

Summary
Although the intrinsic mechanisms that control whether stem cells divide symmetrically or asymmetrically underlie tissue growth and homeostasis, they remain poorly defined. We report that the RNA-binding protein, Fragile X Mental Retardation Protein (FMRP), limits the symmetric division, and resulting expansion, of the stem cell population during adaptive intestinal growth in Drosophila. The elevated insulin sensitivity that FMRP-deficient progenitor cells display contributes to their accelerated expansion, which is suppressed by depletion of insulin signaling components. This FMRP activity is mediated solely via a second, conserved RNA-binding protein, LIN-28, known to boost insulin signaling in stem cells. Via LIN-28, FMRP controls progenitor cell behavior by post-transcriptionally repressing the level of Insulin Receptor (InR). This study identifies the stem cell based mechanism by which FMRP controls tissue adaptation, and raises the possibility that defective adaptive growth underlies the accelerated growth, gastrointestinal, and other symptoms that affect Fragile X Syndrome patients.
eTOC Blurb
Luhur et al. report that FMRP acts via LIN-28 in progenitor cells to dampen the adaptive expansion of intestinal tissue in the fruit fly, raising the possibility that defective LIN28-mediated adaptive growth underlies some of the symptoms that affect Fragile X Syndrome patients.
Introduction
The behavior of adult stem cells underlies the homeostasis and growth of tissues (Klein and Simons, 2011, Simons and Clevers, 2011). In the adult mouse and Drosophila intestine, stem cell homeostasis is achieved via population asymmetry renewal, in which population expansion is matched by loss via differentiation (de Navascues et al., 2012, Lopez-Garcia et al., 2010). Tissue remodeling and adaptive response biases these stem cell population dynamics: after starvation, for example, intestinal stem cells (ISCs) are lost, but they amplify with nutrient availability (de Navascues et al., 2012, McLeod et al., 2010, O’Brien et al., 2011). This population behavior is a net result of individual ISC decisions either to divide asymmetrically, regenerating one ISC and one transient progenitor known as an enteroblast (EB), to symmetrically amplify, in which two ISCs are generated, or to symmetrically differentiate, in which the ISC is lost. While the mechanisms of asymmetric division are well understood, including the role of Notch-Delta signaling in ensuring asymmetric cell fate outcomes (Goulas et al., 2012, Micchelli and Perrimon, 2006, Ohlstein and Spradling, 2006), the intrinsic pathways controlling ISC division mode are, for the most part, unknown.
Despite the lack of clarity regarding intrinsic ISC mechanisms, insulin/IGF-like signaling (IIS), a well-known link connecting nutrient status to animal and organ size, has been implicated in controlling ISC division mode (Chen et al., 2015, Foronda et al., 2014, O’Brien et al., 2011). After feeding, insulin-like peptides (Ilps) are released locally from the visceral muscles surrounding the intestinal epithelium as well as systemically, leading to ISC expansion, presumably by activating the Insulin Receptor (InR) (O’Brien et al., 2011). Genetic depletion of InR blocks ISC division (Choi et al., 2011, O’Brien et al., 2011), however, confounding the ability to directly analyze the role of InR in stem cell division pattern but suggesting that careful adjustment of the insulin sensitivity of ISCs above the level needed for cell division could bias division mode. Consistent with this notion, there is evidence that in many systems, IIS components including InR are regulated post-transcriptionally (reviewed in Panda et al., 2013, and Luhur et al., 2013): in vitro studies in cultured Drosophila cells, for example, indicate that InR is the subject of an unusual form of such regulation that increases its levels during nutrient deprivation when most mRNA translation is blocked (Marr et al., 2007). Prior identification of the microRNA, miR-305, and the RNA-binding protein, LIN-28, as regulators of IIS in ISCs highlights the adult Drosophila intestinal epithelium as a powerful model to decipher how intrinsic post-transcriptional pathways control stem cell behavior (Chen et al., 2015, Foronda et al., 2014). Here, we report that a second, broadly conserved RNA binding protein, Fragile X Mental Retardation Protein (FMRP), functions via LIN-28 but not miR-305 to post-transcriptionally limit InR level within ISCs and, as a result, their expansion during adaptive growth.
Results and Discussion
FMRP and LIN-28 are enriched in intestinal progenitor cells
We previously found that LIN-28 localized to cytoplasmic granules in intestinal progenitor cells (Chen et al., 2015, Luhur and Sokol, 2016), suggesting that messenger ribonucleoprotein complexes (mRNPs) may play an important role in controlling ISC behavior. We therefore screened for other stress granule components expressed in intestinal progenitors and identified another RNA-binding protein, FMRP (Fig 1A). Functionally, FMRP represses mRNA translation while LIN-28 promotes it (Polesskaya et al., 2007, Shyh-Chang and Daley, 2013). Both FMRP and LIN-28 were enriched in intestinal progenitor cells though with differences: LIN-28 was present at approximately equal levels in all progenitor cells, while FMRP appeared to be slightly higher in EBs relative to ISCs (Fig 1A, B, S1A). In addition, while both LIN-28 and FMRP had punctate patterns, few punctae contained both LIN-28 and FMRP under normal conditions. However, stress-inducing arsenite treatment of intestines or cultured embryonic cells caused extensive LIN-28 and FMRP co-localization (Fig S1B, C). Thus, LIN-28- and FMRP-granules were dynamic and responsive to environmental perturbations.
