

Abstract
Hematopoietic stem cells (HSCs) produce most cellular energy through glycolysis rather than through mitochondrial respiration. Consistent with this notion, mitochondrial mass has been reported to be low in HSCs. However, we found that staining with mitotracker green, a commonly used dye to measure mitochondrial content, leads to artifactually low fluorescence specifically in HSCs because of dye efflux. Using mtDNA quantification, enumeration of mitochondrial nucleoids and fluorescence intensity of a genetically encoded mitochondrial reporter we unequivocally show here that HSCs and multipotential progenitors (MPPs) have higher mitochondrial mass than lineage-committed progenitors and mature cells. Despite similar mitochondrial mass, respiratory capacity of MPPs exceeds that of HSCs. Furthermore, although elevated mitophagy has been invoked to explain low mitochondrial mass in HSCs, we observed that mitochondrial turnover capacity is comparatively low in HSCs. We propose that the role of mitochondria in HSC biology may have to be revisited in light of these findings.
Graphical abstract
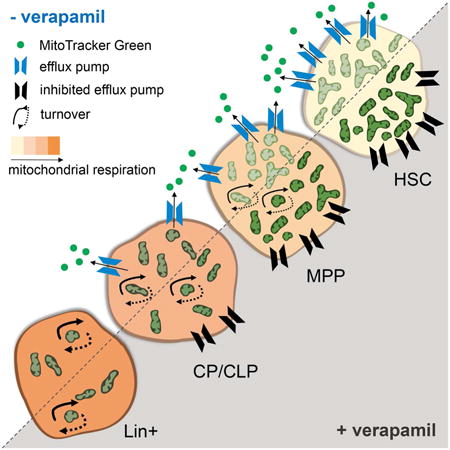
Introduction
It is currently not understood how the functional features of hematopoietic stem cells (HSCs), which can self renew and generate all lineages of the hematopoietic system, are coordinately regulated (Rossi et al., 2012). A particular gap in our understanding is the organellar cell biology of HSCs, particularly the role and function of the mitochondrion. In contrast to most mature cells, HSCs rely predominantly on glycolytic ATP production (Ito and Suda, 2014; Shyh-Chang et al., 2013; Simsek et al., 2010; Takubo et al., 2013). While shown to be important for HSC maintenance (Anso et al., 2017; Bejarano-Garcia et al., 2016; Guitart et al., 2017), some experimental data suggest that mitochondrial respiration may indeed be more dispensable for HSCs than for progenitors (Norddahl et al., 2011; Yu et al., 2013). Consistent with their reduced mitochondrial respiration, HSCs have been reported to be endowed with low mitochondrial mass based on staining with mitochondrial dyes (Mantel et al., 2012; Mohrin et al., 2015; Romero-Moya et al., 2013; Simsek et al., 2010; Takubo et al., 2013; Vannini et al., 2016; Xiao et al., 2012). It has furthermore been suggested that elimination through mitophagy explains low mitochondrial mass in HSCs and plays a role in their maintenance (Ho et al., 2017; Ito et al., 2016; Vannini et al., 2016). However, elevated mitophagy leading to low mitochondrial mass implies that mitophagy is not balanced by biogenesis. This dynamic would ultimately lead to depletion of mitochondria, a condition only known to occur in erythrocyte precursors (Chen et al., 2008; Sandoval et al., 2008). We therefore revisited the ideas of low mitochondrial mass and elevated mitophagy in HSCs.
Results
Most published data on mitochondrial mass in HSCs rely on flow cytometric analysis after staining with Mitotracker dyes (Ho et al., 2017; Ito et al., 2016; Mantel et al., 2012; Mohrin et al., 2015; Romero-Moya et al., 2013; Simsek et al., 2010; Takubo et al., 2013; Vannini et al., 2016; Xiao et al., 2012). We confirmed that after staining with Mitotracker Green (MTG), HSCs were MTGlo compared to multipotential progenitors (MPPs), committed progenitors (CPs, comprising myeloid, erythroid and megakaryocyte progenitors), common lymphoid progenitors (CLPs) and lineage positive (Lin+) cells (Figure 1a, analysis gates in Figure S1a). However, comparative DNA qPCR for select mitochondrial and nuclear genes in the top and bottom 10% of BM cells in terms of MTG fluorescence intensity showed similar mtDNA content in both fractions (Figure 1b), suggesting that MTG fluorescence does not reflect mitochondrial mass.
Figure 1. High mitochondrial mass in HSCs.
