

SUMMARY
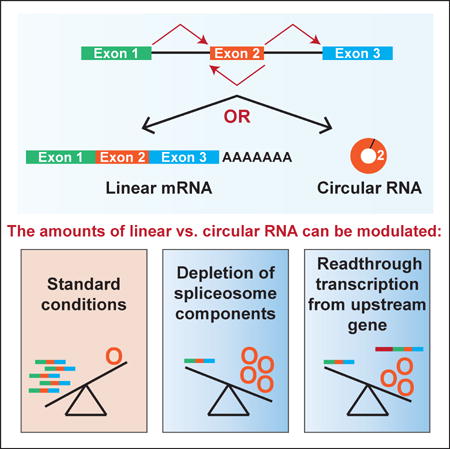
Many eukaryotic genes generate linear mRNAs and circular RNAs, but it is largely unknown how the ratio of linear to circular RNA is controlled or modulated. Using RNAi screening in Drosophila cells, we identify many core spliceosome and transcription termination factors that control the RNA outputs of reporter and endogenous genes. When spliceosome components were depleted or inhibited pharmacologically, the steady-state levels of circular RNAs increased while expression of their associated linear mRNAs concomitantly decreased. Upon inhibiting RNA polymerase II termination via depletion of the cleavage/polyadenylation machinery, circular RNA levels were similarly increased. This is because readthrough transcripts now extend into downstream genes and are subjected to backsplicing. In total, these results demonstrate that inhibition or slowing of canonical pre-mRNA processing events shifts the steady-state output of protein-coding genes towards circular RNAs. This is in part because nascent RNAs become directed into alternative pathways that lead to circular RNA production.
Keywords: circRNA, SF3b1, exon definition, spliceosome, transcription termination, cleavage/polyadenylation, pre-mRNA splicing, Pladienolide B, backsplicing
eTOC Blurb
Many protein-coding genes produce linear mRNAs and circular RNAs. Liang et al. find that circular RNAs become the preferred gene output when core spliceosome or transcription termination factors are depleted from cells. This is in part because nascent RNAs are directed into alternative pathways that lead to circular RNA biogenesis.
INTRODUCTION
Upon initiation of gene transcription, a set of molecular machines are coordinately assembled on the nascent RNA which act to process the transcript into a mature mRNA that is exported to the cytoplasm for translation (reviewed in Bentley, 2014). On most nascent mRNAs, the capping machinery modifies the 5′ end, the pre-mRNA splicing machinery removes introns, and the cleavage/polyadenylation machinery generates the mature 3′ end as well as promotes transcription termination. If these steps do not occur appropriately, surveillance mechanisms are often triggered and the RNA is degraded (reviewed in Jensen et al., 2013; Kilchert et al., 2016). Surprisingly, large-scale RNA profiling efforts have indicated that >75% of the human genome is transcribed to yield a highly complex network of overlapping RNAs (Djebali et al., 2012; Hangauer et al., 2013; Wilusz et al., 2009), which includes many noncoding RNAs as well as readthrough transcripts that extend beyond annotated gene boundaries (Akiva et al., 2006; Maher et al., 2009; Parra et al., 2006; Vilborg et al., 2015). These readthrough transcripts can contain genetic information from multiple genes, although the ultimate fate of most readthrough transcripts remains unclear. One hypothesis is that some may be used for the production of stable RNA species.
Circular RNAs are a recently described class of stable RNAs that are naturally resistant to degradation by exonucleases and are generated from thousands of protein-coding genes (reviewed in Chen, 2016; Ebbesen et al., 2016; Wilusz, 2016). The mature transcripts contain almost exclusively exonic sequences, have covalently linked ends, and are generated when the pre-mRNA splicing machinery “backsplices” to join a downstream splice donor to an upstream splice acceptor (e.g. the end of exon 2 is joined to the beginning of exon 2). Backsplicing is often initiated when complementary sequences from different introns base pair and bring the intervening splice sites close together (Ivanov et al., 2015; Kramer et al., 2015; Liang and Wilusz, 2014; Zhang et al., 2014), although there are also examples in which circular RNA production is promoted by RNA binding proteins (Ashwal-Fluss et al., 2014; Conn et al., 2015) or tied to exon skipping events (Barrett et al., 2015; Kelly et al., 2015). While some circular RNAs are expressed at higher levels than their associated linear mRNAs, most are expressed at much lower levels (<10% of the associated linear mRNA) and accumulate to only a few molecules per cell (Guo et al., 2014; Jeck et al., 2013; Salzman et al., 2012). This suggests that backsplicing is generally inefficient in most cells (Zhang et al., 2016), and it is remains unclear what purpose (if any) most backsplicing events serve.
At the Drosophila Muscleblind (Mbl) locus, backsplicing serves an auto-regulatory function and prevents Mbl protein from accumulating to excess amounts (Ashwal-Fluss et al., 2014). Given that the levels of circular RNAs and linear mRNAs from most genes are not correlated (Salzman et al., 2013), the regulatory strategy used at the Mbl gene seems to be a unique case. Two other circular RNAs (CDR1as/ciRS-7 and Sry) function to sponge specific microRNAs (Hansen et al., 2013; Memczak et al., 2013), but this again does not appear to be a general function for circular RNAs as most are not enriched in microRNA binding sites (Guo et al., 2014). It has instead been suggested that circular RNAs may help form large RNA-protein complexes, e.g. at neuronal synapses (Rybak-Wolf et al., 2015; You et al., 2015), be translated (Abe et al., 2015; Chen and Sarnow, 1995; Kramer et al., 2015; Legnini et al., 2017; Pamudurti et al., 2017; Yang et al., 2017), or be involved in innate immune responses (Chen et al., 2017; Li et al., 2017).
Given this lack of consensus about how and why circular RNAs are produced, we set out to better understand how cells modulate the relative amounts of linear vs. circular RNA that are expressed from protein-coding genes. Using minigene expression plasmids based on the Drosophila laccase2 gene, we found that canonically spliced linear mRNAs are more efficiently generated than circular RNAs. However, the circular RNAs accumulate due to their long half-lives and can ultimately become the dominant isoform observed in cells. Using RNAi screening, we surprisingly found that depletion of many core spliceosomal components, including the SF3b and SF3a complexes, resulted in specific increases in the steady-state levels of circular RNAs from reporters and endogenous Drosophila genes. Upon treating cells with the SF3b inhibitor Pladienolide B, canonical linear mRNA splicing was inhibited while circular RNAs continued to accumulate. This suggests that backsplicing becomes a preferred outcome when spliceosome activity is limiting. Our RNAi screening additionally revealed that circular RNAs can be generated from readthrough transcripts that extend past annotated poly(A) signals. Upon depleting cleavage/polyadenylation factors or the torpedo exonuclease Rat1, failures in transcription termination were observed and the backsplicing machinery processed these transcripts once they extended into the downstream gene. Circular RNA expression can thus be uncoupled from its host gene promoter, a result we confirmed at the endogenous human PAIP2 locus. In total, we propose that slowing or inhibiting canonical pre-mRNA processing events, including splicing or 3′ end processing, shifts the outputs of protein-coding genes towards circular RNAs. As circular RNAs are naturally resistant to degradation by exonucleases, slight changes in the efficiency of backsplicing can result in large changes in the steady-state levels of these transcripts, thereby allowing circular RNAs to be the dominant isoform expressed from some protein-coding genes.
RESULTS
The Laccase2 circular RNA is inefficiently generated, but accumulates due to its long half-life
To characterize the cis-acting regulatory elements and trans-acting factors that dictate the amounts of linear and circular RNA that accumulate from protein-coding genes, we generated a three-exon minigene for the Drosophila laccase2 gene (Figure 1A, top). This was done by modifying our previously described Hy_pMT Laccase2 Exon 2 plasmid (Figure 1A, bottom), which expresses laccase2 exon 2 and its immediate intronic sequences under the control of the metallothionein A promoter (pMT) (Kramer et al., 2015). Exon 1 (with 187-nt of the downstream intron) and exon 3 (with 214-nt of the upstream intron) of the laccase2 gene were added, thereby placing backsplicing in direct competition with canonical splicing (Figures 1A and S1A). Importantly, KpnI and XmaI sites were also inserted into exon 2 so that plasmid-derived transcripts could be distinguished on Northern blots from endogenous Laccase2 transcripts.
Figure 1. The Laccase2 circular RNA accumulates to high levels due to its long half-life.