Figure 1. fmr1 limits the expansion of the intestinal progenitor cell population.
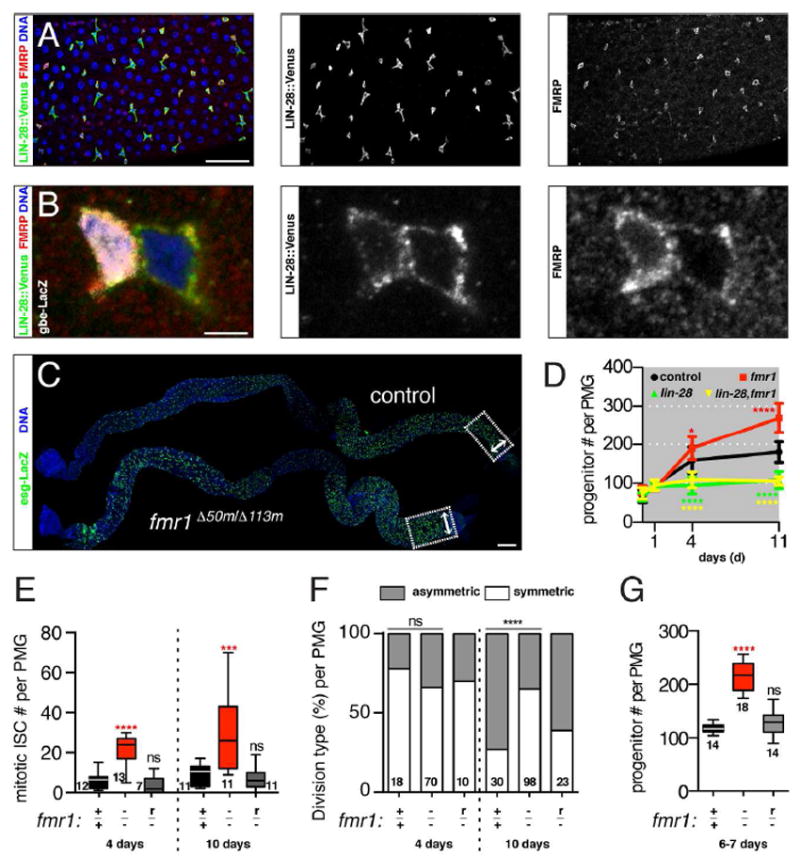
(A) Intestines stained for LIN-28∷Venus (green), FMRP (red), and DNA (blue). Scale bar: 50 μm. (B) Intestinal progenitors stained for EB reporter gbe-LacZ (white), LIN-28∷Venus (green), FMRP (red), and DNA (blue). Scale bar: 3 μm. (C) Control and fmr1Δ50m/Δ113m mutant intestines stained for progenitor marker esg-LacZ (green) and DNA (blue). Areas used to quantify gut width (arrowheads) and progenitor number (boxes) are shown. Scale bar: 100 μm. (D) Line plots of progenitor numbers in the posterior midguts of control, fmr1Δ50m/Δ113m mutant, lin-28Δ1 mutant or lin-28Δ1, fmr1Δ50m/Δ113m double mutant at eclosion as well as 1, 4 and 11 days thereafter. Error bars indicate 95% CI. (E) Number of mitotic, pH3+ cells per control (+/+), fmr1Δ50m/Δ113m (-/-), or rescued Δ50m/Δ113m (r/-) posterior midgut at 4 and 10 days after eclosion. (F) Percentages of asymmetric and symmetric divisions in control (+/+), fmr1Δ50m/Δ113m (-/-), and rescued Δ50m/Δ113m (r/-) posterior midgut at 4 and 10 days after eclosion. (G) Numbers of esg+, progenitor cells per control (+/+), fmr1Δ50m/Δ113m (-/-), or rescued Δ50m/Δ113m (r/-) posterior midgut of 6-7 day old females. (D-G) Significance of control versus mutant comparisons as well as numbers of samples indicated, except for (D) where numbers of samples are listed in methods section. See also Figure S1.
fmr1 limits the expansion of the adult intestinal progenitor cell population
To investigate the role of FMRP in the intestine, we analyzed adults trans-heterozygous for two well characterized fmr1 null alleles (Zhang et al., 2001). These fmr1 mutants had a larger than normal number of progenitors as well as an enlarged intestine (Fig 1C). We focused on the posterior midgut (PMG) to quantify the progression of this phenotype and to compare it to lin-28 null intestines. In contrast to lin-28-/- nulls, which have reduced numbers of progenitor cells (Chen et al., 2015), aged fmr1-/- mutants displayed accelerated progenitor expansion, with normal progenitor number at eclosion but higher than normal numbers in 4- and 11- day old animals (Fig 1D). This enhanced expansion of the progenitor population was triggered by nutrient uptake, since fmr1-/- mutants displayed normal progenitor number when fed a sucrose diet (Fig S1D). The respective reduced and accelerated progenitor expansion of lin-28-/- and fmr1-/- mutants correlated with other intestinal features, including diameter and total cell numbers (S1E-G). Thus, while the intestines of recently eclosed control, lin-28-/- mutant, and fmr1-/- mutants were grossly similar in size, lin-28-/- mutant intestines failed to grow while fmr1-/- mutant intestines grew faster and reached a larger final size than control intestines.