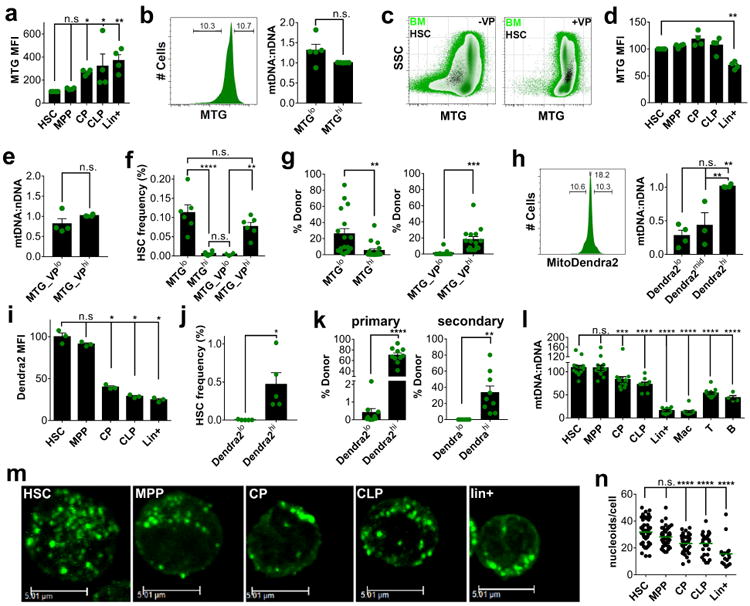
a. MTG MFI of mouse BM hematopoietic populations normalized to HSCs (mean ± s.e.m., n=4). b. Fluorescence histogram (left) of mouse BM stained with MTG and relative mtDNA:nDNA ratio (right) within the 10% MTGlo and MTGhi fractions normalized to MTGhi cells (mean ± s.e.m., n=5). c. Flow cytometric profile of mouse BM (green) and HSCs (black) stained with MTG in the absence (left) or presence (right) of VP. d. MTG MFI of mouse BM hematopoietic populations in the presence of VP normalized to HSCs (mean ± s.e.m., n=4). e. Relative mtDNA:nDNA ratio within the 10% MTG_VPlo and MTG_VPhi BM fractions stained in the presence of VP, normalized to MTGhi (mean ± s.e.m., n=4). f. HSC frequency within the 10% MTGlo and MTGhi mouse BM fractions stained with MTG in the absence or presence of VP (mean ± s.e.m., n=6). g. Donor contribution of BM MTGlo and MTGhi fractions stained in the absence (left) or presence (right) of VP in the PB of recipients 16 weeks after competitive transplantation (mean ± s.e.m., n>15 recipients pooled from three independent transplants). h. Fluorescence histogram of BM (left) from mitoDendra2 mice and relative mtDNA:nDNA ratio (right) within the 10% Dendra2lo, Dendra2mid and Dendra2hi fractions normalized to Dendra2hi (mean ± s.e.m., n>3). i. Dendra2 MFI of BM populations of mitoDendra2 mice normalized to HSCs (mean ± s.e.m., n=3). j. HSC frequency within the 10% Dendra2lo and Dendra2hi BM fractions of mitoDendra2 mice (mean ± s.e.m., n=5). k. Donor contribution of BM Dendra2lo and Dendra2hi fractions in the PB of recipients 16 weeks after competitive transplantation (left) or serial transplantation (right) (mean ± s.e.m, n≥9 recipients pooled from two independent transplants). l. Relative mtDNA:nDNA ratio in mouse BM hematopoietic populations (mean ± s.e.m., n≥4) normalized to HSCs. m. Representative images of mitochondrial nucleoids visualized with immunostaining for Tfam in mouse BM hematopoietic populations (scale bar, 5.01μM). n. Quantification of mitochondrial nucleoids (mean, n≥17 cells pooled from three independent experiments, scale bar 5.01μm). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, n.s.- not significant. See also Figures S1 and S2.