(A) Schematics of the Hy_pMT Laccase2 Exons 1–3 and Hy_pMT Laccase2 Exon 2 plasmids. Upon induction of the metallothionein promoter (pMT) by addition of CuSO4, the ~3.6-kb pre-mRNA derived from the Hy_pMT Laccase2 Exons 1–3 plasmid can be canonically spliced to generate a ~1.8-kb linear mRNA that is subsequently polyadenylated (top) or backspliced to generate a 514-nt circular RNA from exon 2 (bottom). Inverted DNAREP1_DM repeat sequences (green) are present in the introns flanking exon 2. See also Figure S1A. (B) Laccase2 plasmids were transfected into Drosophila DL1 cells, CuSO4 added for 14 h, and Northern blots performed. *Concatenated and/or intertwined circular RNA. β-actin was used as a loading control. See also Figure S1B. (C) A DL1 cell line stably expressing the Hy_pMT Laccase2 Exons 1–3 plasmid was treated with CuSO4 for the indicated amounts of time, and Northern blots performed. See also Figure S1F. (D) To measure RNA half-lives, the stable cell line was treated with CuSO4 for 3 h followed by chelation of the metal with BCS for the indicated amounts of time. Cross-hybridization of the Northern probe with ribosomal RNAs is responsible for the ~2-kb transcripts weakly detected after 12 h. See also Figure S1G.
The Hy_pMT Laccase2 Exon 2 and Hy_pMT Laccase2 Exons 1–3 plasmids were then transfected into Drosophila DL1 cells, copper (CuSO4) was added for 14 h to induce pMT transcription, and total RNA was isolated. Consistent with prior results (Kramer et al., 2015), Northern blots revealed that the pre-mRNA derived from the Hy_pMT Laccase2 Exon 2 plasmid was efficiently backspliced to generate a 514-nt circular RNA (Figure 1B). The Hy_pMT Laccase2 Exons 1–3 plasmid likewise generated the circular RNA, which was resistant to digestion by the 3′-5′ exonuclease RNase R (Figure S1B), as well as a three-exon linear mRNA (Figure 1B). An additional linear mRNA with exon 2 skipped could also be detected, but this transcript accumulated to only ~5% the level of the fully spliced mRNA (Figure 1B). Most notably, the plasmid-derived circular RNA accumulated to higher levels than the fully spliced mRNA (Figure 1B), similar to what is observed from the endogenous laccase2 gene in DL1 cells (Kramer et al., 2015). This three-exon minigene thus accurately recapitulates the endogenous splicing pattern, suggesting it could reveal mechanisms that regulate how much linear vs. circular RNA accumulates from the laccase2 gene.
Base pairing between highly complementary transposable elements in the flanking introns facilitates backsplicing from many eukaryotic genes (Ivanov et al., 2015; Jeck et al., 2013; Liang and Wilusz, 2014; Zhang et al., 2014). Indeed, a pair of inverted DNAREP1_DM family transposons flank exon 2 of the Drosophila laccase2 gene (Figure 1A). Prior mutational analysis showed that ~100 nt of each repeat (which are highly complementary over a ~55-nt region) is sufficient for exon circularization from the Hy_pMT Laccase2 Exon 2 plasmid (Kramer et al., 2015). To determine if the presence of flanking exons alters the extent of intronic base pairing required for backsplicing, we progressively deleted the DNAREP1_DM repeats from the Hy_pMT Laccase2 Exons 1–3 plasmid (Figure S1C). Interestingly, even when there is competition between canonical splicing and backsplicing, ~100 nt of each repeat was sufficient for the circular RNA to accumulate to high levels (Figures S1D and S1E). This supports a model in which base pairing between short complementary regions is a major driver of exon circularization.
The strong accumulation of the Laccase2 circular RNA could be due to the splicing machinery preferring to backsplice or due to differences in half-lives between the mature linear and circular RNAs. To distinguish between these models, we first took advantage of the hygromycin resistance (HygroR) cassette (Figure S1A) to generate a DL1 cell line stably expressing the inducible Hy_pMT Laccase2 Exons 1–3 plasmid. CuSO4 was then added to induce transcription for short periods of time such that the levels of the linear and circular transcripts should largely reflect their synthesis rates (Figure 1C). Unlike what was observed at 14 h of CuSO4 induction (Figure 1B), the linear RNA was the predominant transcript that accumulated at early time points (e.g. 2 h) (Figures 1C and S1F). This suggests that backsplicing is slow and not the preferred splicing outcome, a result that is consistent with recent genome-wide nascent RNA sequencing data in human cells (Zhang et al., 2016). To measure the linear and circular RNA half-lives, a 3 h transcription pulse was induced and shut-off by chelating copper with bathocuproine disulphonate (BCS) (Djuranovic et al., 2012). Whereas the linear RNA had a half-life of ~2 h, the circular RNA remained largely stable after 24 h (Figures 1D and S1G). These results thus indicate that the reporter circular RNA is inefficiently generated, but its long half-life allows the transcript to accumulate to levels greater than the linear mRNA.
Depleting components of the U2 snRNP shifts gene outputs towards circular RNAs
To identify trans-acting factors that control the ratio of linear to circular RNA expressed from Hy_pMT Laccase2 Exons 1–3, we took advantage of the stable cell line and used double-stranded RNAs (dsRNAs) to individually knock down proteins with well-established roles in pre-mRNA splicing for 3 d. CuSO4 was added for the last 14 h and expression of the linear and circular RNAs were measured using Northern blots (Figures 2A, 2B, and S2A). It was previously demonstrated that the SR protein SF2 (homolog of human SRSF1) inhibits backsplicing from the endogenous laccase2 gene (Kramer et al., 2015) and thus we first asked if the Hy_pMT Laccase2 Exons 1–3 reporter recapitulated this regulation. Compared to cells treated with a control (β-gal) dsRNA, depletion of SF2 indeed resulted in a >50-fold increase in circular RNA levels and a corresponding >4-fold decrease in spliced linear RNA levels (Figure 2A, Lane 4). With these proof-of-concept experiments in hand, we then assayed the effect of depleting other pre-mRNA splicing factors, including core spliceosomal components.
Figure 2. The SF3b and SF3a complexes regulate Laccase2 circular RNA levels.

(A and B) A DL1 cell line stably expressing the Hy_pMT Laccase2 Exons 1–3 plasmid was treated with the indicated dsRNAs for 3 d and, where noted, CuSO4 was added for the last 14 h. Northern blots were used to examine expression of reporter-derived transcripts. Representative blots are shown. See also Figure S2. (C) Linear mRNA and circular RNA levels were quantified using ImageQuant from three independent Northern blot experiments. Data are normalized to the β-gal dsRNA samples and are shown as mean±SD. * p<0.01. (D) DL1 cells were treated with the indicated dsRNAs for 3 d. qRT-PCR was then used to quantify expression of the endogenous Laccase2 linear mRNA (Exon 2/3 junction) and Laccase2 circular RNA. Data from four independent experiments were normalized to β-actin (Act42a) and are shown as mean±SD. * p<0.01. (E) Northern blots were used to measure expression of the endogenous laccase2 gene in DL1 cells that had been treated with dsRNAs for 3 d.
Upon depleting SF3b1 (CG2807) or SF3a1 (CG16941), components of the U2 snRNP that are required for pre-spliceosome assembly (Wahl et al., 2009), we found that expression of the mature linear mRNA was largely eliminated (Figure 2A, Lanes 6 and 8). Surprisingly, however, expression of the circular RNA was only slightly decreased upon SF3b1 depletion and, in fact, increased upon SF3a1 depletion (Figures 2A and 2C). The steady-state output of the reporter gene thus became predominately circular RNA when early steps in spliceosome assembly are slowed or inhibited. SF3b1 is the largest subunit of the seven-member SF3b protein complex (Wahl et al., 2009) and individually depleting the other members of this complex (Figure 2B, Lanes 2–8) caused an increase in circular RNA levels coupled to a reduction in linear mRNA levels (Figure 2C). SF3a1 is likewise the largest subunit of the heterotrimeric SF3a complex (Lin and Xu, 2012) and depletion of SF3a2 or SF3a3 also resulted in increased levels of circular RNA (Figure 2B, Lanes 10–11).
Upon examining expression of the endogenous laccase2 locus by qPCR (Figures 2D) or Northern blots (Figure 2E), we found that depleting the SF3b or SF3a complexes also affected endogenous splicing patterns. When any of these ten factors were depleted from DL1 cells, endogenous Laccase2 circular RNA levels increased and linear Laccase2 mRNA levels decreased (Figures 2D and 2E). To next determine if the expression of circular RNAs may generally increase when SF3b or SF3a are depleted from Drosophila cells, we examined the outputs of additional endogenous genes that are known to generate circular RNAs (Ashwal-Fluss et al., 2014). Unextended (Uex) generates a 334-nt circular RNA from exon 4 (Figure 3A) and Plexin A (PlexA) generates a 1439-nt circular RNA from exon 2 (Figure 3B). Like the laccase2 locus, complementary transposable elements are present in the introns flanking each of these exons. Depletion of SF3b or SF3a components caused significant increases in the expression of both the Uex and PlexA circular RNAs (Figures 3A and 3B).
Figure 3. Depleting or pharmacologically inhibiting SF3b results in increased expression of endogenous circular RNAs.