The increase in the intestinal progenitor population, which is composed of both mitotic ISCs and their daughter EBs (Micchelli and Perrimon, 2006, Ohlstein and Spradling, 2006), could be due to alterations not only in ISC division rate but also division pattern. For example, ISCs tend to divide symmetrically during early adulthood to expand in number and drive intestinal tissue growth (O’Brien et al., 2011). Thus, to understand the cellular basis of the fmr1 mutant phenotype, we measured both the amount and type of ISC division. fmr1-/- intestines from 4- and 10-day old animals contained significantly more mitotic ISCs than age-matched controls (Fig 1E). In addition, fmr1-/- ISCs displayed a bias toward symmetric division at 10 days, a time-point when controls had switched to a predominantly asymmetric division pattern (Fig 1F). These phenotypes were all rescued by resupplying fmr1 with a verified rescuing transgene (Dockendorff et al., 2002), confirming that loss of FMRP was responsible for these defects (Fig 1E-G). Three additional observations indicated a cell autonomous role for FMRP in regulating ISC behavior: (i) knockdown of FMRP using a verified RNAi line in ISCs but not EBs enhanced progenitor numbers (Fig S1H-I), (ii) fmr1-/- mutant clones contained more ISCs as well as more total numbers of cells than control clones while, conversely (Fig S1J-L), and (iii) overexpression of FMRP reduced ISC and mitotic cell number (Fig S1J-L). Thus, while LIN-28 promoted ISC expansion (Chen et al., 2015), FMRP acted in ISCs to limit their expansion during adaptive intestinal growth.
fmr1 regulates insulin signaling via lin-28
Since lin-28 and fmr1 encode RNA-binding proteins with overlapping expression profiles but opposite phenotypes, we used epistasis to test whether they acted in the same pathway. In contrast to the elevated progenitor number displayed by fmr1-/- mutants, lin-28-/- fmr1-/- double mutants not only had fewer progenitors (Fig 2A) but also phenocopied the lin-28-/- single mutants with respect to intestine diameter and total cell numbers (S1E, F). Thus, lin-28 is epistatic to fmr1, indicating that fmr1 repressed the expansion of progenitor cell number via lin-28. Since lin-28-/- progenitor phenotypes are caused by reduced IIS (Chen et al., 2015), these results predicted elevated IIS in fmr1-/- progenitors. Confirming this prediction, two established IIS reporters were significantly higher in fmr1-/- mutant progenitors: the tGPH transgenic fluorescent reporter of PI3K activity (Britton et al., 2002), and antisera that detected the activated, phosphorylated form of AKT, a target of InR (Fig 2B-C, S2A-B). Importantly, these fmr1-/- phenotypes were reversed when combined with lin-28 alleles (Fig 2B-C). To test whether elevated IIS caused the fmr1-/- phenotype, we analyzed genetic interactions between fmr1 and two IIS components, InR and chico. In both cases, null mutations in InR and chico dominantly suppressed the increase in fmr1-/- progenitors (Fig 2D, E). These results collectively indicated that fmr1 limits IIS signaling via lin-28 to dampen progenitor number expansion.
Figure 2. fmr1 regulates insulin signaling via lin-28.
