

Abstract
Polyoxygenated hydrocarbons that bear one or more hydroxyl groups comprise a large set of natural and synthetic compounds, often with potent biological activity. In synthetic chemistry, alcohols are important precursors to carbonyl groups, which then can be converted into a wide range of oxygen- or nitrogen-based functionality. Therefore, the selective conversion of a single hydroxyl group in natural products into a ketone would enable the selective introduction of unnatural functionality. However, the methods known to convert a simple alcohol, or even an alcohol in a molecule that contains multiple protected functional groups, are not suitable for selective reactions of complex polyol structures. We present a new ruthenium catalyst with a unique efficacy for the selective oxidation of a single hydroxyl group among many in unprotected polyol natural products. This oxidation enables the introduction of nitrogen-based functional groups into such structures that lack nitrogen atoms and enables a selective alcohol epimerization by stepwise or reversible oxidation and reduction.
The controlled modification of natural products enables chemists to alter the physical properties and resulting functions of these complex molecules. Selective reaction at a specific hydroxyl group in a polyhydroxylated molecule, particularly one that is less reactive towards classical reagents, is a synthetic challenge caused by competing, undesirable side reactions and the lack of selectivity for a reaction at one of the multiple identical functional groups. Catalysts or reagents that differentiate between the same functional groups in different environments are needed to achieve selective, direct functionalization of such structures. To this end, catalysts based on peptides have been developed that selectively acylate, phosphorylate or sulfonate one alcohol group over the others in complex polyols1,2, and rhodium complexes catalyse carbene insertions into the hydroxyl groups in polyol structures3, but these reactions attach substituents to the oxygen atom. They do not form bonds to the carbon at which the hydroxyl group is bound1–3. If a single alcohol among many could be oxidized selectively to a ketone, then a wide range of reactions that would replace the original hydroxyl group, restructure the rings of the natural product or add substituents at the position of the C–H bond alpha to the hydroxyl group could be conducted4.
However, catalysts are not known that selectively oxidize one hydroxyl group among many in a range of polyol natural products. Scattered examples of the oxidation of polyol natural products have been published, but no catalyst or conditions have been broadly applicable to this synthetic goal. Classical methods lead to mixtures of products and existing catalysts oxidize unhindered primary C–H bonds to form aldehydes over hindered secondary C–H bonds in the core of the structures to form ketones (vide infra). Thus, we sought a combination of catalyst and reagent that would be sufficiently mild and selective for the oxidation of secondary alcohols in polyol natural products.
Although many catalysts for the oxidation of alcohols are known, few catalyse the oxidation of hindered secondary alcohols. We envisioned that the oxidation of secondary alcohols over primary alcohols could be achieved by conducting a transfer dehydrogenation with a ketone as the acceptor. Base-activated Ru(p-cymene)(Ts-DPEN) (Ts, tosyl; DPEN, diphenylethylenedia-mine)5, Shvo’s catalyst6,7, Ru(PPh3)Cl2 with KOH7 and Ru-MACHO with KOH8 have been reported to catalyse the dehydrogenation of simple unhindered secondary alcohols, such as cycloalkanols with no beta substituents, or secondary alcohols with one beta substituent in some cases, but are not reported to catalyse the dehydrogenation of more-hindered alcohols that contain two beta substituents. We found that Ru-MACHO with KOH and Shvo’s dimer are able to oxidize this type of hindered model substrate under forcing conditions (Supplementary Information gives details), but not at temperatures that are likely to tolerate highly functionalized molecules. Thus, to achieve the formation of ketone units in polyol natural products, a complex that catalyses the oxidation of hindered secondary alcohols over primary alcohols is needed.
To create such a catalyst, we investigated ruthenium complexes that contain a combination of weakly coordinating anions and unhindered, strongly donating phosphines. The ruthenium centre would be a relatively soft, mild Lewis acid that would not lead to indiscriminant decomposition, the labile anions would allow the formation of an alkoxide from the alcohol with a weak base at low temperatures, and the small phosphines would allow a reaction at the hindered secondary alcohols. By using acetone as the oxidant, the oxidation of primary alcohols into aldehydes would be uphill thermodynamically and the selectivity of the process could favour the oxidation of a hindered secondary alcohol over a primary alcohol. Moreover, it would do so without the radical intermediates of classical methods for preferential oxidation of secondary alcohols but would be unselective for one alcohol over others in a complex structure. Here we show that such a ruthenium complex catalyses the selective dehydrogenation of secondary alcohols in a set of more than a dozen natural products comprising a wide range of structures and that the resulting ketones can be converted by a subsequent catalytic processes into final products that contain nitrogen-based functionality and restructured ring systems.
Results and discussion
Development of catalysts and conditions for the selective oxidation of hindered secondary alcohols
Several sequential phases of catalyst development described in the Supplementary Information led to a new ruthenium complex (Fig. 1a and Supplementary Section 2). The precursor Ru(DMSO)4(OTf)2 (DMSO, dimethylsulfoxide; OTf, SO2CF3) was generated in acetone solvent and treated with various phosphine ligands to give potential catalysts. The complexes that result from the addition of unhindered alkylphosphines PEt3, PMePh2 and PEt2( p-Me2N–Ph) all gave active catalysts for the dehydrogenation of the hindered model alcohol diisopropylcarbinol. Thereafter, a synthesis of a series of catalyst precursors lacking DMSO ligands was developed (Ru-1, Ru-2 and Ru-3), and the activity of these complexes was found to be an order of magnitude greater than that of the system generated in situ. The initial results of the oxidation of a model hindered alcohol are shown in Fig. 1b. In contrast to prior transition-metal catalysts that do not oxidize such hindered secondary alcohols, this ruthenium complex converted dicyclohexylcarbinol into the corresponding ketone at room temperature in only three hours. The same oxidation also occurred when 1,4-cyclohexanedione or trifluoroacetophenone was used as a stoichiometric oxidant and the reaction was run in trifluoroethanol solvent.