(A) DL1 cells were treated with the indicated dsRNAs for 3 d. Northern blots (top) and qRT-PCR (bottom) were then used to quantify expression of the endogenous Uex linear mRNA and circular RNA. A representative blot is shown. Data from four independent qRT-PCR experiments were normalized to β-actin (Act42a) and are shown as mean±SD. * p<0.05. (B) DL1 cells were treated as in A. Northern blots (top) and qRT-PCR (bottom) were used to quantify expression of the endogenous PlexA linear mRNA and circular RNA. A representative blot is shown. Data from four independent qRT-PCR experiments were normalized to β-actin (Act42a) and are shown as mean±SD. * p<0.05. (C) A DL1 cell line stably expressing the Hy_pMT Laccase2 Exons 1–3 plasmid was treated with increasing concentrations of Pladienolide B (Plad B) and, where noted, CuSO4 for 6 h. Northern blots were used to examine expression of reporter-derived transcripts. (D) DL1 cells were treated with DMSO or 5 nM Plad B for 6 h. Total RNA was then isolated and subjected to qRT-PCR to measure the expression of transcripts derived from the PlexA, Uex, and laccase2 genes. Data from three independent experiments were normalized to β-actin (Act42a) and are shown as mean±SD. * p<0.05.
To rule out potential off-target effects that may be caused by 3 days of dsRNA treatment, we first measured the toxicity of the various dsRNAs. Depleting SF3b6, Phf5a, or SF3a2 did not markedly affect cell viability, while depleting other SF3b and SF3a components inhibited growth to some extent (Figure S2B). Nevertheless, when we treated cells with unrelated dsRNAs (targeting unrelated genes) that caused similar levels of toxicity (Figure S2B), the relative ratio of linear vs. circular RNA that accumulated from the Hy_pMT Laccase2 Exons 1–3 reporter was largely unchanged (Figure S2C). This strongly indicates that the changes in circular RNA levels that are observed upon depletion of the SF3b and SF3a complexes are not due to general toxicity of the RNAi, but because these factors are key regulators of protein-coding gene outputs.
Canonical splicing is more sensitive to pharmacological inhibition of SF3b than backsplicing
To further determine if the changes in the steady-state amounts of linear vs. circular RNA are due to differences in their biogenesis efficiencies, we treated the Hy_pMT Laccase2 Exons 1–3 stable cell line for 6 h with CuSO4 and Pladienolide B (Plad B), a naturally occurring anti-tumor drug that binds and inhibits SF3b (Kotake et al., 2007). When cells were treated with 5 or 10 nM Plad B, linear reporter mRNAs failed to accumulate, while circular RNAs were strikingly still produced (Figure 3C). As the dose of Plad B was further increased, circular RNA production decreased, indicating that some amount of active SF3b is required for backsplicing. Nevertheless, circular RNA production is clearly more resistant to SF3b inhibition than canonical splicing. To determine if endogenous backsplicing events are similarly resistant to Plad B treatment, we treated DL1 cells with 5 nM Plad B for 6 h and measured the outputs of the endogenous PlexA, Uex, and laccase2 genes (Figure 3D). Compared to control treated cells, the levels of each of the linear mRNAs were all significantly reduced by Plad B treatment (Figure 3D). In contrast, expression of each of the endogenous circular RNAs increased by 2–4 fold. These data thus mirror the RNAi results and suggest that the relative efficiency of backsplicing increases when spliceosome assembly is slowed or when spliceosomes are in limited quantities.
Endogenous circular RNA levels increase upon depleting a variety of core spliceosomal components
As the spliceosome is formed by a dynamic assembly of five snRNAs and ~170 proteins (reviewed in Wahl et al., 2009), we next addressed if depletion of other core spliceosomal components would similarly affect circular RNA levels. For this analysis, we focused on the endogenous PlexA gene because the PlexA circular RNA was most affected by depletion of SF3b and SF3a components (Figure 3B). 35 proteins that function at different stages of spliceosome assembly or catalysis (Mount and Salz, 2000) were individually depleted from DL1 cells, and Northern blots (Figure 4A) or qPCR (Figure S3) used to measure the output of the PlexA gene. Remarkably, depletion of 25 of these core spliceosomal proteins resulted in >2-fold increases in circular RNA expression. A number of factors with very different annotated functions caused greater than 10-fold increases, including: (i) snRNP-U1-70K and snRNP-U1-C, which are involved in 5′ splice site recognition; (ii) Prp8, the largest protein component in the spliceosome which crosslinks with U5 and U6 snRNAs as well as all the critical sites of chemistry (the 5′ splice site, branch point sequence, and 3′ splice site); and (iii) Slu7 and CG6015 (CDC40), which act in the second catalytic step of splicing. This thus indicates that changing the levels of many core spliceosomal components can have profound impacts on circular RNA levels.
Figure 4. Depleting many core spliceosomal components results in increased expression of endogenous circular RNAs.

(A) DL1 cells were treated with the indicated dsRNAs for 3 d and Northern blots used to quantify expression of the endogenous PlexA linear mRNA and circular RNA. Core spliceosomal components that function at different stages of spliceosome assembly were individually depleted. See also Figure S3. (B) Total RNA was isolated from DL1 cells treated with dsRNAs for 3 d and subjected to qRT-PCR to measure the expression of endogenous circular RNAs. Data from four independent experiments were normalized to β-actin (Act42a) and are shown as mean±SD.
For the core spliceosomal components that caused the largest increases in PlexA circular RNA levels, we then asked if these factors similarly regulate other endogenous circular RNAs (Figure 4B). In general, depleting core spliceosomal components resulted in global increases in circular RNA levels, although the effect sizes varied depending on the factor that was depleted and the circular RNA examined. For example, depletion of snRNP-U1-70K or snRNP-U1-C caused PlexA and pasilla (ps) circular RNA levels to increase >10-fold, but had a smaller effect on expression of the Laccase2, Uex, minibrain (mnb), CG42663, and ectoderm-expressed 4 (Ect4) circular RNAs (Figure 4B). Consistent with these data, modulating core spliceosomal components has been previously demonstrated to affect only a subset of canonical splicing events in Drosophila (Brooks et al., 2015; Park et al., 2004). In total, our data suggest a model in which backsplicing becomes a preferred mode of pre-mRNA processing when spliceosomal components are in limited quantities, thereby causing the steady-state output of genes to be biased towards circular RNAs.
Circular RNAs can be generated from readthrough transcription events
Beyond regulation of circular RNA levels by the splicing machinery, recent work suggests that backsplicing may often occur post-transcriptionally after RNA polymerase II has reached the end of the parent gene (Liang and Wilusz, 2014; Zhang et al., 2016). This led us to predict that inhibiting co-transcriptional 3′ end processing may increase circular RNA levels. Indeed, depleting the pre-mRNA 3′ end processing endonuclease Cpsf73 (Figure 5A, Lane 4 and Figure S4A) caused expression of the reporter circular RNA to increase almost 50-fold while linear RNA levels decreased by ~2-fold (Figure 5B). We initially suspected that Cpsf73 was acting at the 3′ end of the laccase2 minigene, and thus we replaced the downstream SV40 poly(A) signal with a self-cleaving hammerhead ribozyme (HhRz) sequence (Figure S4B). Surprisingly, depletion of Cpsf73 caused Laccase2 circular RNA levels to increase even when the HhRz was present (Figure S4C).
Figure 5. Readthrough transcription from the upstream HygroR gene enables circular RNA production from the minigene reporter.