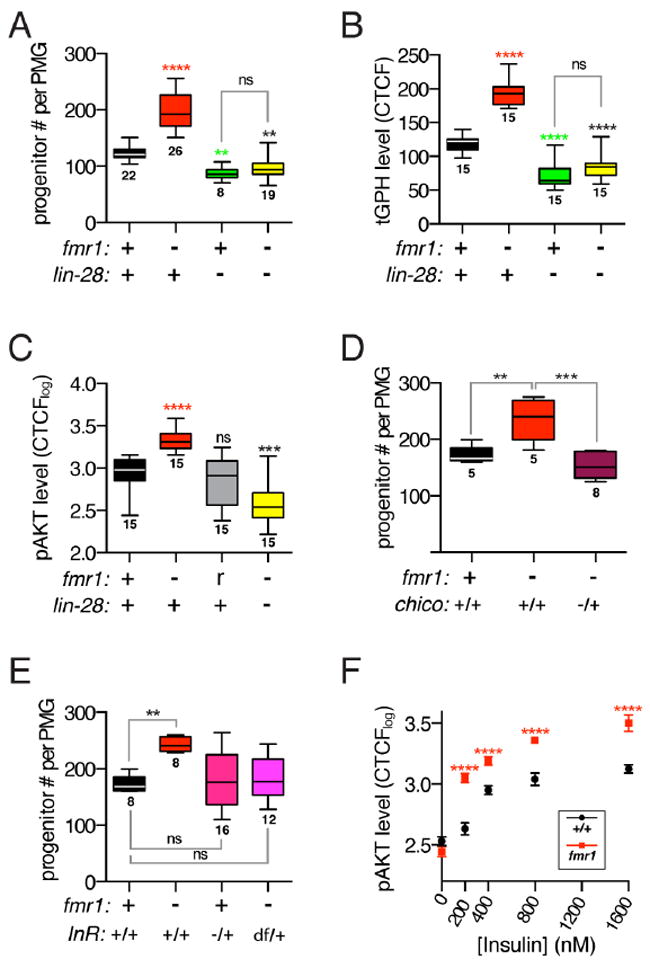
(A) Number of esg+, progenitor cells per control, fmr1Δ50m/Δ113m mutant, lin-28Δ1 mutant or lin-28Δ1, fmr1Δ50m/Δ113m double mutant intestine. (B) Membrane intensity of tGPH reporter in control, fmr1Δ50m/Δ113m mutant, lin-28Δ1 mutant and lin-28Δ1, fmr1Δ50m/Δ113m double mutant progenitor cells. (C) Intensity of p-AKT signal in control, fmr1Δ50m/Δ113m mutant, rescued fmr1Δ50m/Δ113m mutant, and lin-28Δ1, fmr1Δ50m/Δ113m double mutant progenitor cells. (D) Number of esg+, progenitor cells per control, fmr1Δ50m/Δ113m mutant, or fmr1Δ50m/Δ113m, chico1/+ mutant intestine. (E) Number of esg+, progenitor cells per control, fmr1Δ50m/Δ113m mutant, fmr1Δ50m/Δ113m; InRE19/+ mutant, or fmr1Δ50m/Δ113m; InR Df(3R)Exel6186/+ mutant intestine. (F) Intensity of p-AKT signal in control and fmr1Δ50m/Δ113m mutant progenitor cells in intestines treated with increasing concentrations of insulin. Error bars indicate SEM. (A-F) Significance of control versus mutant comparisons as well as numbers of samples are indicated except for (F) where numbers of samples are listed in methods section. See also Figure S2.
fmr1 and lin-28 have opposite effects on InR level
FMRP could limit IIS by repressing InR, a hypothesis suggested by our previous finding that LIN-28 promotes InR level and signaling (Chen et al., 2015) and evidence that fly and human FMRP physically interacts with InR transcript (Ascano et al., 2012, McMahon et al., 2016). Consistent with this hypothesis, progenitor cells in both fmr1-/- mutant intestines as well as clones in otherwise heterozygous tissue were significantly more sensitive to insulin exposure in an ex vivo assay that measured progenitor p-AKT level in dissected intestines (Fig 2F, Fig S2C). Importantly, fmr1-/- mutant intestines did not display elevated p-AKT levels in the absence of insulin in this assay (Fig 2F, S2D), indicating that the fmr1-/- required the presence of the InR ligand and suggesting that the elevated response may be due to insulin hypersensitivity caused by excessive levels of InR.
To directly analyze InR levels in progenitor cells, we used CRISPR-based genome engineering to tag the intracellular domain of InR with a V5 epitope for staining and Luciferase for quantitative measurement of protein levels (Fig 3A), since available InR antibodies were not effective for tissue staining (not shown). This tagged InR was enriched in progenitor cells (Fig S3A) and effectively reported InR levels: it increased after starvation (Fig S3B, C), consistent with the known nutrient-dependent regulation of InR in cell culture (Marr et al., 2007), and was abolished by UAS-InR-RNAi expression (S3D, E). We therefore tested whether changes in FMRP and LIN-28 levels altered InR∷V5 level. UAS-fmr1 repressed InR∷V5 in progenitors while UAS-lin-28 elevated InR∷V5 (Fig 3B,C, Fig S3G). Conversely, fmr1 loss resulted in elevated InR∷V5 while lin-28 loss had the opposite effect (Fig 3D,E, Fig S3H). InR∷V5 transcript levels were not affected by these alterations in LIN-28 and FMRP levels, indicating post-transcriptional regulation of InR∷V5 (Fig S3F). A reporter of miR-305, a microRNA known to regulate InR (Foronda et al., 2014) was also not affected in fmr1-/- or lin-28-/- progenitors (Fig S3I-J), indicating that FMRP and LIN-28 do not regulate InR via this microRNA. However, epistasis analysis revealed that InR∷V5 levels were reduced in lin-28-/- fmr1-/- double mutant progenitors, indicating that fmr1 required lin-28 to repress InR∷V5 (Fig 3D,E). Thus, FMRP and LIN-28 formed a post-transcriptional pathway that controlled InR level, insulin sensitivity, and division behavior of intestinal progenitors.
Figure 3. fmr1 and lin-28 have opposing effects on InR levels.