Figure 1. Model alcohol oxidation and catalyst synthesis.

a, Synthesis of the ruthenium catalyst for alcohol-transfer dehydrogenation. b, Mild oxidation of a hindered secondary alcohol is enabled by the catalyst Ru-2. c, Reactions of primary and secondary alcohols show the selectivity for the oxidation of secondary alcohols over primary alcohols. Conversions are shown in parentheses. d, The mild reduction of a hindered ketone is enabled by Ru-2 with iPrOH as the reductant. r.t., room temperature. NMM; N-methylmorpholine.
The reactions of a series of primary and secondary alcohols and mixtures of primary and secondary alcohols with acetone as the hydrogen acceptor and Ru-2 as the catalyst are shown in Fig. 1c. These results show that the oxidation of secondary alcohols occurs over the oxidation of primary alcohols. The individual reactions of primary alcohols occur to a low conversion over a time that leads to the full conversion of secondary alcohols, and reactions with a mixture of primary and secondary alcohols preferentially convert the secondary alcohol into the ketone.
Selective oxidation of polyol natural products to ketone derivatives
Figure 2 summarizes the transfer dehydrogenation of 13 polyol natural products and three methyl esters of natural products. This figure shows the products from the oxidation reactions and the configuration of the alcohol unit that underwent oxidation. These reactions occur with several classes of natural products, including steroids, diterpenoids, sesquiterpenoids, iridoids, carbohydrates, alkaloids and macrolides, which shows the broad applicability of the catalyst system. The ruthenium catalysts we developed display a remarkable selectivity for the oxidation of a single secondary alcohol within each natural product in Fig. 2 and for a reaction at the alcohol over auxiliary functionality. The reactions of these natural products revealed guidelines to predict the site selectivity of the oxidations. The reactions catalysed by Ru-2 with acetone as the hydrogen acceptor led to the oxidation of secondary alcohols in these structures over the primary alcohols. This catalyst also senses subtle differences between the steric and electronic properties of secondary alcohols, which leads to a remarkable selectivity for the oxidation of one secondary alcohol over other secondary alcohols.
Figure 2. Examples of the site-selective oxidation of polyol natural products.

a, The oxidations of this series of natural products shows that the reactions occur at secondary over primary alcohols in complex structures. b, The oxidations of this series of natural products that contain multiple secondary alcohols show that the reactions occur at one secondary alcohol over other secondary alcohols. c, Thermodynamically less-favourable oxidations and selective dehydrogenations with stoichiometric amounts of acceptor occur with trifluoroacetophenone as the acceptor. Allylic isomerization occurs over oxidation in the presence of a ruthenium catalyst that contains the bidentate phosphine.
In general, the ruthenium catalyst was selective for the more electron rich of the secondary alcohols and in most cases for the less hindered of the secondary alcohols. For example, the catalyst reacts with secondary alcohols that contain fewer than two hydroxy or alkoxy groups on the carbon atoms beta to the hydroxyl group, but it does not react with alcohols that contain two or more oxygen-based substituents at these positions. Primary alcohols that contain a single oxygen atom on the beta carbon are not oxidized by this system. Owing to this effect of electron-withdrawing substituents, the catalyst tolerates polyoxygenated moieties, such as sugars, attached to molecules via glycosidic bonds. In contrast, deoxy sugars are oxidized selectively to give the corresponding ketones by the catalyst system. In general, dione products from the oxidation of multiple alcohols were not observed because of the high sensitivity of the catalyst to differences in the properties of various alcohols. The use of a dehydrogenation process with a ketone as the acceptor, rather than a conventional oxidation with oxygen, peroxide or amine oxide, creates a tolerance for olefins, amides, esters, enoates, epoxides, acetals, amines, phenols and carbohydrates in elaborate topological settings.
The examples in Fig. 2a show the selectivity for oxidation of secondary alcohols over primary alcohols and the selectivity for one secondary alcohol over the others in polyol structures. The dehydrogenation of andrographolide with Ru-2 occurred selectively at the hindered secondary alcohol at the C3 position to form the corresponding ketone in a 65% yield (1a). This secondary alcohol reacts over the secondary alcohol at the C14 position because of the electron-withdrawing groups near C14. The iridoid glycoside aucubin underwent oxidation catalysed by Ru-2 at the secondary alcohol of the cyclopentenol to form ketone 1b in an 87% yield; the primary allylic alcohol remained untouched, and the glycoside unit was stable because each alcohol in a glycoside has two or more beta oxygen atoms. Catalyst Ru-1, which is apparently more oxidizing than Ru-2, generates about ~4% aldehyde, but Ru-2 is fully selective for the formation of the ketone. The oxidation of D-glucal occurred selectively to give a vinylogous ester in an 88% yield (1c). In contrast to those in a fully oxygenated carbohydrate, one hydroxyl group in D-glucal bears only one beta-oxygen atom and, therefore, undergoes oxidation by Ru-2. The oxidation of kirenol formed the natural product 15-dehydrokirenol (1d) in a single step in a 54% yield9. This selectivity appears to be controlled by a combination of the relative reactivity of secondary versus primary alcohols and the reactivity of one secondary alcohol over another. The alpha hydrogen of the cyclohexanol portion of this molecule experiences 1,3-eclipsing interactions with axial methyl and hydroxymethyl groups (Fig. 3), which leads to oxidation at the less-electron-rich secondary alcohol at C15. Chelation of the 1,2-diol could also favour selective oxidation at the C15 position, which helps to overcome the electronic deactivation of this secondary alcohol by the proximal primary alcohol. As is usually observed in our system, the secondary alcohol was oxidized preferentially over the primary alcohols.