HSCs possess xenobiotic efflux pumps that might extrude MTG. We confirmed selective expression in HSCs of Bcrp1 and Mdr1a/b, pumps that are blocked by verapamil (VP) (Goodell et al., 1997; Schinkel et al., 1997; Sorrentino et al., 1995; Zhou et al., 2002; Zhou et al., 2001) (Figure S2a). In the presence of VP, MTG staining increased in all hematopoietic populations tested (see Figure S1a for analysis gates), except for Lin+ cells (Figures 1c and 1d). Consistent with the expression pattern of efflux pumps, this increase was most pronounced in HSCs, and correlated inversely with differentiation stage (Figures 2b and 2c). Although within a select and more homogenous population such as CPs brighter MTG fluorescence was associated with higher mtDNA content (Figure S2d), in total BM stained in the presence of VP MTGhi and MTGlo cells had similar mtDNA content (Figure 1e), indicating that even in the presence of VP MTG fluorescence intensity does not reliably report mitochondrial mass. Previous reports suggested that MTG fluorescence is not only dependent on mitochondrial mass, but also on mitochondrial function and membrane potential (Keij et al., 2000). Nevertheless, phenotypically defined HSCs were highly enriched in the MTGhi population and depleted in the MTGlo population after staining BM cells with MTG in the presence of VP, whereas the opposite was observed after staining in its absence (Figure 1f). To functionally verify these findings, we performed competitive transplantation experiments. After staining without VP, most repopulation capacity was found in the MTGlo fraction (Figure 1g left panel, Figure S1b). In contrast, after staining in the presence of VP, virtually all repopulation capacity was present in MTGhi fraction (Figure 1g right panel, Figure S1b). We next assessed MTG staining in human cord blood. Similar to the mouse, enriched HSCs (lin-CD34+CD38-CD45RA-CD90+, Figure S1c for gates) (Doulatov et al., 2012) were MTGlo in the absence of VP, whereas MTG fluorescence increased after staining in its presence (Figures S2e and S2f). At variance with the mouse, MTG staining in the absence of VP was equally dim in enriched MPPs and myeloid progenitors (MPs) and the effect of VP on MTG staining was as pronounced in MPs as it was in MPPs and HSCs (Figure S2f). Taken together, our observations show that MTG staining is not informative for mitochondrial mass in HSCs.
Figure 2. Metabolism and mitochondrial turnover.
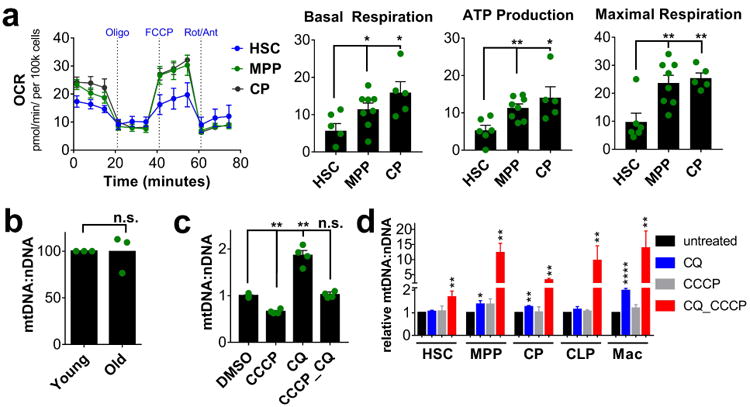
a. Real time analysis (left) of mitochondrial oxygen consumption (OCR) during mitochondrial stress test and quantitative analysis (right) of basal respiration, ATP production and maximal respiration in HSCs (LSKCD48-), MPPs (LSKCD48+) and CPs (mean ± s.e.m., n=5). b. Relative mtDNA:nDNA ratio in HSCs from young and old mice normalized to young (mean ± s.e.m., n=3) c. Effect of mitophagy inducer CCCP and autophagy inhibitor CQ on the relative mtDNA:nDNA ratio of 3T3 and d. hematopoietic populations normalized to untreated (mean ± s.e.m., n˃3). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, n.s.- not significant.
To more rigorously assess mitochondrial mass, we first examined fluorescence intensity in mice that express a mitochondrially targeted Dendra2 (Pham et al., 2012). MitoDendra2hi (top 10% of MFI) cells contained approximately threefold more mtDNA than mitoDendra2lo cells (bottom 10% of MFI), while mitoDendra2mid cells (middle 10% of MFI) had intermediate mtDNA content (Figure 1h). MitoDendra2 fluorescence therefore reports mitochondrial mass. HSCs and MPPs had the highest MFI and fluorescence decreased in more differentiated populations (Figures 1i and S2g). Phenotypically defined HSCs show nearly absolute enrichment in the mitoDendra2hi (top 10% of MFI) population (Figure 1j). Competitive transplantation and serial transplantation studies (Figure 1k) confirmed that almost all long-term repopulating activity was in the mitoDendra2hi fraction. These data indicate high mitochondrial mass in HSCs.
Measurement of mtDNA content was consistent with these observations. mtDNA content was the highest in HSCs and MPPs and declined during differentiation (Figure 1l). To further confirm the decline in mtDNA with differentiation, we cultured purified HSCs in the presence of KL, TPO and IL6, conditions where HSCs invariably differentiate into predominantly myeloid cells. mtDNA content decreased progressively over 7 days of culture (Figure S2h), supporting the notion that mtDNA content declines during hematopoietic differentiation. In human cord blood, mtDNA content was higher in progenitors and HSCs than in mature cells (Figure S2i). However, human HSCs and progenitors had similar mtDNA content, in contrast to their murine counterparts. Nevertheless, the data show that also in the human, HSCs are not characterized by low mitochondrial mass, despite previous reports to the contrary based on MTG staining (Romero-Moya et al., 2013).