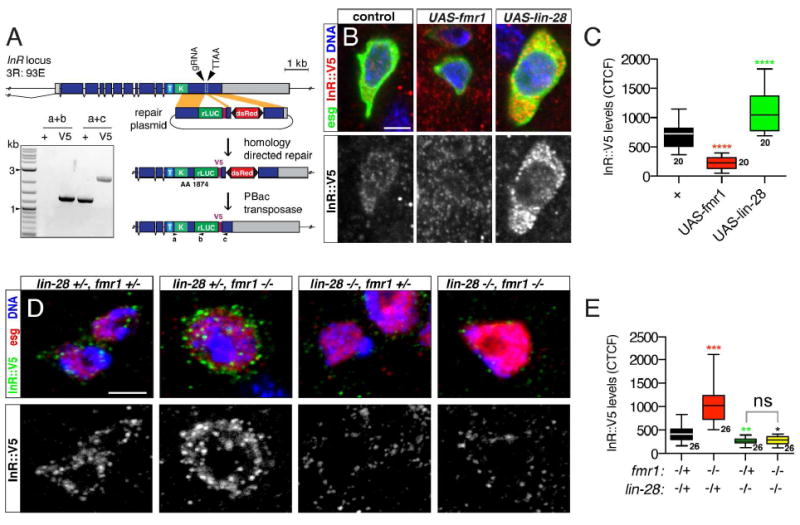
(A) InR∷V5 construction scheme. See methods for full description. Inset: PCR-based confirmation of InR tagging. (B) Intestinal progenitors from esgTS (control), esgTS/UAS-fmr1 and esgTS/UAS-lin-28 intestines stained for esg>GFP (green), InR∷V5 (red), and DNA (blue). Scale bar: 3 μm. (C) InR∷V5 fluorescence intensity of samples shown in (B). (D) Intestinal progenitors stained for InR∷V5 (green), esg-LacZ (red) and DNA (blue) from adults harboring mutations in one or both copies of lin-28 and fmr1. Scale bar: 3 μm. (E) InR∷V5 fluorescence intensity of samples shown in (D). Significance of control versus mutant comparisons as well as numbers of samples indicated. See also Figure S3.
LIN-28 is sensitive to alterations in FMRP level
To investigate the basis for the functional interaction between FMRP and LIN-28, we first tested whether they bound each other directly. However, we found no evidence of physical interaction between LIN-28 and FMRP in either wildtype or arsenite-treated embryos or cells (Fig S4A-C), suggesting that their co-localization in cells was indirect. Therefore, we next analyzed the effect of fmr1 loss or overexpression on LIN-28 expression and localization in progenitor cells. When fmr1 level was reduced, either in fmr1-/- mutant intestines or in fmr1-/- mutant clones, LIN-28 protein level was lower and detected in a few small, bright, cytoplasmic punctae in progenitor cells (Fig 4A-C, Fig S4D). Conversely, when fmr1 was overexpressed, either in progenitor cells throughout the intestine or in clones, LIN-28 levels increased and LIN-28 protein appeared to coalesce into larger cytoplasmic aggregates (Fig 4A-C, Fig S4D). This effect was not reciprocal, since alterations in LIN-28 level did not affect the subcellular distribution or level of FMRP (Fig S4E), nor did mutations in the RNA-binding domains of LIN-28 that we previously found led to aberrant LIN-28 accumulation in the nucleus (Fig S4F). Taken together, these observations that FMRP affected LIN28 level and distribution, but not vice versa, supported our genetic evidence that fmr1 functions upstream of lin-28.
Figure 4. LIN-28 is sensitive to alterations in fmr1 dosage.

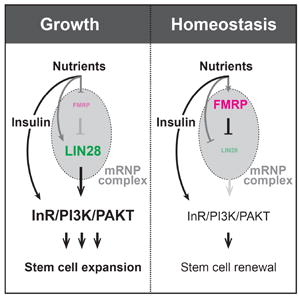
(A) Intestinal progenitors stained for LIN-28∷Venus (green) and DNA (blue) from control (left), fmr1Δ50m/Δ113m mutant (middle), and esgTS/UAS-fmr1 (right) intestines. Scale bar: 10 μm. (B) LIN-28∷Venus fluorescence intensity of samples shown in (A). Significance of control versus mutant comparisons as well as numbers of samples are indicated. (C)Tub-Gal4, UAS-gfp-labeled adult control (left), fmr1Δ50m (middle), and UAS-fmr1 (right) clones generated in 7-day old adults and stained for GFP (green), LIN-28∷mCherry (red), and DNA (blue). Scale bar: 3 μm. (D) In our model, LIN-28 predominates during adaptive growth to promote the mRNA translation needed for symmetric ISC division and expansion of the progenitor population. FMRP is activated later, during the transition from growth to homeostasis, in order to recruit LIN-28 repressive mRNPs and switch the ISC division pattern from symmetric to asymmetric. See also Figure S4.