Figure 3. Examples of the 1,3-eclipsing interactions that influence the selectivity of the dehydrogenation of certain polyols.
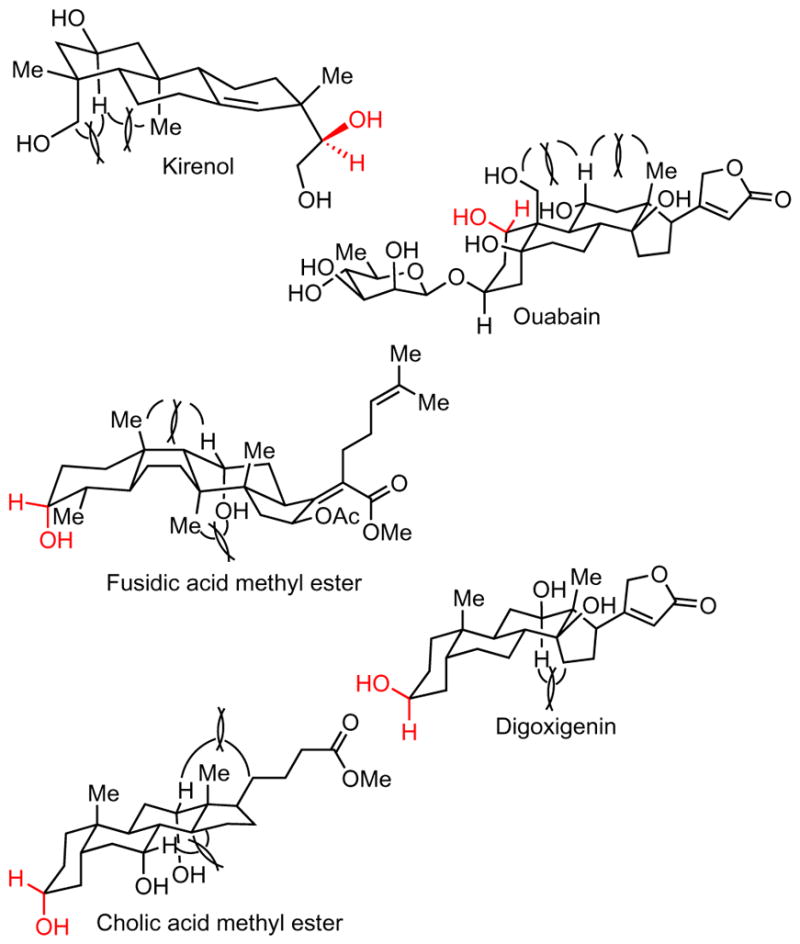
Steric occlusion of the alpha hydrogen or the hydroxyl group of the alcohols present in complex molecules by distal substituents with an eclipsing 1,3-relationship decreases the rate of oxidation at these sites. The site of dehydrogenation is shown in red.
Oxidation of ouabain occurred with a remarkable selectivity for one of many hydroxyl groups. Ouabain contains eight hydroxyl groups, one in a primary alcohol, five in secondary alcohols and two in tertiary alcohols. Under the conditions with acetone as the hydrogen acceptor and Ru-2 as the catalyst, the oxidation of ouabain occurred at a single secondary alcohol to give a mono-ketone product in a 92% yield (1e). The glycosidic functionality was fully tolerated because of the deactivation of each of these hydroxyl groups by an adjacent glycosidic oxygen atom or hydroxyl substituents. The primary alcohol, again, was less reactive than the secondary alcohol with acetone as the acceptor and Ru-2 as the catalyst. The alpha hydrogen on the secondary alcohol of ring C has 1,3-diaxial relationships with the hydroxymethyl substituent and a methyl substituent and, therefore, is less accessible sterically than the hydrogen that reacts (Fig. 3). Treatment of the keto-ouabain product with NH2OSO3H led to deglycosylation, generating 1-keto-ouabagenin (3a) in an 80% yield (Supplementary Information). This reaction exposes a less-hindered alcohol and provides a route to a useful keto-cardenolide core structure.
The examples in Fig. 2b show that small differences in the electronic and steric environments of the hydroxyl groups in polyols that contain only secondary alcohols lead to large effects on the site selectivity. For example, the dehydrogenation of fusidic acid methyl ester yielded a single ketone derivative in a 97% yield (1f). Both hydroxyl groups of this natural product are sterically hindered, but one alcohol is slightly more hindered partly because of a 1,3-eclipsing arrangement of the alpha hydrogen of the hydroxy group on the C ring with a gamma methyl group, and oxidation occurs at the less hindered of the two alcohols (Fig. 3). Steric effects also control the selectivities for the oxidation of digoxigenin, although the difference in steric properties in this case is more pronounced. In this structure, the alcohol that lacks vicinal substituents undergoes oxidation (1g), whereas the alpha hydrogen of the other hydroxyl group bears a gauche 1,2 interaction and an eclipsing 1,3 interaction that blocks the site (Fig. 3). Cholic acid methyl ester is dehydrogenated selectively at the hydroxy group that lacks beta substituents or 1,3-eclipsing interactions between the alpha hydrogen and another substituent (1h and Fig. 3). Finally, the reaction of for-skolin illustrates the strong effect of the electronic properties of two secondary alcohols on selectivity. One of the secondary alcohols of forskolin is flanked by a quaternary carbon beta to the alcohol, and the other is flanked by a vicinal acetate. In this case, oxidation occurs at the more electron-rich alcohol, even though it is more sterically hindered (1i).