In a third approach we stained with antibodies against Tfam, the mtDNA-binding protein that compacts the organelle genome into punctiform structures termed nucleoids (Figure 1m). The number of nucleoids per cell was highest in HSCs, and declined throughout differentiation (Figures 1m and 1n), paralleling mitoDendra2 fluorescence and mtDNA data. Collectively, these observations demonstrate that, in contrast to what is currently accepted in the field, mitochondrial mass is comparatively high in HSCs.
If ATP production were the prime goal of maintaining elevated mitochondrial mass in HSCs, then respiratory capacity should be similar in HSCs and MPPs, as both populations have comparable mitochondrial mass. We examined mitochondrial function using Seahorse in enriched HSCs, as defined by the LSKCD48- phenotype, MPPs (LSKCD48+) and CPs (Lin-Sca1-kit+). Baseline oxygen consumption, mitochondrial ATP production and maximal respiratory capacity, as measured after addition of the uncoupler, FCCP, were significantly higher in MPPs and CPs than in enriched HSCs (Figure 2a). The elevated mitochondrial mass in HSCs is therefore not used for respiration or for maintaining high respiratory capacity.
Next, we examined mitochondrial turnover. HSC mitochondrial mass is stable over the lifespan of the mouse (Figure 2b), thus mitophagy must be balanced by biogenesis to avoid depletion of mitochondria. We incubated cells with Chloroquine (CQ) and CCCP. CQ inhibits lysosomal acidification, blocks autophagic flux and mitophagy and increases mitochondrial mass if balanced mitophagy and biogenesis are ongoing (Pickrell and Youle, 2015). CCCP induces maximal mitochondrial depolarization and in doing so induces mitophagy, resulting in a decline in mitochondrial mass (Pickrell and Youle, 2015). In 3T3 cells, CQ increased mtDNA, indicative of mitophagy balanced by biogenesis, while CCCP reduced mtDNA content, as expected (Figure 2c). Combining CQ and CCCP brought mtDNA back to control levels, showing that additional mitophagy induced by CCCP is blocked by CQ (Figure 2c).
Hematopoietic cells behaved very differently however (Figure 2d). CQ did not significantly affect mtDNA content in HSCs, but increased mtDNA content in progenitors and mature cells, except for CLPs. Progenitors, with the exception of CLPs, therefore undergo steady-state mitochondrial turnover, while HSCs do not detectably do so. Surprisingly, CCCP alone did not change mtDNA content in any population. However, in the presence of CCCP and CQ a strong increase (5 to 20-fold) in mtDNA content was observed in all populations, except for HSCs where this increase was much less pronounced (1.7-fold), indicating a compensatory increase in biogenesis in response to CCCP. Taken together, these data suggest extensive mitochondrial turnover and high mitophagic and biogenetic capacity in MPPs, while turnover capacity and mitophagy are low in HSCs. These findings argue against a role for mitophagy in reducing mitochondrial mass in HSCs,.
Discussion
Using three complementary approaches we show that, in contrast to what is currently accepted in the field, mitochondrial mass is high in HSCs and MPPs. The differentiation-associated decline in mitochondrial mass in human stem and progenitor cells appeared less pronounced than in the mouse, which may be due to biological differences or the less well defined nature of progenitor populations in human compared to mouse (Doulatov et al., 2012). Nevertheless, in the human model too, the highest mitochondrial mass was observed in the most primitive hematopoietic cells, including enriched HSCs.
These findings raise the question why HSCs are endowed with a high mitochondrial mass and which mechanism underlies this elevated mitochondrial mass. Active mitophagy to maintain presumed low mitochondrial mass in HSCs, as has been proposed (Ho et al., 2017; Ito et al., 2016; Vannini et al., 2016), would ultimately lead to depletion of mitochondria, which does not occur. HSCs therefore have a high set point for mitochondrial mass and low turnover. As respiratory capacity is low in HSCs, while extensive remodeling appears to take place in MPPs, is unlikely that mitochondria in HSCs are paused but poised to rapidly ramp up respiration upon differentiation. Mitochondria are also required for several biosynthetic pathways and intermediary metabolism (Vander Heiden et al., 2009), apoptosis (Youle and van der Bliek, 2012) and intracellular calcium homeostasis (Rizzuto et al., 2012). We showed previously that the mitochondrial fusion protein, Mitofusin 2 (Mfn2), is required for the maintenance of HSCs with extensive lymphoid potential, and that this effect was mediated through enhanced buffering of intracellular calcium by mitochondria (Luchsinger et al., 2016). Though recent publications do indicate an important role for respiration in HSC maintenance as well (Anso et al., 2017; Bejarano-Garcia et al., 2016; Guitart et al., 2017), these findings suggest that mitochondria perform specific and essential roles in HSCs, including but likely not limited to calcium buffering, that may not be directly dependent on ATP production. We therefore suggest that the role of mitochondria in the biology of HSCs may need to be revisited.