In conclusion, we report that FMRP functions via LIN-28 to control the behavior of progenitor cells that underlies the adaptive growth of the adult intestine in response to nutrition. In particular, we find that FMRP controls the insulin sensitivity of progenitor cells by post-transcriptionally regulating the levels of InR via LIN-28. Based on our results, we favor a model in which FMRP promotes the formation of cytoplasmic mRNP complexes that contain both translationally stalled mRNAs and inactive LIN-28, and that loss of fmr1 both disperses and activates LIN-28, leading to mRNA translation (see Fig 4D). Since our data clearly identifies an antagonism between FMRP and LIN-28, we were surprised by observations that FMRP also seems to promote LIN-28 levels (Fig 4A-C, Fig S4D). We interpret this as a homeostatic cellular response to a depletion in LIN-28 activity, akin to the upregulation of LIN-28 in response to starvation (Luhur and Sokol 2016). However, we cannot rule out the possibility that FMRP promotes a repressive activity of LIN-28, and expect that future studies will uncover the mechanistic basis for the functional interaction between FMRP and LIN-28.
Loss of FMRP in humans causes Fragile X Syndrome (FXS). The work presented here suggests that dysregulated, stem cell based, tissue growth could be at the root of a series of understudied and poorly understood FXS symptoms, including elevated brain size and weight at birth, accelerated height growth in childhood, macroorchidism in adolescence, gastrointestinal problems, and heightened susceptibility to obesity (Kidd et al., 2014, McLennan et al., 2011, Nowicki et al., 2007), whose cellular bases are completely uncharacterized. Elevated IIS contributes is known to contribute to macroorchidism (Wise, 2017), suggesting that it may underlie many of these symptoms. Elevated IIS is also associated with fmr1 loss in the nervous system: loss of FMRP leads to elevated levels of p-AKT in mouse neurons and brains (Zhu et al., 2011, Gross et al., 2010, Hoeffer et al., 2012, Sharma et al., 2010) and the memory deficits of fmr1 mutant flies are corrected by either genetic or pharmacological reduction of insulin signaling (Monyak et al., 2016). Thus, this work raises the possibility that the LIN-28/FMRP pathway is broadly conserved, operates in multiple tissues, and is directly relevant to both the neural and non-neural symptoms displayed by FXS patients.
Experimental Procedures
Drosophila strains and husbandry
Female flies were used in all experiments. Flies were aged between 0 to 14 days, as indicated. Newly eclosed animals were unmated, but flies aged more than a day were mated. Strains were cultured on standard Bloomington Drosophila Stock Center media (http://fly.bio.indiana.edu/Fly_Work/media-recipes/bloomfood.htm) supplemented with 30% (w/v) yeast paste. Starved flies were reared in empty glass vials containing a tissue wetted with 1% sucrose solution that was refreshed daily. Flies were cultured in 18°C, 25 °C, or 29°C incubators set for a 12-hour light/dark schedule and 65% humidity. Flies were transferred to new vials every two days. Flies were reared in groups of 15-20 (typically 3 males and up to 17 females) and aged until ready to be sacrificed for experiments. Genotypes of all flies are listed in Table S1, and details regarding the construction of new transgenic animals generated for this study can be found in the supplemental material.
Temperature and ex vivo treatments
For temporal and regional gene expression targeting experiments (TARGET), flies were grown at 18°C, collected over 1-2 days, and then reared in 29°C for up to 7 days before being sacrificed. For clonal analysis, clones were induced by heatshock and labeled with the mosaic analysis with repressible cell marker (MARCM) method. For these experiments, animals were reared at 25°C until eclosion, collected over 1 day, aged for 5 days, and then heatshocked at 37°C for 20 minutes in a Lauda circulating water bath. After heatshock, flies were reared at 25°C for 10 or 60 days.
For ex vivo insulin assays, intestines from animals starved for 4-days were incubated in Grace’s insect media supplemented with 200-1600 nM human insulin (Sigma) for 10 minutes and then fixed. For ex vivo arsenite treatment, intestines from females aged for 8-10 days on normal diet were incubated in Krebs-Ringer media supplemented with 1mM arsenite (Sigma) for 60 minutes and then fixed.
Measurements, Analyses, and Statistical Methods
Detailed descriptions of methods used to quantify intestinal features, including size, cell numbers, and cell division patterns, can be found in the supplemental material. Statistical analyses were performed using Prism (GraphPad; version 5.0). Datasets were tested for normality using the D’Agostino-Pearson test. For comparisons of two data sets with parametric distribution, p-values were determined using the two-tailed unpaired t-test or, when the data were determined to follow non-parametric distribution, the Mann-Whitney test. For multiple comparisons of three or more data sets following parametric distribution, p-values were determined using one-way ANOVA, followed by post-hoc tests with Tukey’s multiple comparison correction. Unless otherwise noted, n.s not significant, *p < 0.05, **p < 0.01, ***p < 0.001, and **** p < 0.0001.
Supplementary Material
Highlights.