Sterically accessible vicinal diols react when they lack additional proximal oxygen atoms. When reactive, they undergo dehydrogenation to form hydroxy ketones without epimerization of the adjacent alcohol. For example, the oxidation of oestriol with Ru-2 yielded the α-hydroxyketone product 1j, which results from the dehydrogenation of the more sterically accessible alcohol, in a 97% yield. However, the polyketide mupirocin methyl ester reacted in a 70% yield (1k) at the methyl carbinol over the alcohols in the vicinal diol. This vicinal diol unit of mupirocin methyl ester is less electron rich than that in oestriol because of the oxygen of the tetrahydropyran ring, and the methyl carbinol is sterically accessible.
For the oxidation of deacetylbaccatin III, which is the core of taxol and a precursor to docetaxel, an α-hydroxyketone unit inhibited the catalyst activity under our standard conditions. The α-hydroxyketone unit bound to the catalyst in a pure acetone solvent to form a ruthenium alkoxide, which was characterized by single-crystal X-ray diffraction (Supplementary Section 3). However, reaction in a mixture of acetone and trifluoroethanol (added to labilize the alkoxide) led to the selective oxidation of the secondary allylic alcohol to give a single ketone product in a 70% yield (1l). In concert, the secondary alcohol of the β-hydroxyketone underwent full epimerization, presumably from a reversible retro-aldol process10.
The catalyst Ru-2 reacts with remarkable tolerance for primary allylic alcohols and α-hydroxyenoates. The oxidation of the iridoid glycoside genipin and the macrolide brefeldin demonstrate the ability to oxidize a secondary alcohol without isomerization of a primary or secondary allylic alcohol in the same molecule to give the corresponding aldehyde or ketone. Brefeldin underwent selective oxidation in 87% yield at the alcohol on the five-membered ring to form 1m. In contrast, reaction of this same compound with the catalyst generated from Cy2P(CH2)4PCy2 and Ru (DMSO)4(OTf)2 led to a selective isomerization of the allylic alcohol to the corresponding ketone 1q. Genipin underwent oxidation by Ru-2 at the secondary alcohol over the primary allylic alcohol to form lactone 1n directly; prior oxidation to the lactone required protection and deprotection of the primary allylic alcohol11.
Figure 2c shows oxidations that were conducted with ketones that have higher oxidation potentials than that of acetone. Trifluoroacetophenone can be used in stoichiometric quantities as the oxidant in various solvents in place of acetone when a more strongly oxidizing acceptor is required. For example, selective lactonization of the diterpenoid lagochiline to give the natural product lagochirsine (1o) occurred in a 40% yield with 2 equiv. trifluoroacetophenone in a dioxane solvent and Ru-2 as the catalyst, whereas the reaction with acetone as the oxidant gave an acetonide product derived from the 1,3-diol moiety. The lactonization occurred after dehydrogenation of the relatively exposed primary alcohol, driven by the formation of a five-membered ring. Previously, the oxidation of lagochiline formed products from the oxidation of multiple hydroxyl groups12. Also, trifluoroacetophenone in excess quantities can be used to increase the rate of some slow and thermodynamically less favourable oxidations. For example, the oxidation of ivermectin with acetone as the hydrogen acceptor occurred in a <10% yield, but the same reaction with 20 equiv. tri-fluoroacetophenone in dioxane formed ketone 1p in a 54% isolated yield. The other secondary alcohol in this structure bears multiple oxygen atoms on adjacent carbon atoms, so it is unreactive to our system.
Selective transformations of the keto derivatives of polyol natural products
The catalyst we discovered for the selective generation of a ketone unit in these natural products provides the potential to create derivatives of these natural products from modification at a single site. For example, we sought to use the ketone to incorporate nitrogen-containing functionality by reductive amination. However, catalytic aminations of hindered alcohols in complex polyols have not been reported, and the array of functionality in most natural products would be unlikely to tolerate the reaction temperatures, oxidants and bases used in the catalytic oxidation and amination with published catalytic systems. Therefore, we sought to identify simple catalytic conditions for this process. We also sought to modify the ring systems by insertions of nitrene units by a Beckmann rearrangement to form lactams. However, the lack of Beckmann rearrangements of such hindered and functionalized structures necessitated the development of the conditions for this process.
Figure 4 shows the formation of products that contain nitrogen-based functionality from selected examples of the ketones formed by our alcohol dehydrogenation. To demonstrate the potential for the oxidation and amination to occur on complex structures, we studied reactions of 3-keto-andrographolide. The arylamine derivative 2f was prepared from ketone 2e by retro-aldol cleavage of the hydroxy-methyl group, followed by reductive amination with the iridium catalyst Cp*IrCl(N-(4-dimethylaminophenyl)-2-pyridylcarboxamide) (Ir-1)13 (Cp*, pentamethyl-cyclopentadienyl) and arylammonium formate. Classical reductive amination conditions with 2e gave diastereomeric mixtures of the amination product or side reactions at the enoate functionality, which included isomerization, reduction or conjugate addition. The seven-membered lactam 2d was prepared by reaction of the ketone with NH2OSO3H in trifluoroethanol (TFE) (aqueous (aq.)). This process is driven by the excellent leaving-group ability of sulfate in aqueous trifluoroethanol, which dissolves nonpolar substrates. In other solvents, the oxime was obtained as the main product. Again, more classical methods that involve the initial formation of the oxime gave isomerization of the enoate functionality, elimination of the hydroxy group on the five-membered ring and other decomposition reactions. Yet, the initial formation of the oxime did allow us to isolate a product 2c that contained a fused isoxazole group after the formation of an oxime and subsequent treatment with TsCl.