(A) A DL1 cell line stably expressing the Hy_pMT Laccase2 Exons 1–3 plasmid was treated with the indicated dsRNAs for 3 d and, where noted, CuSO4 was added for the last 14 h. Northern blots were used to examine expression of reporter-derived transcripts. See also Figure S4A. (B) Linear mRNA and circular RNA levels were quantified using ImageQuant from four independent Northern blot experiments and were normalized to the “β-gal, No copper” samples. Data are shown as mean±SD. * p<0.05. (C) Schematic of readthrough transcription model. In control treated cells (top), only the upstream copia transposon LTR promoter is active when no CuSO4 is present. This results in the production of the HygroR mRNA, which is cleaved/polyadenylated, and the downstream RNA is rapidly degraded by the Rat1 exonuclease to facilitate transcription termination. When Cpsf73 is depleted (bottom), 3′ processing of the HygroR mRNA is inefficient, resulting in readthrough transcription into the downstream laccase2 minigene. When the intronic repeats (green) base pair to one another, a circular RNA is produced. See also Figure S4. (D) DL1 cells stably expressing the Hy_pMT Laccase2 Exons 1–3 plasmid were treated with the indicated dsRNAs for 3 d and total RNA isolated. Northern blots using probes complementary to Laccase2 exon 2 (Lanes 1–4) or the HygroR mRNA (Lanes 5–8) were performed. (E) The laccase2 minigene from the Hy_pMT Laccase2 Exons 1–3 plasmid was replaced with an eGFP open reading frame. In addition, the downstream polyadenylation signal was replaced with the hammerhead ribozyme (HhRz) preceded by a 74-nt sequence from the 3′ end of the Rift Valley Fever Virus (RVFV) NSs mRNA (brown). A DL1 cell line stably expressing this plasmid was then generated and treated with the indicated dsRNAs for 3 d. Depletion of Cpsf73 resulted in the production of a fusion HygroR-eGFP mRNA that is stabilized at its 3′ end by the RVFV NSs sequence.
Further work showed that depletion of Cpsf73 caused the reporter-derived circular RNA to be expressed at a high level even when CuSO4 was not added (Figure 5A, Lane 3 and Figure 5B), suggesting that the circular RNA was transcribed from a constitutive promoter. As depletion of Cpsf73 did not cause the endogenous metallothionein A promoter (pMT) to be active in the absence of copper (Figure S4D), we reasoned that the reporter-derived circular RNA was likely not being transcribed from its expected promoter. To rule out the existence of a cryptic promoter within the laccase2 minigene, this sequence was replaced with a portion of the Drosophila dati pre-mRNA that can generate a circular RNA independently of inverted intronic repeats (Figure S4E). Expression of the dati circular RNA could be induced upon CuSO4 treatment in control treated cells, but Cpsf73 depletion caused the reporter-derived dati circular RNA to be constitutively produced (Figure S4E). We thus concluded that a constitutive promoter upstream of pMT was likely driving expression of the Laccase2 and dati circular RNAs from the reporters.
Located upstream is a constitutive copia transposon LTR promoter that drives expression of the hygromycin resistance (HygroR) gene (Figures 5C and S1A). In control treated DL1 cells that stably maintain the Hy_pMT Laccase2 Exons 1–3 plasmid, the hygromycin mRNA is constitutively transcribed and efficiently cleaved/polyadenylated using the SV40 poly(A) signal at its 3′ end (Figure 5C, top). As laccase2 minigene transcripts do not appreciably accumulate when CuSO4 is absent (Figures 2A and 5A), RNA polymerase II derived from the copia promoter rarely (if ever) transcribes far into the downstream gene. However, when Cpsf73 is depleted, we reasoned that the HygroR pre-mRNA fails to be cleaved at its 3′ end, resulting in transcriptional readthrough into the downstream laccase2 minigene (Figure 5C, bottom). Once both inverted repeats have been transcribed, the intervening exon can be backspliced to generate a circular RNA. As predicted by this model, depletion of Symplekin (Symp), a scaffolding protein for the cleavage/polyadenylation machinery, also resulted in generation of the circular RNA (Figure 5D). Furthermore, we detected a >4-kb linear transcript that was produced regardless of whether CuSO4 was added when Cpsf73 or Symp was depleted (Figure 5A). This transcript corresponds to a fusion HygroR-Laccase2 linear mRNA that has been polyadenylated downstream of the laccase2 minigene (Figure 5D).
The torpedo model of transcription termination posits that cleavage by Cpsf73 provides an entry site for a 5′-3′ exonuclease (Rat1 in Drosophila), which degrades the downstream RNA in order to catch up to the elongation complex and cause it to terminate (Connelly and Manley, 1988). Consistent with the torpedo model, we found that depletion of Rat1 caused expression of the reporter circular RNA to become constitutive (Figures 5A, Lanes 5–6 and 5B). A fusion HygroR-Laccase2 linear mRNA did not accumulate upon Rat1 depletion, indicating that Rat1 functions only after 3′ cleavage (Figures 5A and 5D).
As a final test of the readthrough transcription model, we replaced the laccase2 minigene with an eGFP open reading frame followed by the self-cleaving HhRz (Figure 5E, top). Because ribozyme cleavage is not sufficient to stabilize the 3′ end of a mature linear mRNA (Figure S4C), we added a 74-nt sequence from the 3′ end of the Rift Valley Fever Virus (RVFV) NSs mRNA immediately upstream of the HhRz sequence (Molleston et al., 2016) (Figure 5E, top). This NSs sequence is able to block 3′-5′ degradation, allowing the non-polyadenylated eGFP mRNA to accumulate to high levels in cells (Figure 5E, Lane 2). After generating a stable DL1 cell line expressing the eGFP plasmid, we predicted that Cpsf73 depletion would result in generation of a fusion HygroR-eGFP linear mRNA. Indeed, a ~3500-nt transcript was detected in cells depleted of Cpsf73, irrespective of CuSO4 addition (Figure 5E, Lanes 3–4). These data demonstrate that depletion of Cpsf73, Symp, or Rat1 causes readthrough transcription from the hygromycin resistance gene, which can result in the production of stable circular RNAs and fusion linear RNAs. This suggests that circular RNAs do not have to be made from their host gene promoters and that modulating transcription termination efficiencies can alter the steady-state levels of circular RNAs.
Production of endogenous circular RNAs by readthrough transcription
Having established that transcription termination defects can lead to circular RNA accumulation from reporters, we asked if endogenous circular RNAs are produced from readthrough transcription events. Human embryonic carcinoma PA1 cells were treated with 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB) for 3 h to arrest transcription, followed by pulse labeling with the uridine analog 4-thiouridine (4sU) and purification of newly synthesized RNAs at different time points after DRB removal (Zhang et al., 2016) (Figure 6A). 39 potential polycistronic transcripts (indicative of readthrough transcription) were detected upon quantifying expression in intergenic regions between gene pairs (Figures S5A and S5B). We focused our validation efforts on the human MATR3 and PAIP2 genes, which are in the same transcriptional orientation, with the downstream PAIP2 gene generating a circular RNA (circPAIP2) from exons 2 and 3 (Figures 6A and S5C). The transcription elongation rate of the MATR3 gene is faster than average in PA1 cells (Zhang et al., 2016) and significant amounts of nascent RNAs were continuously detected throughout the ~10-kb intergenic region between MATR3 and PAIP2 (Figures 6A and S5C). This suggested that MATR3 primary transcripts may often extend into the downstream PAIP2 gene, and some of these RNAs may be backspliced to yield the PAIP2 circular RNA.
Figure 6. Readthrough transcription generates endogenous circular RNAs from the human PAIP2 gene.

(A) Readthrough transcription from MATR3 as shown by nascent RNA-seq in human PA1 cells (Zhang et al., 2016). The downstream PAIP2 gene generates a circular RNA from exons 2 and 3 (purple). See also Figure S5. (B) Immunoblot to confirm depletion of CPSF3 (top) or CPSF4 (bottom) by shRNAs in PA1 cells. scr, scrambled shRNA control. Actin was used as a loading control. (C) Schematic showing qRT-PCR primers used to quantify transcripts from the MATR3-PAIP2 locus. (D and E) qRT-PCR quantification of transcripts from the MATR3-PAIP2 intergenic region (D) or the PAIP2 gene (E) in PA1 cells depleted of CPSF3 or CPSF4. Data are shown as mean±SD from three independent experiments. * p<0.05, ** p<0.01 (F) PA1 cells were treated for 8 h with a phosphorothioate-modified antisense oligodeoxynucleotide (ASO) complementary to the MATR3-PAIP2 intergenic region. (G) qRT-PCR was then used to quantify transcripts from the MATR3-PAIP2 intergenic region or the PAIP2 gene. Locations of primers are shown in F. Data are shown as mean±SD from three independent experiments. * p<0.05, ** p<0.01
shRNAs were first used to deplete CPSF3 (CPSF73) or CPSF4 (CPSF30) from PA1 cells (Figure 6B) and quantitative RT-PCR used to quantify RNAs derived from the MATR3-PAIP2 intergenic region or the PAIP2 gene (Figure 6C). Consistent with a defect in MATR3 transcription termination, depletion of CPSF3 or CPSF4 resulted in increased expression of transcripts from the MATR3-PAIP2 intergenic region (Figure 6D). Expression of circPAIP2 also increased, suggesting that these MATR3 readthrough transcripts are likely used to generate stable circular RNAs from the downstream PAIP2 gene (Figure 6E). To directly confirm that readthrough transcription from MATR3 is required for the increase in circular RNA levels, PA1 cells were treated with a phosphorothioate-modified antisense oligonucleotide (ASO) that targeted the MATR3-PAIP2 intergenic region (Figure 6F). Compared to cells treated with a scrambled (scr) oligo, ASO treatment reduced intergenic RNA levels by ~80% and simultaneously caused significant decreases in the levels of the PAIP2 pre-mRNA, linear mRNA, and circular RNA (Figure 6G). Of note, ASO treatment reduced PAIP2 circular RNA levels by >30%, indicating that production of this endogenous circular RNA is often not from its host gene promoter and instead from a readthrough transcript. In total, these results indicate that failures to co-transcriptionally cleave at the 3′ ends of genes, such as human MATR3, can result in the production of circular RNAs from downstream genes.
DISCUSSION
Thousands of circular RNAs can be generated from protein-coding regions, but we are only beginning to understand how, when, and why these RNAs are made. In the current study, we demonstrated that backsplicing is normally less efficient than canonical splicing, but the steady-state levels of Drosophila circular RNAs significantly increase upon inhibition or slowing of multiple canonical pre-mRNA processing events. While linear mRNA levels were decreased upon depletion of many core spliceosomal components, circular RNAs became remarkably enriched under these conditions (Figure 7A). Some of this effect is due to the long half-lives of circular RNAs, but our data also indicate that backsplicing becomes a preferred splicing outcome when active spliceosomes are limiting (Figures 2–4). Inhibition of 3′ end processing and transcription termination likewise resulted in increased circular RNA levels (Figures 5–6). This is because circular RNA biogenesis can occur on readthrough transcripts that extend into downstream genes (Figure 7B). Altogether, these results suggest that inhibition or slowing of co-transcriptional processing events allows increased opportunity for backsplicing reactions to occur. As mature circular RNAs have long half-lives, they are able to accumulate to high levels and become the predominant output of some genes.
Figure 7. Proposed models for how inhibition or slowing of canonical pre-mRNA processing events can result in increased circular RNA levels.