FMRP limits adaptive tissue expansion in the adult Drosophila intestine
FMRP represses symmetric division and insulin sensitivity of intestinal stem cells
FMRP mediates its effect in intestinal stem cells solely via LIN-28
FMRP acts via LIN-28 to post-transcriptionally repress Insulin Receptor
Acknowledgments
We thank Kendal Broadie, Stephen Cohen, Tom Jongens, Norbert Perrimon, the Bloomington Drosophila Stock Centers, the Developmental Studies Hybridoma Bank, and the Drosophila Genome Resource Center for reagents; the Indiana Statistical Consulting Center for math advice; Brian Calvi and Andrew Zelhof and two anonymous reviewers for comments that improved the manuscript; and the National Institutes of Health (Awards R21OD019916 and R01GM124220) and Indiana University for financial support.
Footnotes
Author Contributions:
Conceptualization, A.L. and N.S.; Methodology, A.L., and N.S.; Investigation, A.L, K.B, I.A.; Software, S.C; Writing – Original Draft, A.L. and N.S.; Funding Acquisition, N.S.; Resources, N.S.; Supervision, A.L. and N.S.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- ASCANO M, JR, MUKHERJEE N, BANDARU P, MILLER JB, NUSBAUM JD, CORCORAN DL, LANGLOIS C, MUNSCHAUER M, DEWELL S, HAFNER M, WILLIAMS Z, OHLER U, TUSCHL T. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. 2012;492:382–6. doi: 10.1038/nature11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRITTON JS, LOCKWOOD WK, LI L, COHEN SM, EDGAR BA. Drosophila’s insulin/PI3-kinase pathway coordinates cellular metabolism with nutritional conditions. Dev Cell. 2002;2:239–49. doi: 10.1016/s1534-5807(02)00117-x. [DOI] [PubMed] [Google Scholar]
- CHEN CH, LUHUR A, SOKOL N. Lin-28 promotes symmetric stem cell division and drives adaptive growth in the adult Drosophila intestine. Development. 2015;142:3478–87. doi: 10.1242/dev.127951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOI NH, LUCCHETTA E, OHLSTEIN B. Nonautonomous regulation of Drosophila midgut stem cell proliferation by the insulin-signaling pathway. Proc Natl Acad Sci U S A. 2011;108:18702–7. doi: 10.1073/pnas.1109348108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE NAVASCUES J, PERDIGOTO CN, BIAN Y, SCHNEIDER MH, BARDIN AJ, MARTINEZ-ARIAS A, SIMONS BD. Drosophila midgut homeostasis involves neutral competition between symmetrically dividing intestinal stem cells. EMBO J. 2012;31:2473–85. doi: 10.1038/emboj.2012.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOCKENDORFF TC, SU HS, MCBRIDE SM, YANG Z, CHOI CH, SIWICKI KK, SEHGAL A, JONGENS TA. Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron. 2002;34:973–84. doi: 10.1016/s0896-6273(02)00724-9. [DOI] [PubMed] [Google Scholar]
- FORONDA D, WENG R, VERMA P, CHEN YW, COHEN SM. Coordination of insulin and Notch pathway activities by microRNA miR-305 mediates adaptive homeostasis in the intestinal stem cells of the Drosophila gut. Genes Dev. 2014;28:2421–31. doi: 10.1101/gad.241588.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOULAS S, CONDER R, KNOBLICH JA. The Par complex and integrins direct asymmetric cell division in adult intestinal stem cells. Cell Stem Cell. 2012;11:529–40. doi: 10.1016/j.stem.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GROSS C, NAKAMOTO M, YAO X, CHAN CB, YIM SY, YE K, WARREN ST, BASSELL GJ. Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J Neurosci. 2010;30:10624–38. doi: 10.1523/JNEUROSCI.0402-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOEFFER CA, SANCHEZ E, HAGERMAN RJ, MU Y, NGUYEN DV, WONG H, WHELAN AM, ZUKIN RS, KLANN E, TASSONE F. Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav. 2012;11:332–41. doi: 10.1111/j.1601-183X.2012.00768.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIDD SA, LACHIEWICZ A, BARBOUTH D, BLITZ RK, DELAHUNTY C, MCBRIEN D, VISOOTSAK J, BERRY-KRAVIS E. Fragile X syndrome: a review of associated medical problems. Pediatrics. 2014;134:995–1005. doi: 10.1542/peds.2013-4301. [DOI] [PubMed] [Google Scholar]
- KLEIN AM, SIMONS BD. Universal patterns of stem cell fate in cycling adult tissues. Development. 2011;138:3103–11. doi: 10.1242/dev.060103. [DOI] [PubMed] [Google Scholar]
- LOPEZ-GARCIA C, KLEIN AM, SIMONS BD, WINTON DJ. Intestinal stem cell replacement follows a pattern of neutral drift. Science. 2010;330:822–5. doi: 10.1126/science.1196236. [DOI] [PubMed] [Google Scholar]
- LUHUR A, SOKOL N. Starving for more: Nutrient sensing by LIN-28 in adult intestinal progenitor cells. Fly (Austin) 2016 doi: 10.1080/19336934.2016.1158366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUHUR A, CHAWLA G, SOKOL N. MicroRNAs as components of systemic signaling pathways in Drosophila melanogaster. Curr Top Dev Biol. 2013;105:97–123. doi: 10.1016/B978-0-12-396968-2.00004-X. [DOI] [PubMed] [Google Scholar]
- MARR MT, 2ND, D’ALESSIO JA, PUIG O, TJIAN R. IRES-mediated functional coupling of transcription and translation amplifies insulin receptor feedback. Genes Dev. 2007;21:175–83. doi: 10.1101/gad.1506407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCLENNAN Y, POLUSSA J, TASSONE F, HAGERMAN R. Fragile x syndrome. Curr Genomics. 2011;12:216–24. doi: 10.2174/138920211795677886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCLEOD CJ, WANG L, WONG C, JONES DL. Stem cell dynamics in response to nutrient availability. Curr Biol. 2010;20:2100–5. doi: 10.1016/j.cub.2010.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCMAHON AC, RAHMAN R, JIN H, SHEN JL, FIELDSEND A, LUO W, ROSBASH M. TRIBE: Hijacking an RNA-Editing Enzyme to Identify Cell-Specific Targets of RNA-Binding Proteins. Cell. 2016;165:742–53. doi: 10.1016/j.cell.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MICCHELLI CA, PERRIMON N. Evidence that stem cells reside in the adult Drosophila midgut epithelium. Nature. 2006;439:475–9. doi: 10.1038/nature04371. [DOI] [PubMed] [Google Scholar]
- MONYAK RE, EMERSON D, SCHOENFELD BP, ZHENG X, CHAMBERS DB, ROSENFELT C, LANGER S, HINCHEY P, CHOI CH, MCDONALD TV, BOLDUC FV, SEHGAL A, MCBRIDE SM, JONGENS TA. Insulin signaling misregulation underlies circadian and cognitive deficits in a Drosophila fragile X model. Mol Psychiatry. 2016 doi: 10.1038/mp.2016.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NOWICKI ST, TASSONE F, ONO MY, FERRANTI J, CROQUETTE MF, GOODLIN-JONES B, HAGERMAN RJ. The Prader-Willi phenotype of fragile X syndrome. J Dev Behav Pediatr. 2007;28:133–8. doi: 10.1097/01.DBP.0000267563.18952.c9. [DOI] [PubMed] [Google Scholar]
- O’BRIEN LE, SOLIMAN SS, LI X, BILDER D. Altered modes of stem cell division drive adaptive intestinal growth. Cell. 2011;147:603–14. doi: 10.1016/j.cell.2011.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OHLSTEIN B, SPRADLING A. The adult Drosophila posterior midgut is maintained by pluripotent stem cells. Nature. 2006;439:470–4. doi: 10.1038/nature04333. [DOI] [PubMed] [Google Scholar]
- PANDA AC, GRAMMATIKAKIS I, YOON JH, ABDELMOHSEN K. Posttranscriptional regulation of insulin family ligands and receptors. Int J Mol Sci. 2013;14:19202–29. doi: 10.3390/ijms140919202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POLESSKAYA A, CUVELLIER S, NAGUIBNEVA I, DUQUET A, MOSS EG, HAREL-BELLAN A. Lin-28 binds IGF-2 mRNA and participates in skeletal myogenesis by increasing translation efficiency. Genes Dev. 2007;21:1125–38. doi: 10.1101/gad.415007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHARMA A, HOEFFER CA, TAKAYASU Y, MIYAWAKI T, MCBRIDE SM, KLANN E, ZUKIN RS. Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci. 2010;30:694–702. doi: 10.1523/JNEUROSCI.3696-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHYH-CHANG N, DALEY GQ. Lin28: primal regulator of growth and metabolism in stem cells. Cell Stem Cell. 2013;12:395–406. doi: 10.1016/j.stem.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIMONS BD, CLEVERS H. Strategies for homeostatic stem cell self-renewal in adult tissues. Cell. 2011;145:851–62. doi: 10.1016/j.cell.2011.05.033. [DOI] [PubMed] [Google Scholar]
- WISE TL. Changes in insulin-like growth factor signaling alter phenotypes in Fragile X Mice. Genes Brain Behav. 2017;16:241–249. doi: 10.1111/gbb.12340. [DOI] [PubMed] [Google Scholar]
- ZHANG YQ, BAILEY AM, MATTHIES HJ, RENDEN RB, SMITH MA, SPEESE SD, RUBIN GM, BROADIE K. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell. 2001;107:591–603. doi: 10.1016/s0092-8674(01)00589-x. [DOI] [PubMed] [Google Scholar]
- ZHU H, SHYH-CHANG N, SEGRE AV, SHINODA G, SHAH SP, EINHORN WS, TAKEUCHI A, ENGREITZ JM, HAGAN JP, KHARAS MG, URBACH A, THORNTON JE, TRIBOULET R, GREGORY RI, ALTSHULER D, DALEY GQ CONSORTIUM, D., INVESTIGATORS, M. The Lin28/let-7 axis regulates glucose metabolism. Cell. 2011;147:81–94. doi: 10.1016/j.cell.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.