Figure 4. Oxidation and amination of structurally complex polyol natural products.

a, The incorporation of nitrogen-based functional groups into andrographolide occurs by the formation of oxime derivatives and catalytic reductive amination. (i) NH2OH–HCl, pyridine, 40 °C, 4 h; (ii) TsCl, NEt3, THF, 40 °C, 2 d; (iii) NH2OSO3H, 1:1 0.01% TFA:TFE, 50 °C, 24 h; (iv) NH2OSO3H, 1:1 2.5% NaHCO3(aq):TFE, 50 °C, 40 min, then 80 °C, 60 min; (v) 2SmI2, methyl acrylate, 4:1 THF:TFE, 65 °C, 25 min; (vi) 3% Ir-1, C8H9NH2, HCO2H, MeOH, 65 °C. b, The incorporation of nitrogen-based functional groups into fusidic acid methyl ester is achieved by a direct conversion of the ketone formed by oxidation into the corresponding lactam and by catalytic and diastereoselective reductive amination of this ketone to the free amine. (vii) NH2OSO3H, 1:1 H2O:hexafluoroisopropanol, 50 °C, 20 min, then 80 °C, 30 min, then 1:1 H2O:Me2CO, 80 °C, 10 min; (viii) 2.5% Ir-1, NH4O2CH, MeOH, 65 °C, 4 h. c, The incorporation of nitrogen-based functional groups into D-glucal is achieved by catalytic and diastereoselective reductive amination. The conjugation of two natural products was achieved by this approach. (ix) 2.5% Ir-1, C5H4F2NH3O2CH, MeOH, 65 °C, 10 h; (x) Ir-1, lithocholic amine, HCO2H, MeOH, 65 °C, 20 h. d, The incorporation of nitrogen-based functional groups into oestriol is achieved by a one-step catalytic conversion of the hydroxyketone to give the corresponding lactam or by the combination of Baeyer–Villiger oxidation and reductive amination to yield N-substituted lactams. (xi) 3% Pt-1, H2O2(aq), THF, r.t., 2 d or 3% Pt-1, H2O2(aq), THF, 45 °C, 6 h (77% yield); (xii) R = H, 2.5% Ir-1, NH4O2CH, MeOH, 65 °C, 4 h; R = Ph, 2.5% Ir-1, PhNH3O2CH, MeOH, 12 h; (xiii) 2.5% Ir-1, NH4O2CH, MeOH, 65 °C, 4 h.
This sequence of oxidation and amination was applied to several additional natural products. Figure 4b shows the reaction of keto-fusidic acid methyl ester with NH2OSO3H to form the seven-membered lactams 2h and 2i directly. Reaction of the same ketone with ammonium formate catalysed by Ir-1 led to a diastereoselective reductive amination to form the free primary amine 2j. Figure 4c shows that the reductive amination of the dehydrogenation product of D-glucal with the ammonium salt of 2,6-difluoroaniline, as well as with the amine derivative of lithocholic acid, catalysed by Ir-1 gave diastereomerically pure amine products 2l and 2m in one step from the concomitant reduction of the carbon–carbon double bond and reductive amination of the ketone.
In addition to these reactions that involve a single ketone formed by alcohol oxidation, we developed a route from complex vicinal diols to lactams. As shown in Fig. 4d, the hydroxyketone derived from the oxidation of oestriol was directly converted into lactam 2p in a redox-neutral reaction under reductive amination conditions with Ir-1 and ammonium formate. As an alternative, stepwise oxidation and amination of hydroxyketone 1j allowed the parallel synthesis of multiple lactams. Baeyer–Villiger oxidation of the keto-oestriol occurred with H2O2 as the oxidant and [Pt(dppb) (OH)]2[BF4]2 (Pt-1)14 (dppb, 1,4-bis(diphenylphosphino)butyl) as the catalyst to generate acetal 2o in an 84% yield. Treatment of the acetal product with Ir-1 and ammonium formate salts (NH3RO2CH for R = H, Ph) led to N-substituted lactam derivatives 2p and 2q in 78 and 77% yields, respectively.
Epimerization of alcohol units in polyol natural products
Our system not only catalyses the transfer of hydrogen from hindered alcohols to acetone with isopropanol as the by-product, but it also catalyses the reverse transfer of hydrogens from isopropanol to hindered ketones, such as pinacolone (Fig. 1d). This result suggested the potential to epimerize a single hydroxy group in complex polyols with a single catalyst. For example, the ketone generated from the dehydrogenation of fusidic acid methyl ester was reduced by catalytic Ru-2 and isopropanol to give 3-epi-fusidic acid methyl ester (4a) in an 80% yield, with the remaining material identified as the parent fusidic acid methyl ester (Fig. 5a). Similarly, ouabain, which was oxidized selectively with acetone, was reduced with isopropanol catalysed by Ru-2 to form 1-epi-ouabain (4b) in 90% yield. With other polyols, such as D-glucal and andrographolide, reduction of the ketone regenerated the starting natural product.
Figure 5. The reduction of complex ketones and one-step epimerization of complex alcohols.

a, Reduction of complex ketones with isopropanol enables site-selective epimerization within polyol natural products. b, Site-selective epimerization of complex polyols is achieved in one step with Ru-2 as the catalyst in the absence of an acceptor. S.M., starting material. NMM; N-methylmorpholine.