(A) In wild-type (WT) cells (left), exons within pre-mRNAs are first defined and spliceosomal components assemble across each exon. U1 snRNP recognizes the downstream 5′ splice site, U2 snRNP binds the upstream polypyrimidine tract and branch point sequence, and factors such as SR proteins mediate cross-exon interactions. These cross-exon interactions are subsequently replaced with cross-intron interactions to enable full assembly of the spliceosome and generation of a linear mRNA. When spliceosome activity is limiting (e.g. due to depletion of core spliceosome components) (right), we propose that cross-exon interactions are not easily replaced with cross-intron interactions. The full spliceosome thus assembles across an exon, resulting in backsplicing and the generation of a circular RNA. (B) Failure to efficiently terminate transcriptional units (e.g. due to depletion of cleavage/polyadenylation factors) can cause nascent RNAs to be extended into downstream genes. If the appropriate signals are present in this readthrough transcript (such as inverted intronic repeats), backsplicing can occur to release a mature circular RNA. The remaining nascent RNA likely consists of a Y-shaped structure with a 2′-5′ phosphodiester bond at the upstream branch site. This structure may be debranched, thereby providing an entry site for exonucleases, including Rat1, that enable transcription termination.
Canonical splicing and backsplicing have different sensitivities to spliceosome inhibition
As a nascent RNA is being transcribed, 5′ and 3′ splice sites at the ends of exons are recognized and spliceosomes begin to be assembled in a stepwise manner. Depending on what splice sites are paired together, a variety of mature RNAs can be produced, including circular RNAs. In many cases, pre-mRNA splicing outcomes are dictated by cis-acting sequence elements (namely, intronic and exonic splicing enhancers or silencers) that serve as binding sites for trans-acting factors, such as SR proteins or hnRNPs, that act positively or negatively to recruit snRNPs to nearby splice sites (reviewed in Busch and Hertel, 2012; Smith and Valcarcel, 2000). Indeed, depletion of SR proteins or hnRNPs results in significant changes to alternative splicing patterns (Brooks et al., 2015), including effects on circular RNA levels (Figure 2A) (Kramer et al., 2015). In the current work, we surprisingly demonstrated that circular RNA levels also significantly increase upon alterations in the amounts or activities of core spliceosomal components (Figures 2–4). Modulating the levels of spliceosomal components has previously been shown to affect individual splicing decisions in yeast (Pleiss et al., 2007), Drosophila (Brooks et al., 2015; Park et al., 2004), and mammalian cells (Papasaikas et al., 2015; Saltzman et al., 2011), but the effects were often quite small and gene-specific. In contrast, we demonstrated here that the expression of circular RNAs from multiple endogenous loci increased when spliceosomal components were depleted (Figure 4B). The long half-lives of circular RNAs likely magnify the changes in biogenesis efficiencies, resulting in the large effect sizes observed.
Considering that both canonical splicing and backsplicing require splice sites (Starke et al., 2015), why do these processes display such distinct sensitivities to spliceosome inhibition? We propose that this difference may be due, at least in part, to the process by which exonic sequences are first identified in most pre-mRNAs (Berget, 1995). During exon definition, initial splicing complexes are formed across exons: U1 and U2 snRNPs bind at opposite ends of each exon, stabilized by additional factors, such as SR proteins, that establish a network of protein-protein interactions (Figure 7A, left). These cross-exon interactions must then somehow be converted to cross-intron interactions (thereby pairing splice sites from separate exons) in order to yield a canonically spliced linear mRNA. However, to generate a circular RNA, these initial exon definition complexes likely do not have to be disrupted and, in fact, may be stabilized by base pairing between complementary sequences in the flanking introns (Figure 7A, right). Furthermore, these exon definition complexes may help directly promote backsplicing as they can contain the U4/U6.U5 tri-snRNP and potentially be converted to B-like pre-catalytic splicing complexes (Schneider et al., 2010). We thus propose that when core spliceosomal components are depleted from cells, cross-exon interactions may be slowly or rarely converted into productive cross-intron interactions. This allows catalytically competent backsplicing complexes to be assembled more frequently, thus resulting in production of more circular RNAs and fewer canonical linear mRNAs (Figure 7A).
It remains possible that backsplicing only requires a subset of the factors that are normally required for canonical linear splicing (or, conversely, that backsplicing requires factors that are dispensable for canonical linear splicing), but further detailed mechanistic studies are required to clarify this point. Regardless of the exact molecular details, the current work clearly demonstrates that backsplicing and canonical splicing are differentially affected when core spliceosomal components are inhibited or depleted from cells. Our study thus predicts that endogenous tissues expressing limiting amounts of spliceosomal components should produce higher levels of circular RNAs. It will be particularly informative to determine if this model holds true in vivo in neuronal or aging tissues, which produce the highest amounts of annotated circular RNAs (Rybak-Wolf et al., 2015; Westholm et al., 2014; You et al., 2015).
Readthrough transcription can be exploited to generate circular RNAs from downstream regions
RNA polymerase II sometimes precisely terminates after reaching a poly(A) signal, but termination at most genes occurs at variable distances (sometimes as much as 10-kb) downstream from the poly(A) signal (Proudfoot, 2016; Shi and Manley, 2015). This allows many RNAs to be generated from intergenic regions (Vilborg et al., 2015) and, in fact, the global efficiency of termination is known to change in cancer (Grosso et al., 2015; Kannan et al., 2011; Maher et al., 2009), viral infection (Nemeroff et al., 1998; Rutkowski et al., 2015), and osmotic stress (Vilborg et al., 2015). Once produced, some readthrough transcripts have been proposed to help maintain the nuclear scaffold during osmotic stress (Vilborg et al., 2015). Others serve as precursors to stable RNAs, including Drosophila piRNAs (Chen et al., 2016; Mohn et al., 2014) and human SPA long noncoding RNAs (Wu et al., 2016) that lack their own promoter sequences.
In the current study, we found that circular RNAs can be efficiently generated from readthrough transcription in a manner highly analogous to how piRNAs and SPA RNAs are generated. Once both flanking intronic repeats have been transcribed, backsplicing can occur, thereby physically separating the mature circular RNA from the nascent transcript (Figure 7B). Such backsplicing events may be fairly common in wild-type cells as we found that at least 30% of the endogenous PAIP2 circular RNA is made from readthrough transcription from the upstream MATR3 gene (Figure 6). On the other hand, readthrough transcription can interfere with the activation of downstream promoters, and it is thus likely that the expression of a number of circular RNAs will decrease when the global efficiency of termination is altered.
Once generated, mature circular RNAs likely have their own unique functions (Hansen et al., 2013; Memczak et al., 2013), but we further suggest that the backsplicing events responsible for their biogenesis may also serve a role in aiding in transcription termination. This is because backsplicing likely leaves a Y-shaped structure with a 2′-5′ phosphodiester bond at the branch site in the remaining nascent RNA (Figure 7B). The lariat debranching enzyme may act upon this structure, thereby creating an entry site for Rat1 or other 5′-3′ exonucleases to terminate downstream transcription. It will be very informative in the future to determine if backsplicing events truly help repress gene transcription levels as well as prevent polymerases from continuing to transcribe unchecked.
In summary, our findings provide key insights into how the spliceosome and pre-mRNA 3′ end processing machineries regulate gene outputs and dictate the relative amounts of linear vs. circular RNA that accumulate from protein-coding genes. In particular, we demonstrated that inhibition or slowing of pre-mRNA processing events can profoundly increase the steady-state levels of circular RNAs, in part by increasing their biogenesis. We thus propose that the global differences in circular RNA levels that are observed across cell types (Westholm et al., 2014) may be due to differences in the concentrations or activities of pre-mRNA processing components. When such factors are limiting, canonical pre-mRNA processing events may not occur quickly, thereby allowing nascent RNAs to be directed towards alternative pathways that lead to circular RNA production. As factors like SF3b1 and U2AF1 (homolog of Drosophila U2af38) are commonly mutated in multiple human cancers (Yoshida et al., 2011), an attractive hypothesis is that these mutant proteins may be altering the amounts of linear vs. circular RNAs made from protein-coding genes, thereby resulting in disease phenotypes.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jeremy E. Wilusz (wilusz@pennmedicine.upenn.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Drosophila DL1 cells were grown at 25°C in Schneider’s Drosophila medium (ThermoFisher Scientific 21720024), supplemented with 1% (v/v) penicillin-streptomycin (ThermoFisher Scientific 15140122), 1% (v/v) L-glutamine (ThermoFisher Scientific 35050061), and 10% (v/v) fetal bovine serum (HyClone SH30910.03). DL1 stable cell lines were generated and maintained by selection with 150 μg/mL hygromycin B.
PA1 cells were grown at 37°C, 5% CO2 in MEMa supplemented with 10% (v/v) fetal bovine serum, 1% (v/v) L-glutamine, and 1% (v/v) penicillin-streptomycin.
Microbe strains
One Shot TOP10 E. coli competent cells (ThermoFisher Scientific C404006) were stored at −80°C and grown in LB medium at 37°C.
METHOD DETAILS
Expression plasmid construction
To generate expression plasmids for transfection into Drosophila cells, the indicated sequences were inserted into a pMK33/pMtHy-based plasmid between the metallothionein promoter (pMT) and the SV40 polyadenylation signal as further described in the Plasmid Details Section.
Drosophila cell culture and transfections
For transient transfections into Drosophila DL1 cells, 2 × 106 cells were plated in complete media in 6-well dishes and 2 μg of each expression plasmid was transfected using Effectene (Qiagen 301427; 16 μL Enhancer and 8 μL Effectene Reagent). On the following day, a final concentration of 500 μM copper sulfate (Fisher BioReagents BP346-500) was added for 14 h (where indicated) to induce transcription from the metallothionein promoter. Total RNA was then isolated using Trizol (ThermoFisher Scientific 15596018) as per the manufacturer’s instructions.
To measure RNA half-lives in DL1 cells stably expressing the Hy_pMT Laccase2 Exons 1–3 plasmid, 500,000 cells/well were seeded in 12-well dishes and incubated for 2 days. 500 μM copper sulfate was added for 3 h followed by washing the cells twice with media containing 500 μM bathocuproine disulphonate (BCS; Sigma B1125-500MG). New media containing 50 μM BCS was then added and samples collected at the indicated time points using Trizol.
Pladienolide B (EMD Millipore Calbiochem 5.30196.0001) was added to cells for 6 h and total RNA isolated using Trizol.
Drosophila RNAi
Double-stranded RNAs from the DRSC (Drosophila RNAi Screening Center) were generated by in vitro transcription (MEGAscript kit, ThermoFisher Scientific AMB13345) of PCR templates containing the T7 promoter sequence on both ends. Primer sequences are provided in Table S1. Knockdown experiments in 12-well dishes were then performed by bathing 500,000 cells with 2 μg of dsRNA. Cells were incubated for 3 days and a final concentration of 500 μM copper sulfate was added for the final 14 h (where indicated). Total RNA was isolated using Trizol.
To quantify toxicity caused by the dsRNAs, cells were re-suspended 2 h prior to collection and 10% of the sample was re-plated (50 μl/well, 3 wells per dsRNA) in 96-well plates (Corning 3904). After 2 h, cells were fixed in 5% formaldehyde and counterstained with Hoechst 33342 (Sigma B2261) to visualize nuclei. Four images per well were captured at 20x using an automated microscope (ImageXpress Micro, Molecular Devices) and analyzed with MetaXpress software to measure total cells. The average cell number for each set of images was calculated and normalized to β-gal dsRNA treated cells.
Mammalian cell culture and transfections
Metabolic labeling of nascent RNAs in human cells with 4sU and RNA-seq analysis were previously described (Zhang et al., 2016). shRNAs to deplete CPSF3 or CPSF4 from PA1 cells and Western blot procedure were previously described (Wu et al., 2016). Phosphorothioate-modified antisense oligodeoxynucleotides (ASOs) were synthesized at BioSune (Shanghai, China) and introduced to PA1 cell by nucleofection (Lonza) according to the manufacturer’s protocol. After 8 h ASO treatment, total RNA was isolated using Trizol and analyzed by qRT-PCR. ASO sequences are provided in Table S1.
Northern blotting
Northern blots using NorthernMax reagents (ThermoFisher Scientific) and oligonucleotide probes were performed as previously described (Tatomer et al., 2017). All oligonucleotide probe sequences are provided in Table S1. Blots were viewed and quantified with the Typhoon 9500 scanner (GE Healthcare) and quantified using ImageQuant (GE Healthcare). Representative blots are shown. For RNase R treatments, 20 μg of total RNA was treated with 10 U RNase R (Epicentre RNR07250) at 37°C for 10 min.
qRT-PCR
Total RNAs were isolated using Trizol (ThermoFisher Scientific 15596018), treated with DNase I (DNA-free Kit, ThermoFisher Scientific AM1906), and cDNA was reversed transcribed using SuperScript III (ThermoFisher Scientific 18080051). For all qRT-PCRs, actin mRNA was used as an internal control to normalize the data. qRT-PCR primer sequences are provided in Table S1.
Computational identification of polycistronic transcripts from nascent RNA-seq
Circular RNAs were identified by CIRCexplorer (Zhang et al., 2014) and potential polycistronic transcripts identified as follows. All adjacent gene pairs that have the same transcriptional direction (with no additional annotated genes in between) were first selected from RefSeq. Gene pairs without a circRNA derived from the downstream gene were then filtered out. The remaining interval regions were split into bins of 500 bp with 50 bp overhangs to generate a bin queue I.
RPKM values from the RNA-seq were used to define the expression level of each bin and the longest continuous queue Q was defined from the interval bins.
Bins in the queue Q were required to meet the following two conditions:
- Expression of each bin in Q is greater than 1.
- There is no significant expression changes between two adjacent bins from the queue Q.
The continuity of the interval region was then estimated by calculating the CI (Continuity Index).
Gene pairs with CI greater than 0.5 were considered as potential polycistronic transcripts.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance for comparisons of means was assessed by Student’s t test for qRT-PCRs and Northern blots. Statistical details and error bars are defined in each figure legend.
PLASMID DETAILS
All Drosophila expression plasmids were generated from the Hy_pMT EGFP SV40 pA Sense plasmid (https://www.addgene.org/69911/), which is a modified form of the pMK33/pMtHy plasmid. In brief, the metallothionein promoter (marked in blue) drives expression of the EGFP ORF (marked in green) that terminates in the SV40 polyadenylation signal (marked in pink). HygroR (marked in red) is driven by a copia transposon LTR promoter and terminates with an SV40 late poly(A) signal (marked in gray). An Amp selectable marker is also present. The full plasmid sequence is as follows:


The following eight plasmids were generated by inserting the indicated sequences into the XhoI and NotI sites of Hy_pMT EGFP SV40 pA Sense, thereby replacing the EGFP ORF with portions of Laccase2 or dati.
Hy_pMT Laccase2 Exon 2 (Exon 2 in yellow)
Note: This plasmid was previously named Hy_pMT Laccase2 MCS-Laccase2 608-1097 (Kramer et al. 2015; https://www.addgene.org/69890/)
Used in Figure 1B
Used in Figure S1E

Hy_pMT Laccase2 Exons 1–3 (Exon 1 in green, Exon 2 in yellow, Exon 3 in blue)
Used in Figures 1B, 1C, 1D, 2A, 2B, 3C, 5A, 5D
Used in Figures S1B, S1D, S1E, S2C

Hy_pMT Laccase2 Exons 1–3 Δ300 (Exon 1 in green, Exon 2 in yellow, Exon 3 in blue)
Used in Figure S1D

Hy_pMT Laccase2 Exons 1–3 Δ 400 (Exon 1 in green, Exon 2 in yellow, Exon 3 in blue)
Used in Figure S1D

Hy_pMT Laccase2 Exons 1–3 Δ450 (Exon 1 in green, Exon 2 in yellow, Exon 3 in blue)
Used in Figure S1D

Hy_pMT Laccase2 Exons 1–3 Δ 500 (Exon 1 in green, Exon 2 in yellow, Exon 3 in blue)
Used in Figure S1D

Hy_pMT Laccase2 Exons 1–3 450–1245 (Exon 1 in green, Exon 2 in yellow, Exon 3 in blue)
Used in Figure S1E


Hy_pMT dati (Exon 2 in yellow)
Used in Figure S4E

The following plasmid was generated by inserting the indicated sequence into the NsiI and BamHI sites of Hy_pMT Laccase2 Exons 1–3, thereby replacing the last 100 nt of exon 3 and the SV40 pA signal with a hammerhead ribozyme (HhRz) sequence.
Hy_pMT Laccase2 Exons 1–3 HhRz
Used in Figure S4C
![]()
The following plasmid was generated by inserting the indicated sequence into the NotI and SpeI sites of Hy_pMT EGFP SV40 pA Sense, thereby replacing the SV40 pA signal with an alternative 3′ end processing sequence.
Hy_pMT eGFP-RVFV NSs HhRz
Used in Figure 5E
![]()
Supplementary Material
Highlights.
RNAi screen identifies pre-mRNA processing factors that modulate circular RNA levels
Circular RNA expression increases when core spliceosomal components are depleted
Backsplicing can occur in conditions that inhibit canonical mRNA splicing events
Readthrough transcription enables production of circular RNAs from downstream genes
Acknowledgments
We thank Gideon Dreyfuss and all members of the Wilusz, Chen, Cherry, and Yang labs for discussions and advice. This work was supported by National Institutes of Health grants R00-GM104166, R35-GM119735, R01-NS099371, start-up funds from the University of Pennsylvania, and grants 91540115, 91440202 and 31471241 from NSFC. J.E.W. is a Rita Allen Foundation Scholar. S.C. is a recipient of the Burroughs Wellcome Investigators in the Pathogenesis of Infectious Disease Award.
Footnotes
AUTHOR CONTRIBUTIONS
J.E.W. conceived and designed the project. D.L., D.C.T., and J.E.W. designed and performed experiments with help from S.C.; Z.L. and L.Y. performed nascent RNA-seq bioinformatics analyses; H.W. and L.-L.C. designed and performed PAIP2 experiments. D.L., D.C.T., and J.E.W. wrote the paper with input from the authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe N, Matsumoto K, Nishihara M, Nakano Y, Shibata A, Maruyama H, Shuto S, Matsuda A, Yoshida M, Ito Y, et al. Rolling Circle Translation of Circular RNA in Living Human Cells. Sci Rep. 2015;5:16435. doi: 10.1038/srep16435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiva P, Toporik A, Edelheit S, Peretz Y, Diber A, Shemesh R, Novik A, Sorek R. Transcription-mediated gene fusion in the human genome. Genome Res. 2006;16:30–36. doi: 10.1101/gr.4137606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N, Kadener S. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell. 2014;56:55–66. doi: 10.1016/j.molcel.2014.08.019. [DOI] [PubMed] [Google Scholar]
- Barrett SP, Wang PL, Salzman J. Circular RNA biogenesis can proceed through an exon-containing lariat precursor. eLife. 2015;4 doi: 10.7554/eLife.07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DL. Coupling mRNA processing with transcription in time and space. Nat Rev Genet. 2014;15:163–175. doi: 10.1038/nrg3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berget SM. Exon recognition in vertebrate splicing. J Biol Chem. 1995;270:2411–2414. doi: 10.1074/jbc.270.6.2411. [DOI] [PubMed] [Google Scholar]
- Brooks AN, Duff MO, May G, Yang L, Bolisetty M, Landolin J, Wan K, Sandler J, Booth BW, Celniker SE, et al. Regulation of alternative splicing in Drosophila by 56 RNA binding proteins. Genome Res. 2015;25:1771–1780. doi: 10.1101/gr.192518.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch A, Hertel KJ. Evolution of SR protein and hnRNP splicing regulatory factors. Wiley Interdiscip Rev RNA. 2012;3:1–12. doi: 10.1002/wrna.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Sarnow P. Initiation of protein synthesis by the eukaryotic translational apparatus on circular RNAs. Science. 1995;268:415–417. doi: 10.1126/science.7536344. [DOI] [PubMed] [Google Scholar]
- Chen LL. The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol. 2016;17:205–211. doi: 10.1038/nrm.2015.32. [DOI] [PubMed] [Google Scholar]
- Chen YC, Stuwe E, Luo Y, Ninova M, Le Thomas A, Rozhavskaya E, Li S, Vempati S, Laver JD, Patel DJ, et al. Cutoff Suppresses RNA Polymerase II Termination to Ensure Expression of piRNA Precursors. Mol Cell. 2016;63:97–109. doi: 10.1016/j.molcel.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YG, Kim MV, Chen X, Batista PJ, Aoyama S, Wilusz JE, Iwasaki A, Chang HY. Sensing Self and Foreign Circular RNAs by Intron Identity. Mol Cell. 2017;67:228–238. e225. doi: 10.1016/j.molcel.2017.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, Roslan S, Schreiber AW, Gregory PA, Goodall GJ. The RNA Binding Protein Quaking Regulates Formation of circRNAs. Cell. 2015;160:1125–1134. doi: 10.1016/j.cell.2015.02.014. [DOI] [PubMed] [Google Scholar]
- Connelly S, Manley JL. A functional mRNA polyadenylation signal is required for transcription termination by RNA polymerase II. Genes Dev. 1988;2:440–452. doi: 10.1101/gad.2.4.440. [DOI] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djuranovic S, Nahvi A, Green R. miRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science. 2012;336:237–240. doi: 10.1126/science.1215691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebbesen KK, Kjems J, Hansen TB. Circular RNAs: Identification, biogenesis and function. Biochim Biophys Acta. 2016;1859:163–168. doi: 10.1016/j.bbagrm.2015.07.007. [DOI] [PubMed] [Google Scholar]
- Grosso AR, Leite AP, Carvalho S, Matos MR, Martins FB, Vitor AC, Desterro JM, Carmo-Fonseca M, de Almeida SF. Pervasive transcription read-through promotes aberrant expression of oncogenes and RNA chimeras in renal carcinoma. eLife. 2015;4 doi: 10.7554/eLife.09214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Agarwal V, Guo H, Bartel DP. Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 2014;15:409. doi: 10.1186/s13059-014-0409-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hangauer MJ, Vaughn IW, McManus MT. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. 2013;9:e1003569. doi: 10.1371/journal.pgen.1003569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–388. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- Ivanov A, Memczak S, Wyler E, Torti F, Porath HT, Orejuela MR, Piechotta M, Levanon EY, Landthaler M, Dieterich C, et al. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep. 2015;10:170–177. doi: 10.1016/j.celrep.2014.12.019. [DOI] [PubMed] [Google Scholar]
- Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19:141–157. doi: 10.1261/rna.035667.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TH, Jacquier A, Libri D. Dealing with pervasive transcription. Mol Cell. 2013;52:473–484. doi: 10.1016/j.molcel.2013.10.032. [DOI] [PubMed] [Google Scholar]
- Kannan K, Wang L, Wang J, Ittmann MM, Li W, Yen L. Recurrent chimeric RNAs enriched in human prostate cancer identified by deep sequencing. Proc Natl Acad Sci U S A. 2011;108:9172–9177. doi: 10.1073/pnas.1100489108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly S, Greenman C, Cook PR, Papantonis A. Exon Skipping Is Correlated with Exon Circularization. J Mol Biol. 2015;427:2414–2417. doi: 10.1016/j.jmb.2015.02.018. [DOI] [PubMed] [Google Scholar]
- Kilchert C, Wittmann S, Vasiljeva L. The regulation and functions of the nuclear RNA exosome complex. Nat Rev Mol Cell Biol. 2016;17:227–239. doi: 10.1038/nrm.2015.15. [DOI] [PubMed] [Google Scholar]
- Kotake Y, Sagane K, Owa T, Mimori-Kiyosue Y, Shimizu H, Uesugi M, Ishihama Y, Iwata M, Mizui Y. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat Chem Biol. 2007;3:570–575. doi: 10.1038/nchembio.2007.16. [DOI] [PubMed] [Google Scholar]
- Kramer MC, Liang D, Tatomer DC, Gold B, March ZM, Cherry S, Wilusz JE. Combinatorial control of Drosophila circular RNA expression by intronic repeats, hnRNPs, and SR proteins. Genes Dev. 2015;29:2168–2182. doi: 10.1101/gad.270421.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legnini I, Di Timoteo G, Rossi F, Morlando M, Briganti F, Sthandier O, Fatica A, Santini T, Andronache A, Wade M, et al. Circ-ZNF609 Is a Circular RNA that Can Be Translated and Functions in Myogenesis. Mol Cell. 2017;66:22–37. e29. doi: 10.1016/j.molcel.2017.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Liu CX, Xue W, Zhang Y, Jiang S, Yin QF, Wei J, Yao RW, Yang L, Chen LL. Coordinated circRNA Biogenesis and Function with NF90/NF110 in Viral Infection. Mol Cell. 2017;67:214–227. e217. doi: 10.1016/j.molcel.2017.05.023. [DOI] [PubMed] [Google Scholar]
- Liang D, Wilusz JE. Short intronic repeat sequences facilitate circular RNA production. Genes Dev. 2014;28:2233–2247. doi: 10.1101/gad.251926.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin PC, Xu RM. Structure and assembly of the SF3a splicing factor complex of U2 snRNP. EMBO J. 2012;31:1579–1590. doi: 10.1038/emboj.2012.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher CA, Kumar-Sinha C, Cao X, Kalyana-Sundaram S, Han B, Jing X, Sam L, Barrette T, Palanisamy N, Chinnaiyan AM. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458:97–101. doi: 10.1038/nature07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- Mohn F, Sienski G, Handler D, Brennecke J. The rhino-deadlock-cutoff complex licenses noncanonical transcription of dual-strand piRNA clusters in Drosophila. Cell. 2014;157:1364–1379. doi: 10.1016/j.cell.2014.04.031. [DOI] [PubMed] [Google Scholar]
- Molleston JM, Sabin LR, Moy RH, Menghani SV, Rausch K, Gordesky-Gold B, Hopkins KC, Zhou R, Jensen TH, Wilusz JE, et al. A conserved virus-induced cytoplasmic TRAMP-like complex recruits the exosome to target viral RNA for degradation. Genes Dev. 2016;30:1658–1670. doi: 10.1101/gad.284604.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mount SM, Salz HK. Pre-messenger RNA processing factors in the Drosophila genome. J Cell Biol. 2000;150:F37–44. doi: 10.1083/jcb.150.2.f37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeroff ME, Barabino SM, Li Y, Keller W, Krug RM. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3′end formation of cellular pre-mRNAs. Mol Cell. 1998;1:991–1000. doi: 10.1016/s1097-2765(00)80099-4. [DOI] [PubMed] [Google Scholar]
- Pamudurti NR, Bartok O, Jens M, Ashwal-Fluss R, Stottmeister C, Ruhe L, Hanan M, Wyler E, Perez-Hernandez D, Ramberger E, et al. Translation of CircRNAs. Mol Cell. 2017;66:9–21. e27. doi: 10.1016/j.molcel.2017.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papasaikas P, Tejedor JR, Vigevani L, Valcarcel J. Functional splicing network reveals extensive regulatory potential of the core spliceosomal machinery. Mol Cell. 2015;57:7–22. doi: 10.1016/j.molcel.2014.10.030. [DOI] [PubMed] [Google Scholar]
- Park JW, Parisky K, Celotto AM, Reenan RA, Graveley BR. Identification of alternative splicing regulators by RNA interference in Drosophila. Proc Natl Acad Sci U S A. 2004;101:15974–15979. doi: 10.1073/pnas.0407004101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra G, Reymond A, Dabbouseh N, Dermitzakis ET, Castelo R, Thomson TM, Antonarakis SE, Guigo R. Tandem chimerism as a means to increase protein complexity in the human genome. Genome Res. 2006;16:37–44. doi: 10.1101/gr.4145906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleiss JA, Whitworth GB, Bergkessel M, Guthrie C. Transcript specificity in yeast pre-mRNA splicing revealed by mutations in core spliceosomal components. PLoS Biol. 2007;5:e90. doi: 10.1371/journal.pbio.0050090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot NJ. Transcriptional termination in mammals: Stopping the RNA polymerase II juggernaut. Science. 2016;352:aad9926. doi: 10.1126/science.aad9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski AJ, Erhard F, L’Hernault A, Bonfert T, Schilhabel M, Crump C, Rosenstiel P, Efstathiou S, Zimmer R, Friedel CC, et al. Widespread disruption of host transcription termination in HSV-1 infection. Nature Commun. 2015;6:7126. doi: 10.1038/ncomms8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak-Wolf A, Stottmeister C, Glazar P, Jens M, Pino N, Giusti S, Hanan M, Behm M, Bartok O, Ashwal-Fluss R, et al. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol Cell. 2015;58:870–885. doi: 10.1016/j.molcel.2015.03.027. [DOI] [PubMed] [Google Scholar]
- Saltzman AL, Pan Q, Blencowe BJ. Regulation of alternative splicing by the core spliceosomal machinery. Genes Dev. 2011;25:373–384. doi: 10.1101/gad.2004811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO. Cell-type specific features of circular RNA expression. PLoS Genet. 2013;9:e1003777. doi: 10.1371/journal.pgen.1003777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One. 2012;7:e30733. doi: 10.1371/journal.pone.0030733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M, Will CL, Anokhina M, Tazi J, Urlaub H, Luhrmann R. Exon definition complexes contain the tri-snRNP and can be directly converted into B-like precatalytic splicing complexes. Mol Cell. 2010;38:223–235. doi: 10.1016/j.molcel.2010.02.027. [DOI] [PubMed] [Google Scholar]
- Shi Y, Manley JL. The end of the message: multiple protein-RNA interactions define the mRNA polyadenylation site. Genes Dev. 2015;29:889–897. doi: 10.1101/gad.261974.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CW, Valcarcel J. Alternative pre-mRNA splicing: the logic of combinatorial control. Trends Biochem Sci. 2000;25:381–388. doi: 10.1016/s0968-0004(00)01604-2. [DOI] [PubMed] [Google Scholar]
- Starke S, Jost I, Rossbach O, Schneider T, Schreiner S, Hung LH, Bindereif A. Exon circularization requires canonical splice signals. Cell Rep. 2015;10:103–111. doi: 10.1016/j.celrep.2014.12.002. [DOI] [PubMed] [Google Scholar]
- Tatomer DC, Liang D, Wilusz JE. Inducible Expression of Eukaryotic Circular RNAs from Plasmids. Methods Mol Biol. 2017;1648:143–154. doi: 10.1007/978-1-4939-7204-3_11. [DOI] [PubMed] [Google Scholar]
- Vilborg A, Passarelli MC, Yario TA, Tycowski KT, Steitz JA. Widespread Inducible Transcription Downstream of Human Genes. Mol Cell. 2015;59:449–461. doi: 10.1016/j.molcel.2015.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- Westholm JO, Miura P, Olson S, Shenker S, Joseph B, Sanfilippo P, Celniker SE, Graveley BR, Lai EC. Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep. 2014;9:1966–1980. doi: 10.1016/j.celrep.2014.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilusz JE. Circular RNAs: Unexpected outputs of many protein-coding genes. RNA Biol. 2016 doi: 10.1080/15476286.2016.1227905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 2009;23:1494–1504. doi: 10.1101/gad.1800909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Yin QF, Luo Z, Yao RW, Zheng CC, Zhang J, Xiang JF, Yang L, Chen LL. Unusual Processing Generates SPA LncRNAs that Sequester Multiple RNA Binding Proteins. Mol Cell. 2016;64:534–548. doi: 10.1016/j.molcel.2016.10.007. [DOI] [PubMed] [Google Scholar]
- Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, Jin Y, Yang Y, Chen LL, Wang Y, et al. Extensive translation of circular RNAs driven by N6-methyladenosine. Cell Res. 2017;27:626–641. doi: 10.1038/cr.2017.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- You X, Vlatkovic I, Babic A, Will T, Epstein I, Tushev G, Akbalik G, Wang M, Glock C, Quedenau C, et al. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nature Neurosci. 2015;18:603–610. doi: 10.1038/nn.3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL, Yang L. Complementary sequence-mediated exon circularization. Cell. 2014;159:134–147. doi: 10.1016/j.cell.2014.09.001. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xue W, Li X, Zhang J, Chen S, Zhang JL, Yang L, Chen LL. The Biogenesis of Nascent Circular RNAs. Cell Rep. 2016;15:611–624. doi: 10.1016/j.celrep.2016.03.058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.