Contact For Reagent And Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lead contact Hans Snoeck (hs2680@cumc.columbia.edu).
Experimental Model And Subject Details
Animals
C57BL/6J mice (CD45.2) and B6.SJL-PtprcaPep3b/BoyJ (CD45.1) were purchased from The Jackson Laboratory (West Grove, PA, USA). Mice with ubiquitous expression of the MitoDendra2 reporter were obtained by crossing conditional Mito-Dendra2 transgenic (Pham) mice (B6;129S-Gt(ROSA)26Sortm1(CAG-COX8A/Dendra2)Dcc/J) with E2A-Cre mice (B6.FVB-Tg(EIIa-cre)C5379Lmgd/J), both purchased from Jackson Laboratory (Luchsinger et al., 2016). Animals were housed in a specific pathogen-free facility. Experiments and animal care were performed in accordance with the Columbia University Institutional Animal Care and Use Committee. All mice were used at age 8–14 weeks and both genders were used for experiments with the exception of old mice that were used at age 2 years.
Human cord blood
De-identified cord blood was obtained from the New York Blood Center and the experiments were performed in accordance with protocol approved by Columbia University Institutional Review Board protocol number AAAR0324.
Cells
NIH-3T3 cells were purchased from ATCC (Manassas, VA, USA) and cultured in 10% FCS/DMEM at 37°C. Hematopoietic populations were isolated from mouse bone marrow and sorted directly into complete media using StemPro34 (Invitrogen, Carlsbad, CA, USA), 100 ng ml-1 SCF, 100 ng ml-1 TPO, 50 ng ml-1 IL-6 (Peptrotech, NJ, USA). Culture of hematopoietic populations was carried out in complete media in 5% O2 at 37°C. When indicated, cells were cultured for 24h in the presence of Chloroquine (50uM, Sigma, St. Louis, MO, USA) or/and 10uM CCCP (10uM, Sigma).
Method Details
Cell Isolation and Preparation
Bone marrow isolation was prepared by crushing the tibia, femur, and pelvis of each mice in 1×PBS. The cell suspension was centrifuged at 1200rpm for 5min at 4°C and the pellet was re-suspended in 1×ACK lysis buffer (Gibco, Carlsbad, CA, USA). After washing in 1×PBS (Corning, NY, USA) the cells were filtered through a 40μm cell strainer, centrifuged, and the pellet re-suspended in buffer only or staining mix. Peripheral blood was collected by submandibular vein puncture with a sterile disposable lancet. Red blood cells were lysed two times with 1×ACK lysis buffer, washed in 1×PBS, and centrifuged at 1200rpm for 5min at 4°C. The pellet was re-suspended in antibody mix. De-identified CB units were diluted at a 1:1 ratio with Iscove's MEM (IMEM, Gibco) and layered in a 2:1 ratio on Histopaq-1077 (Sigma). Samples were centrifuged at 400g for 30min at 25°C without breaking. Mononuclear cells were collected at the plasma:Histopaq interface and washed in IMEM followed by red blood cell lysis with 1×ACK lysis buffer. After washing in 1×PBS, cells were centrifuged at 1200rpm for 5min at 4°C and the pellet re-suspended in staining mix, See Key Resources Table for antibody information.
Flow Cytometry and Cell Sorting
Antibody cocktails were prepared in MACS buffer and incubated with cells for at least 30min at 4°C. For experiments with MitoTracker Green (MTG), cells were incubated with MTG (30nM, Invitrogen) in the presence or absence of verapamil (50μM, Sigma) at 37°C for 20-30min, washed in 1×PBS and centrifuged at 1200rpm for 5min at 4°C. For flow cytometric cell sorting of bone marrow fractions based on MTG and Dendra2 fluorescence, cells were re-suspended in buffer only and top and bottom 10-15% cells of the fluorescence distribution were isolated. For sorting of specific populations, pellet was re-suspended in antibody mix and cells were sorted directly into complete media. Flow cytometry and cell sorting were performed on a LSR II flow cytometer (Becton Dickinson, Mountain View, CA, USA) and on an Influx cell sorter (Becton Dickinson), respectively. Data were analyzed using FlowJo 9.9.3 (TreeStar, Ashland, OR, USA). See Key Resources Table for information on Antibodies and Figure S1 for gating strategies.
Bone marrow transplantation
5×104 donor bone marrow cells sorted based on MTG (CD45.1) or Dendra2 (CD45.2) fluorescence were transplanted by tail vein injection into lethally irradiated (two doses of 478 cGy over 3h using a Rad Source RS-2000 X-ray irradiator (Brentwood, TN, USA) CD45.2 recipients together with 20×104 freshly isolated CD45.1+CD45.2+ F1 (MTG transplants) or CD45.2 (Dendra2 transplants) competitor bone marrow cells. For secondary transplantations 50×104 freshly isolated bone marrow cells from Dendra2 primary recipients were injected into lethally irradiated CD45.2 secondary recipients. Cells were counted manually using an hemocytometer and viability was assessed by trypan blue staining. For two weeks after injection recipient mice were given antibiotics via drinking water. At 8 and 16 weeks post-transplantation, peripheral blood of recipients was analyzed for donor chimerism.
Mitochondrial DNA quantification
Sorted or cultured hematopoietic populations were washed with 1× PBS, centrifuged at 1200rpm for 5min at 4°C and re-suspended in 70μl lysis buffer (10mM Tris-HCl pH7.5, 50mM NaCl, 6.25mM, 0.045% NP40, 0.45% Tween20) with freshly added proteinase K (1mg/ml) followed by incubation for 2h at 56°C and 15min at 95°C in a PCR machine (Bio-Rad, Philadelphia, PA, USA, van der Burg et al., 2011). Quantification was performed by qPCR with SYBR green-based detection (Thermo Fisher Scientific, Boston, MA, USA) using 5μL of the lysate as input in each reaction. Relative mtDNA:nDNA ratio was calculated using the ΔΔCt method upon targeting of nuclear-encoded genes (mouse actB_Fwd: 5′CGGCTTGCGGGTGTTAAAAG3′, mouse actB_Rev: 5′CGTGATCGTAGCGTCTGGTT3′, human B2M_Fwd: 5′TGCTGTCTCCATGTTTGATGTATCT3′, human B2M_Rev: 5′TCTCTGCTCCCCACCTCTAAGT3′) and mitochondrial-encoded genes (mouse cytB_Fwd: 5′CTTCATGTCGGACGAGGCTTA3′, mouse cytB_Rev: 5′TGTGGCTATGACTGCGAACA3′, human tRNALeu_Fwd: 5′CACCCAAGAACAGGGTTTGT3′, human tRNALeu_Rev: 5′TGGCCATGGGTATGTTGTTA3′).
Immunofluorescence
Sorted hematopoietic populations (2-5×103 cells) were collected in complete media and plated on MicroWell 384-well glass-bottom plates (Thermo) coated with 1μg/mL poly-D-lysine (Sigma) overnight in a humidified chamber. Cells were spun at 30g for 5min and fixed with 4%PFA for 10min at room temperature. Cells were then permeabilized with 0.1% TritonX-100/PBS for 10min and blocked with 1% BSA/0.1% TritonX-100/PBS for 1h at room temperature. Cells were incubated in 50μL of anti-TFAM (Abcam, Cambridge, UK) 1:200 rabbit primary antibody in blocking solution overnight at 4°C in a humidified chamber, washed three times with 1×PBS and incubated with anti-rabbit 488 AlexaFluor secondary antibody (Invitrogen) 1:500 for 1h in blocking solution at room temperature. After washing three times with 1×PBS cells were mounted with fluorescent mounting media (Vector Labs, Burlingame, CA, USA). Confocal images were acquired with a Leica SP8 mutli-photon confocal microscope. For nucleoid quantification confocal z-stacks were projected as a z-project, and the number of TFAM punctae tracked with the cell counter plugin in ImageJ (NIH, Bethesda, MD, USA).
Quantitative RT-PCR
Sorted cell populations (5×103 cells) in 0.25mL of complete media were added to 0.75mL of Trizol LS Reagent (Invitrogen) and RNA was isolated according to the manufacturer's instructions. RNA was dissolved in 0.012mL of water and used to synthesize cDNA using Superscript III Reverse Transcriptase (Invitrogen). Target Ct values were determined using inventoried Taqman probes (Applied Biosystems Carlsbad, CA, USA) for bcrp1 (Mm00496364_m1) mdr1a (Mm00440761_m1) and mdr1b (Mm00440736_m1). Relative quantification was calculated using the ΔΔCt method normalized to actin (Mm00496364_m1) and HSC.
Seahorse
For Seahorse metabolic assays, DMEM media was neutralized to pH 7.4 at 37 °C prior to the experiments. XFp flux cartridges were hydrated in XF Calibrant overnight at 37 °C. For all assays, HSCs (LSK CD48-) MPPs (LSK CD48+) and CPs (Lin-Sca1-kit+) were isolated from 6 mice and ≥50,000 cells plated onto one well of a Seahorse XFp culture plate coated overnight with Cell-Tak reagent in a humidified chamber. Cells were immobilized by centrifugation at 200×g for 3min at room temperature and washed twice with DMEM. Cells were equilibrated in a humidified non-CO2 incubator until the start of the assay. Flux cartridges were loaded with drugs prepared with DMEM according to manufacturer's instructions. Oxygen consumption rate values were obtained using the XFp Mito Stress Kit. Metabolic parameters were derived from calculations based on manufacturer's instructions. Due to feasibility of cell number isolation, experiments are represented as one or two technical replicates per cell type, per condition over 5 independent experiments.
Quantification And Statistical Analysis
For statistical comparison of two groups, unpaired two-tailed Student's t test was used. When more than two groups were compared, one-way ANOVA followed by multiple comparisons test was performed using Prism version 7.00 for Windows (GraphPad, La Jolla, CA, USA). Due to variability in the data, statistical comparison of untreated and CQ_CCCP was performed without assumption of normal distribution using Mann-Whitney test. Differences among group means were considered significant when the probability value, p, was less than 0.05. Sample size (n) represents biological replicates. No statistical methods were used to predetermine sample size. The experiments were not randomized, and the investigators were not blinded to allocation during experiments and outcome assessment.
Data and Software Availability
Data have been deposited on Mendeley Data DOI:10.17632/snbcc8bh78.1
Supplementary Material
Supplementary Figure 1. Sort and analysis gates, related to Figure 1 and 2.
Supplementary Figure 2. Mitochondrial mass in hematopoietic cells, related to Figure 1.
Acknowledgments
Supported by grants NIH R01CA167289 and NIH R01AG029626 (HWS). Flow cytometry was performed in the CCTI Flow Cytometry Core, supported in part by the Office of the Director, National Institutes of Health under awards S10RR027050 and S10OD020056. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Author Contributions: Conceptualization, Writing and Visualization, H.W.S. and M.J.A; Methodology, M.J.A and L.L.L.; Investigation, M.J.A, L.L.L., D.J.C. and L.J.W, Supervision and Funding Acquisition, H.W.S.
The authors have no conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anso E, Weinberg SE, Diebold LP, Thompson BJ, Malinge S, Schumacker PT, Liu X, Zhang Y, Shao Z, Steadman M, et al. The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat Cell Biol. 2017;19:614–625. doi: 10.1038/ncb3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejarano-Garcia JA, Millan-Ucles A, Rosado IV, Sanchez-Abarca LI, Caballero-Velazquez T, Duran-Galvan MJ, Perez-Simon JA, Piruat JI. Sensitivity of hematopoietic stem cells to mitochondrial dysfunction by SdhD gene deletion. Cell Death Dis. 2016;7:e2516. doi: 10.1038/cddis.2016.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Sandoval H, Wang J. Selective mitochondrial autophagy during erythroid maturation. Autophagy. 2008;4:926–928. doi: 10.4161/auto.6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: a human perspective. Cell Stem Cell. 2012;10:120–136. doi: 10.1016/j.stem.2012.01.006. [DOI] [PubMed] [Google Scholar]
- Goodell MA, Rosenzweig M, Kim H, Marks DF, DeMaria M, Paradis G, Grupp SA, Sieff CA, Mulligan RC, Johnson RP. Dye efflux studies suggest that hematopoietic stem cells expressing low or undetectable levels of CD34 antigen exist in multiple species. Nat Med. 1997;3:1337–1345. doi: 10.1038/nm1297-1337. [DOI] [PubMed] [Google Scholar]
- Guitart AV, Panagopoulou TI, Villacreces A, Vukovic M, Sepulveda C, Allen L, Carter RN, van de Lagemaat LN, Morgan M, Giles P, et al. Fumarate hydratase is a critical metabolic regulator of hematopoietic stem cell functions. J Exp Med. 2017;214:719–735. doi: 10.1084/jem.20161087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, Figueroa ME, Passegue E. Autophagy maintains the metabolism and function of young and old stem cells. Nature. 2017;543:205–210. doi: 10.1038/nature21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nature reviews Mol Cell Biol. 2014;15:243–256. doi: 10.1038/nrm3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Turcotte R, Cui J, Zimmerman SE, Pinho S, Mizoguchi T, Arai F, Runnels JM, Alt C, Teruya-Feldstein J, et al. Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science. 2016;354:1156–1160. doi: 10.1126/science.aaf5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keij JF, Bell-Prince C, Steinkamp JA. Staining of mitochondrial membranes with 10-nonyl acridine orange, MitoFluor Green, and MitoTracker Green is affected by mitochondrial membrane potential altering drugs. Cytometry. 2000;39:203–210. doi: 10.1002/(sici)1097-0320(20000301)39:3<203::aid-cyto5>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Luchsinger LL, de Almeida MJ, Corrigan DJ, Mumau M, Snoeck HW. Mitofusin 2 maintains haematopoietic stem cells with extensive lymphoid potential. Nature. 2016;529:528–531. doi: 10.1038/nature16500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantel C, Messina-Graham S, Moh A, Cooper S, Hangoc G, Fu XY, Broxmeyer HE. Mouse hematopoietic cell-targeted STAT3 deletion: stem/progenitor cell defects, mitochondrial dysfunction, ROS overproduction, and a rapid aging-like phenotype. Blood. 2012;120:2589–2599. doi: 10.1182/blood-2012-01-404004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrin M, Shin J, Liu Y, Brown K, Luo H, Xi Y, Haynes CM, Chen D. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science. 2015;347:1374–1377. doi: 10.1126/science.aaa2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norddahl GL, Pronk CJ, Wahlestedt M, Sten G, Nygren JM, Ugale A, Sigvardsson M, Bryder D. Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging. Cell Stem Cell. 2011;8:499–510. doi: 10.1016/j.stem.2011.03.009. [DOI] [PubMed] [Google Scholar]
- Pham AH, McCaffery JM, Chan DC. Mouse lines with photo-activatable mitochondria to study mitochondrial dynamics. Genesis. 2012;50:833–843. doi: 10.1002/dvg.22050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nature reviews Mol Cell Biol. 2012;13:566–578. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- Romero-Moya D, Bueno C, Montes R, Navarro-Montero O, Iborra FJ, Lopez LC, Martin M, Menendez P. Cord blood-derived CD34+ hematopoietic cells with low mitochondrial mass are enriched in hematopoietic repopulating stem cell function. Haematologica. 2013;98:1022–1029. doi: 10.3324/haematol.2012.079244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi L, Lin KK, Boles NC, Yang L, King KY, Jeong M, Mayle A, Goodell MA. Less is more: unveiling the functional core of hematopoietic stem cells through knockout mice. Cell Stem Cell. 2012;11:302–317. doi: 10.1016/j.stem.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232–235. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinkel AH, Mayer U, Wagenaar E, Mol CA, van Deemter L, Smit JJ, van der Valk MA, Voordouw AC, Spits H, van Tellingen O, et al. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc Natl Acad Sci U S A. 1997;94:4028–4033. doi: 10.1073/pnas.94.8.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyh-Chang N, Daley GQ, Cantley LC. Stem cell metabolism in tissue development and aging. Development. 2013;140:2535–2547. doi: 10.1242/dev.091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, Schneider JW, Zhang CC, Sadek HA. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 2010;7:380–390. doi: 10.1016/j.stem.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino BP, McDonagh KT, Woods D, Orlic D. Expression of retroviral vectors containing the human multidrug resistance 1 cDNA in hematopoietic cells of transplanted mice. Blood. 1995;86:491–501. [PubMed] [Google Scholar]
- Takubo K, Nagamatsu G, Kobayashi CI, Nakamura-Ishizu A, Kobayashi H, Ikeda E, Goda N, Rahimi Y, Johnson RS, Soga T, et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell. 2013;12:49–61. doi: 10.1016/j.stem.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini N, Girotra M, Naveiras O, Nikitin G, Campos V, Giger S, Roch A, Auwerx J, Lutolf MP. Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nat Comm. 2016;7:13125. doi: 10.1038/ncomms13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao N, Jani K, Morgan K, Okabe R, Cullen DE, Jesneck JL, Raffel GD. Hematopoietic stem cells lacking Ott1 display aspects associated with aging and are unable to maintain quiescence during proliferative stress. Blood. 2012;119:4898–4907. doi: 10.1182/blood-2012-01-403089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu WM, Liu X, Shen J, Jovanovic O, Pohl EE, Gerson SL, Finkel T, Broxmeyer HE, Qu CK. Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell. 2013;12:62–74. doi: 10.1016/j.stem.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Morris JJ, Barnes Y, Lan L, Schuetz JD, Sorrentino BP. Bcrp1 gene expression is required for normal numbers of side population stem cells in mice, and confers relative protection to mitoxantrone in hematopoietic cells in vivo. Proc Natl Acad Sci U S A. 2002;99:12339–12344. doi: 10.1073/pnas.192276999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, Lagutina I, Grosveld GC, Osawa M, Nakauchi H, et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med. 2001;7:1028–1034. doi: 10.1038/nm0901-1028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Sort and analysis gates, related to Figure 1 and 2.
Supplementary Figure 2. Mitochondrial mass in hematopoietic cells, related to Figure 1.