A one-step epimerization also occurred in some cases. For example, the site-selective epimerization of fusidic acid methyl ester at the C3 position catalysed by Ru-2 occurred in an 84% yield, whereas that of ouabain occurred at the C1 position in a 60% yield (Fig. 5b and Supplementary Section 4). Although Shvo’s catalyst, which is widely used for dynamic kinetic resolutions15, also catalysed the epimerization of fusidic acid methyl ester at 70 °C to give 84% of the C3 epimer, it did not react with ouabain up to the decomposition temperature of 100 °C. The combination of (Ph5Cp)Ru(CO)Cl and KOtBu, which also has been used to epimerize simple alcohols16,17, did not react with either of these molecules (Supplementary Section 4).
Comparison with alternative methods for alcohol oxidation applied to polyol natural products
Although the catalytic oxidation of alcohols has been the subject of much research, few studies targeted the oxidation of complex polyols. Instead, most studies focused on the reactions of unhindered benzylic and allylic alcohols, linear alkanols or cyclohexanols18–22. In the most recent and arguably most impressive example, a series of polyglycosides were oxidized by Waymouth’s catalyst in 30–60% yields at the terminal sugar23. Prior to this study, the polyketide antibiotic mupirocin methyl ester was oxidized by the combination of TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl), NaOCl and KBr to give only a 31% yield of a monooxidation product24, and the labdane diterpene forskolin was oxidized by stoichiometric CrO3/pyridine in an 80% yield25, whereas another labdane diterpene, andrographolide, was oxidized to the 3-ketone in only a 5% yield under the optimized conditions26. These studies illustrate that the conditions developed for the reaction of one structure do not translate into the oxidation of another structure of the same or different class, and some molecules have resisted selective oxidation under all known conditions27–29.
To assess in more detail the suitability of known procedures with classical stoichiometric reagents and modern catalysts for the selective oxidation of a broader range of polyols, we conducted reactions on several representative densely functionalized structures shown in Fig. 3: andrographolide, ouabain, mupirocin methyl ester and kirenol (Supplementary Section 5). Each of these four natural products gave mixtures of products with classical reagents, and no conditions gave a significant yield of the ketone product obtained from the reaction with Ru-1 or Ru-2 as the catalyst and acetone as the hydrogen acceptor. The reactions we conducted with simple published substrates occurred as described with both classical reagents. However, the reactions under these conditions with the four selected natural products gave complex mixtures. Andrographolide reacted with Dess Martin periodinane (DMP) to oxidize the primary alcohol over the secondary alcohols to generate a mixture of 49% aldehyde, 35% keto-aldehyde and only 3% of the ketone. N-bromosuccinimide in acetone, tested because of the selective oxidation of simple bile acids28, gave a complex mixture. Swern oxidation converted 65% of andrographolide into an unknown structure that lacks a ketone or aldehyde, and Oppenhauer conditions with trifluoroacetone catalysed by AlEt2OEt gave little to no conversion. The oxidation of mupirocin, ouabain methyl ester and kirenol also gave mixtures of products with these oxidation systems. The reaction of mupirocin with DMP gave an unselective oxidation to a complex mixture of products (at least five), the 1H NMR spectrum of which contained multiple new olefin resonances that signalled elimination processes. The reaction of the same substrate with AlEt2OEt/trifluoroacetone or the Swern reagent formed a mixture of many products with a low selectivity. The oxidation of ouabain with DMP formed one or more aldehyde products in 45% yield from the oxidation of the primary alcohol, along with multiple additional products. The Swern reagent and AlEt2OEt/trifluoroacetone gave full, unselective conversion into multiple products that bear olefins, as determined by 1H NMR spectroscopy. The reaction of kirenol with DMP gave decomposition to a mixture of several products, two of which contain aldehyde functionality, whereas reaction with AlEt2OEt/trifluoroacetone gave full decomposition to multiple products that bear new olefins.
The ability of modern oxidation and alternative transfer-dehydrogenation catalysts to oxidize andrographolide, mupirocin methyl ester and ouabain selectively was also assessed. The reaction of andrographolide with benzoquinone and catalytic [Pd(neocuproine)(OAc)]2[OTf]2 (ref. 30), which was reported to oxidize secondary over primary alcohols in 1,2- and 1,3-diol units, gave products that were similar to those of the reaction of DMP (65% aldehyde, 30% keto-aldehyde and no 3-ketone). The attempted oxidation of the same substrate with Ru-MACHO and KOH led to a low conversion. Shvo’s dimer oxidized andrographolide under optimized conditions to give a 43% yield of the 3-ketone, along with three other products in 32, 19 and 4% yields derived from the oxidation of the primary alcohol with a concomitant side reaction, loss of the hydroxyenoate functionality and loss of the 1,1-disubstituted olefin, respectively. Ru-MACHO and KOH led to decomposition of mupirocin methyl ester. Oxidation of mupirocin methyl ester by Shvo’s dimer occurred concomitantly with multiple side reactions, such that little product from the selective oxidation of the alcohol to the ketone occurred. Waymouth’s catalyst gave a low conversion of ouabain into multiple products, whereas the combination of Ru-MACHO and KOH led to the decomposition of ouabain without oxidation. Shvo’s dimer led to no conversion of ouabain up to temperatures at which autodecomposition occurs.
Mechanistic analysis of kinetic versus thermodynamic origins of the selectivity
The mechanism of ruthenium-catalysed transfer dehydrogenations is generally thought to begin by displacement of one of the labile ligands with alcohol and the generation of a ruthenium alkoxide7,31. When a tertiary amine is included in the reactions, the generation of the alkoxide could occur by deprotonation of the bound alcohol by an amine. In TFE solvent, no base is needed for the alcohol oxidation to proceed (Fig. 1b), and the generation of the alkoxide could occur by elimination of HOTf stabilized by a substrate alcohol. β-hydrogen elimination from the alkoxide would then generate the ketone and a ruthenium hydride. The resulting hydride would insert acetone to form an isopropoxide complex that undergoes protonolysis to initiate a second cycle.
To examine whether the observed selectivity for the oxidation of secondary over primary alcohols results from a kinetic selectivity or whether it results from a thermodynamic selectivity because of the reversible oxidation of the primary alcohol and irreversible oxidation of the secondary alcohol, we conducted a deuterium labelling study (Supplementary Section 7). The dehydrogenations of andrographolide, oestriol, ouabain and fusidic acid methyl ester were conducted with 5 equiv. isopropanol-d8 added as the source of deuteride. No deuterium was incorporated into any positions of the product molecules. An analogous experiment was conducted with a combination of cyclohexanol and 2-methylpentanol in acetone in the presence of 5 equiv. isopropanol-d8 (Supplementary Section 7). Under these conditions, no oxidation of the primary alcohol was observed, and no deuterium was incorporated into the position alpha to oxygen in the primary alcohol that would signal reversible oxidation. Only the oxidation of cyclohexanol was observed, and just 1–2% deuterium was incorporated into the position alpha to oxygen in the unreacted cyclohexanol. These results indicate that, with the catalyst we report, the presence of excess acceptor leads to a kinetic selectivity for the oxidation of alcohols in both complex and simple substrates, even a specific hindered secondary alcohol over a primary alcohol.
Conclusion
The selective oxidation of one secondary alcohol among many can be used strategically in a variety of synthetic contexts to alleviate the need for protective groups to dictate site selectivity. The present catalyst addresses this challenge and provides a foundation for further advancements in hydrogen-transfer reactions involving X–H bonds. Alcohols are the most common functional group in natural products because of the selective hydroxylation of C–H bonds by P450 enzymes, whereas amino groups are less common because of the lack of enzymes for a direct amination. Yet, nitrogen-based functionality can impart favourable properties for biological activity. The catalytic reactions that we have reported create new complex architectures that could provide new leads for medicine, molecular biology and agroscience. The ability to epimerize precisely the site of an OH group, to convert an OH group into an NH group or to incorporate nitrogen into the ring system or chain provides the ability to combine nature’s spectacular synthetic prowess to create complex architectures with modern catalytic synthetic chemistry as a means to create unnatural products with enhanced physical properties and function over those provided solely from biosynthetic pathways.
Methods
General procedure for alcohol oxidation
Under N2, equimolar Ru-2 and N-methylmorpholine (NMM) were combined in acetone or TFE to form a solution. This solution was then added to a solution of the alcohol starting material in acetone or a mixture of acetone and TFE in a vial, along with a magnetic stir bar if the starting alcohol did not fully dissolve at room temperature. Next, the vial was sealed and heated at 65 °C for several hours, with stirring if applicable. The reaction was then cooled to room temperature and evaporated to dryness. Finally, the residue was purified by recrystallization or column chromatography on silica gel.
Data availability
The data reported in this paper are available in the Supplementary Materials. Crystallographic data for the structures reported in this paper are deposited at the Cambridge Crystallographic Data Centre (CCDC) under the deposition numbers CCDC 1556838 (Ru-3-Cl), CCDC 1556837 (Ru-2-DABIII-alkoxide), CCDC 1556836 (Ru-2), CCDC 1556835 (2e), CCDC 1556834 (2d), CCDC 1556833 (2c) and CCDC 1556613 (1l). Copies of these data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif.
Supplementary Material
Acknowledgments
We thank the NIH (GM-55382) and the Berkeley Center for Green Chemistry Systems Approach to Green Energy Integrated Graduate Education and Research Traineeship for financial support. We acknowledge A. DiPasquale for assistance with X-ray crystallographic characterizations and the NIH (SIG S10-RR027172) for facility funding.
Footnotes
Author contributions
C.K.H. and J.F.H. conceived and designed the experiments. C.K.H. performed the experiments. C.K.H. and J.F.H. analysed the data. C.K.H. and J.F.H. co-wrote the paper.
Additional information
Supplementary information and chemical compound information are available in the online version of the paper. Reprints and permissions information is available online at www.nature.com/reprints. Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Correspondence and requests for materials should be addressed to J.F.H.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Lewis CA, Miller SJ. Site-selective derivatization and remodeling of erythromycin A by using simple peptide-based chiral catalysts. Angew Chem Int Ed. 2006;45:5616–5619. doi: 10.1002/anie.200601490. [DOI] [PubMed] [Google Scholar]
- 2.Sun X, Lee H, Lee S, Tan KL. Catalyst recognition of cis-1,2-diols enables site-selective functionalization of complex molecules. Nat Chem. 2013;5:790–795. doi: 10.1038/nchem.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peddibhotla S, Dang Y, Liu JO, Romo D. Simultaneous arming and structure/activity studies of natural products employing O–H insertions: an expedient and versatile strategy for natural products-based chemical genetics. J Am Chem Soc. 2007;129:12222–12231. doi: 10.1021/ja0733686. [DOI] [PubMed] [Google Scholar]
- 4.Dobereiner GE, Crabtree RH. Dehydrogenation as a substrate-activating strategy in homogeneous transition-metal catalysis. Chem Rev. 2010;110:681–703. doi: 10.1021/cr900202j. [DOI] [PubMed] [Google Scholar]
- 5.Noyori R, Hashiguchi S. Asymmetric transfer hydrogenation catalyzed by chiral ruthenium complexes. Acc Chem Res. 1997;30:97–102. [Google Scholar]
- 6.Shvo Y, Czarkie D, Rahamim Y. A new group of ruthenium complexes: structure and catalysis. J Am Chem Soc. 1986;108:7400–7402. [Google Scholar]
- 7.Almeida MLS, Beller M, Wang G, Backvall J. Ruthenium(II)-catalyzed Oppenauer-type oxidation of secondary alcohols. Chem-Eur J. 1996:1533–1536. [Google Scholar]
- 8.Oldenhuis NJ, Dong VM, Guan Z. From racemic alcohols to enantiopure amines: Ru-catalyzed diastereoselective amination. J Am Chem Soc. 2014;136:12548–12551. doi: 10.1021/ja5058482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiang Y, Zhang H, Fan CQ, Yue JM. Novel diterpenoids and diterpenoid glycosides from Siegesbeckia orientalis. J Nat Prod. 2004;67:1517–1521. doi: 10.1021/np0400407. [DOI] [PubMed] [Google Scholar]
- 10.Arnone A, et al. Microbial transformation of 10-deacetylbaccatin III (10-DAB) by Curvularia lunata and Trametes hirsuta. J Mol Catal B. 2006;42:95–98. [Google Scholar]
- 11.Luo J, et al. Synthesis of stable genipin derivatives and studies of their neuroprotective activity in PC12 cells. ChemMedChem. 2012;7:1661–1668. doi: 10.1002/cmdc.201200258. [DOI] [PubMed] [Google Scholar]
- 12.Safaev MA, Zainutdinov UN, Aslanov KA. Continuous method for obtaining lagochirzen. Chem Nat Compd. 1995;31:145. [Google Scholar]
- 13.Watanabe M, Tanaka K, Miki T, Murata K. Process for preparing amine compound. 2012/0065426 A1. US patent. 2012
- 14.Gavagnin R, Cataldo M, Pinna F, Strukul G. Diphosphine–palladium and –platinum complexes as catalysts for the Baeyer–Villiger oxidation of ketones: effect of the diphosphine, oxidation of acyclic ketones, and mechanistic studies. Organometallics. 1998;17:661–667. [Google Scholar]
- 15.Pàmies O, Bäckvall JE. Combination of enzymes and metal catalysts. A powerful approach in asymmetric catalysis. Chem Rev. 2003;103:3247–3261. doi: 10.1021/cr020029g. [DOI] [PubMed] [Google Scholar]
- 16.Warner MC, Backvall JE. Mechanistic aspects on cyclopentadienylruthenium complexes in catalytic racemization of alcohols. Acc Chem Res. 2013;46:2545–2555. doi: 10.1021/ar400038g. [DOI] [PubMed] [Google Scholar]
- 17.Långvik O, Mavrynsky D, Leino R. Selective ruthenium-catalyzed epimerization of chiral sec-alcohols. Catal Today. 2015;241:255–259. [Google Scholar]
- 18.Mallat T, Baiker A. Oxidation of alcohols with molecular oxygen on solid catalysts. Chem Rev. 2004;104:3037–3058. doi: 10.1021/cr0200116. [DOI] [PubMed] [Google Scholar]
- 19.Kwon MS, et al. Palladium nanoparticles entrapped in aluminum hydroxide: dual catalyst for alkene hydrogenation and aerobic alcohol oxidation. Org Lett. 2005;7:1077–1079. doi: 10.1021/ol047381w. [DOI] [PubMed] [Google Scholar]
- 20.Steves JE, Stahl SS. Stable TEMPO and ABNO catalyst solutions for user-friendly (bpy)Cu/nitroxyl-catalyzed aerobic alcohol oxidation. J Org Chem. 2015;80:11184–11188. doi: 10.1021/acs.joc.5b01950. [DOI] [PubMed] [Google Scholar]
- 21.Kim WH, Park IS, Park J. Acceptor-free alcohol dehydrogenation by recyclable ruthenium catalyst. Org Lett. 2006;8:2543–2545. doi: 10.1021/ol060750z. [DOI] [PubMed] [Google Scholar]
- 22.Yi CS, Zeczycki TN, Guzei IA. Highly cooperative tetrametallic ruthenium-μ-oxo-μ-hydroxo catalyst for the alcohol oxidation reaction. Organometallics. 2006;25:1047–1051. doi: 10.1021/om0510674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eisink NNHM, Lohse J, Witte MD, Minnaard AJ. Regioselective oxidation of unprotected 1,4 linked glucans. Org Biomol Chem. 2016;14:4859–4864. doi: 10.1039/c6ob00608f. [DOI] [PubMed] [Google Scholar]
- 24.Scott RW, et al. Mupirocin F: structure elucidation, synthesis and rearrangements. Tetrahedron. 2011;67:5098–5106. [Google Scholar]
- 25.Bhat SV, Bajwa BS, Dornauer H, de Souza NJ. Reactions of forskolin, a biologically active diterpenoid from coleus forskohlii. J Chem Soc Perkin Trans. 1982;1:767–771. [Google Scholar]
- 26.Mathad VT, Kumar S, Raj K. Oxidation studies on andrographolide. Nat Prod Res. 2006;20:1053–1058. doi: 10.1080/14786410500441466. [DOI] [PubMed] [Google Scholar]
- 27.Hayashi M, Yamada K, Nakayama S. