJeunesse CM, Talmage KE, et al. . Fibrin(ogen) exacerbates inflammatory joint disease through a mechanism linked to the integrin alphaMbeta2 binding motif. J Clin Invest. 2007;117(11):3224-3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raghu H, Jone A, Cruz C, et al. . Plasminogen is a joint-specific positive or negative determinant of arthritis pathogenesis in mice. Arthritis Rheumatol. 2014;66(6):1504-1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flick MJ, Chauhan AK, Frederick M, et al. . The development of inflammatory joint disease is attenuated in mice expressing the anticoagulant prothrombin mutant W215A/E217A. Blood. 2011;117(23):6326-6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li J, Ny A, Leonardsson G, Nandakumar KS, Holmdahl R, Ny T. The plasminogen activator/plasmin system is essential for development of the joint inflammatory phase of collagen type II-induced arthritis. Am J Pathol. 2005;166(3):783-792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marty I, Péclat V, Kirdaite G, Salvi R, So A, Busso N. Amelioration of collagen-induced arthritis by thrombin inhibition. J Clin Invest. 2001;107(5):631-640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cook AD, Braine EL, Campbell IK, Hamilton JA. Differing roles for urokinase and tissue-type plasminogen activator in collagen-induced arthritis. Am J Pathol. 2002;160(3):917-926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cook AD, Rowley MJ, Mackay IR, Gough A, Emery P. Antibodies to type II collagen in early rheumatoid arthritis. Correlation with disease progression. Arthritis Rheum. 1996;39(10):1720-1727. [DOI] [PubMed] [Google Scholar]
- 8.De Nardo CM, Lenzo JC, Pobjoy J, Hamilton JA, Cook AD. Urokinase-type plasminogen activator and arthritis progression: contrasting roles in systemic and monoarticular arthritis models. Arthritis Res Ther. 2010;12(5):R199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang YH, Carmeliet P, Hamilton JA. Tissue-type plasminogen activator deficiency exacerbates arthritis. J Immunol. 2001;167(2):1047-1052. [DOI] [PubMed] [Google Scholar]
- 10.Busso N, Péclat V, So A, Sappino AP. Plasminogen activation in synovial tissues: differences between normal, osteoarthritis, and rheumatoid arthritis joints. Ann Rheum Dis. 1997;56(9):550-557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muñoz-Valle JF, Ruiz-Quezada SL, Oregón-Romero E, et al. . PAI-1 mRNA expression and plasma level in rheumatoid arthritis: relationship with 4G/5G PAI-1 polymorphism. Rheumatol Int. 2012;32(12):3951-3956. [DOI] [PubMed] [Google Scholar]
- 12.Saxne T, Lecander I, Geborek P. Plasminogen activators and plasminogen activator inhibitors in synovial fluid. Difference between inflammatory joint disorders and osteoarthritis. J Rheumatol. 1993;20(1):91-96. [PubMed] [Google Scholar]
- 13.Busso N, Péclat V, Van Ness K, et al. . Exacerbation of antigen-induced arthritis in urokinase-deficient mice. J Clin Invest. 1998;102(1):41-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cook AD, De Nardo CM, Braine EL, et al. . Urokinase-type plasminogen activator and arthritis progression: role in systemic disease with immune complex involvement. Arthritis Res Ther. 2010;12(2):R37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Apparailly F, Bouquet C, Millet V, et al. . Adenovirus-mediated gene transfer of urokinase plasminogen inhibitor inhibits angiogenesis in experimental arthritis. Gene Ther. 2002;9(3):192-200. [DOI] [PubMed] [Google Scholar]
- 16.Slot O, Brünner N, Locht H, Oxholm P, Stephens RW. Soluble urokinase plasminogen activator receptor in plasma of patients with inflammatory rheumatic disorders: increased concentrations in rheumatoid arthritis. Ann Rheum Dis. 1999;58(8):488-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gálvez J, Sola J, Ortuño G, et al. . Microscopic rice bodies in rheumatoid synovial fluid sediments. J Rheumatol. 1992;19(12):1851-1858. [PubMed] [Google Scholar]
- 18.Mondino A, Blasi F. uPA and uPAR in fibrinolysis, immunity and pathology. Trends Immunol. 2004;25(8):450-455. [DOI] [PubMed] [Google Scholar]
- 19.Yoshida E, Tsuchiya K, Sugiki M, Sumi H, Mihara H, Maruyama M. Modulation of the receptor for urokinase-type plasminogen activator in macrophage-like U937 cells by inflammatory mediators. Inflammation. 1996;20(3):319-326. [DOI] [PubMed] [Google Scholar]
- 20.Liu G, Yang Y, Yang S, et al. . The receptor for urokinase regulates TLR2 mediated inflammatory responses in neutrophils. PLoS One. 2011;6(10):e25843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gueler F, Rong S, Mengel M, et al. . Renal urokinase-type plasminogen activator (uPA) receptor but not uPA deficiency strongly attenuates ischemia reperfusion injury and acute kidney allograft rejection. J Immunol. 2008;181(2):1179-1189. [DOI] [PubMed] [Google Scholar]
- 22.Thornton S, Boivin GP, Kim KN, Finkelman FD, Hirsch R. Heterogeneous effects of IL-2 on collagen-induced arthritis. J Immunol. 2000;165(3):1557-1563. [DOI] [PubMed] [Google Scholar]
- 23.Bezerra JA, Bugge TH, Melin-Aldana H, et al. . Plasminogen deficiency leads to impaired remodeling after a toxic injury to the liver. Proc Natl Acad Sci USA. 1999;96(26):15143-15148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bugge TH, Suh TT, Flick MJ, et al. . The receptor for urokinase-type plasminogen activator is not essential for mouse development or fertility. J Biol Chem. 1995;270(28):16886-16894. [DOI] [PubMed] [Google Scholar]
- 25.Gyetko MR, Sud S, Chen GH, Fuller JA, Chensue SW, Toews GB. Urokinase-type plasminogen activator is required for the generation of a type 1 immune response to pulmonary Cryptococcus neoformans infection. J Immunol. 2002;168(2):801-809. [DOI] [PubMed] [Google Scholar]
- 26.Gyetko MR, Sud S, Chensue SW. Urokinase-deficient mice fail to generate a type 2 immune response following schistosomal antigen challenge. Infect Immun. 2004;72(1):461-467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stuart JM, Townes AS, Kang AH. Nature and specificity of the immune response to collagen in type II collagen-induced arthritis in mice. J Clin Invest. 1982;69(3):673-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshino S, Sagai M. Enhancement of collagen-induced arthritis in mice by diesel exhaust particles. J Pharmacol Exp Ther. 1999;290(2):524-529. [PubMed] [Google Scholar]
- 29.Kinne RW, Bräuer R, Stuhlmüller B, Palombo-Kinne E, Burmester GR. Macrophages in rheumatoid arthritis. Arthritis Res. 2000;2(3):189-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soler Palacios B, Estrada-Capetillo L, Izquierdo E, et al. . Macrophages from the synovium of active rheumatoid arthritis exhibit an activin A-dependent pro-inflammatory profile. J Pathol. 2015;235(3):515-526. [DOI] [PubMed] [Google Scholar]
- 31.Ou Y, Li W, Li X, Lin Z, Li M. Sinomenine reduces invasion and migration ability in fibroblast-like synoviocytes cells co-cultured with activated human monocytic THP-1 cells by inhibiting the expression of MMP-2, MMP-9, CD147. Rheumatol Int. 2011;31(11):1479-1485. [DOI] [PubMed] [Google Scholar]
- 32.Zhu P, Ding J, Zhou J, Dong WJ, Fan CM, Chen ZN. Expression of CD147 on monocytes/macrophages in rheumatoid arthritis: its potential role in monocyte accumulation and matrix metalloproteinase production. Arthritis Res Ther. 2005;7(5):R1023-R1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cook AD, Turner AL, Braine EL, Pobjoy J, Lenzo JC, Hamilton JA. Regulation of systemic and local myeloid cell subpopulations by bone marrow cell-derived granulocyte-macrophage colony-stimulating factor in experimental inflammatory arthritis. Arthritis Rheum. 2011;63(8):2340-2351. [DOI] [PubMed] [Google Scholar]
- 34.Eyles JL, Hickey MJ, Norman MU, et al. . A key role for G-CSF-induced neutrophil production and trafficking during inflammatory arthritis. Blood. 2008;112(13):5193-5201. [DOI] [PubMed] [Google Scholar]
- 35.Krausz S, Garcia S, Ambarus CA, et al. . Angiopoietin-2 promotes inflammatory activation of human macrophages and is essential for murine experimental arthritis. Ann Rheum Dis. 2012;71(8):1402-1410. [DOI] [PubMed] [Google Scholar]
- 36.Toh ML, Bonnefoy JY, Accart N, et al. . Bone- and cartilage-protective effects of a monoclonal antibody against colony-stimulating factor 1 receptor in experimental arthritis. Arthritis Rheumatol. 2014;66(11):2989-3000. [DOI] [PubMed] [Google Scholar]
- 37.Williams RO, Feldmann M, Maini RN. Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc Natl Acad Sci USA. 1992;89(20):9784-9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Williams RO, Ghrayeb J, Feldmann M, Maini RN. Successful therapy of collagen-induced arthritis with TNF receptor-IgG fusion protein and combination with anti-CD4. Immunology. 1995;84(3):433-439. [PMC free article] [PubMed] [Google Scholar]
- 39.Alonzi T, Fattori E, Lazzaro D, et al. . Interleukin 6 is required for the development of collagen-induced arthritis. J Exp Med. 1998;187(4):461-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma Y, Thornton S, Boivin GP, Hirsh D, Hirsch R, Hirsch E. Altered susceptibility to collagen-induced arthritis in transgenic mice with aberrant expression of interleukin-1 receptor antagonist. Arthritis Rheum. 1998;41(10):1798-1805. [DOI] [PubMed] [Google Scholar]
- 41.Li J, Hsu HC, Yang P, et al. . Treatment of arthritis by macrophage depletion and immunomodulation: testing an apoptosis-mediated therapy in a humanized death receptor mouse model. Arthritis Rheum. 2012;64(4):1098-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barrera P, Blom A, van Lent PL, et al. . Synovial macrophage depletion with clodronate-containing liposomes in rheumatoid arthritis. Arthritis Rheum. 2000;43(9):1951-1959. [DOI] [PubMed] [Google Scholar]
- 43.Patel SK, Janjic JM. Macrophage targeted theranostics as personalized nanomedicine strategies for inflammatory diseases. Theranostics. 2015;5(2):150-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Al-Atrash G, Shetty S, Idell S, et al. . IL-2-mediated upregulation of uPA and uPAR in natural killer cells. Biochem Biophys Res Commun. 2002;292(1):184-189. [DOI] [PubMed] [Google Scholar]
- 45.Chen W, Jin WQ, Chen LF, Williams T, Zhu WL, Fang Q. Urokinase receptor surface expression regulates monocyte migration and is associated with accelerated atherosclerosis. Int J Cardiol. 2012;161(2):103-110. [DOI] [PubMed] [Google Scholar]
- 46.Sonoda KH, Nakamura T, Young HA, Hart D, Carmeliet P, Stein-Streilein J. NKT cell-derived urokinase-type plasminogen activator promotes peripheral tolerance associated with eye. J Immunol. 2007;179(4):2215-2222. [DOI] [PubMed] [Google Scholar]
- 47.Sitrin RG, Pan PM, Harper HA, Todd RF III, Harsh DM, Blackwood RA. Clustering of urokinase receptors (uPAR; CD87) induces proinflammatory signaling in human polymorphonuclear neutrophils. J Immunol. 2000;165(6):3341-3349. [DOI] [PubMed] [Google Scholar]
- 48.Söderström K, Stein E, Colmenero P, et al. . Natural killer cells trigger osteoclastogenesis and bone destruction in arthritis. Proc Natl Acad Sci USA. 2010;107(29):13028-13033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blazek K, Eames HL, Weiss M, et al. . IFN-λ resolves inflammation via suppression of neutrophil infiltration and IL-1β production. J Exp Med. 2015;212(6):845-853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chiba A, Mizuno M, Tomi C, et al. . A 4-trifluoromethyl analogue of celecoxib inhibits arthritis by suppressing innate immune cell activation. Arthritis Res Ther. 2012;14(1):R9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu X, Xiao L, Huo R, et al. . Cyr61 is involved in neutrophil infiltration in joints by inducing IL-8 production by fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res Ther. 2013;15(6):R187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Farris SD, Hu JH, Krishnan R, et al. . Mechanisms of urokinase plasminogen activator (uPA)-mediated atherosclerosis: role of the uPA receptor and S100A8/A9 proteins. J Biol Chem. 2011;286(25):22665-22677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fleetwood AJ, Achuthan A, Schultz H, et al. . Urokinase plasminogen activator is a central regulator of macrophage three-dimensional invasion, matrix degradation, and adhesion. J Immunol. 2014;192(8):3540-3547. [DOI] [PubMed] [Google Scholar]
- 54.Sisson TH, Nguyen MH, Yu B, Novak ML, Simon RH, Koh TJ. Urokinase-type plasminogen activator increases hepatocyte growth factor activity required for skeletal muscle regeneration. Blood. 2009;114(24):5052-5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nguyen DH, Catling AD, Webb DJ, et al. . Myosin light chain kinase functions downstream of Ras/ERK to promote migration of urokinase-type plasminogen activator-stimulated cells in an integrin-selective manner. J Cell Biol. 1999;146(1):149-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Webb DJ, Thomas KS, Gonias SL. Plasminogen activator inhibitor 1 functions as a urokinase response modifier at the level of cell signaling and thereby promotes MCF-7 cell growth. J Cell Biol. 2001;152(4):741-752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jo M, Eastman BM, Webb DL, Stoletov K, Klemke R, Gonias SL. Cell signaling by urokinase-type plasminogen activator receptor induces stem cell-like properties in breast cancer cells. Cancer Res. 2010;70(21):8948-8958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Andersson C, Vasan RS. Compiling the complement of genes implicated in coronary artery disease. Circ Cardiovasc Genet. 2014;7(6):738-740. [DOI] [PubMed] [Google Scholar]
- 59.Wang Y, Kristan J, Hao L, Lenkoski CS, Shen Y, Matis LA. A role for complement in antibody-mediated inflammation: C5-deficient DBA/1 mice are resistant to collagen-induced arthritis. J Immunol. 2000;164(8):4340-4347. [DOI] [PubMed] [Google Scholar]
- 60.Yang XS, Liu MY, Zhang HM, Xue BZ, Shi H, Liu DX. Protein kinase C-δ mediates sepsis-induced activation of complement 5a and urokinase-type plasminogen activator signaling in macrophages. Inflamm Res. 2014;63(7):581-589. [DOI] [PubMed] [Google Scholar]
- 61.Kanno Y, Ishisaki A, Kawashita E, Kuretake H, Ikeda K, Matsuo O. uPA Attenuated LPS-induced Inflammatory Osteoclastogenesis through the Plasmin/PAR-1/Ca(2+)/CaMKK/AMPK Axis. Int J Biol Sci. 2016;12(1):63-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kalbasi Anaraki P, Patecki M, Tkachuk S, Kiyan Y, Haller H, Dumler I. Urokinase receptor mediates osteoclastogenesis via M-CSF release from osteoblasts and the c-Fms/PI3K/Akt/NF-κB pathway in osteoclasts. J Bone Miner Res. 2015;30(2):379-388. [DOI] [PubMed] [Google Scholar]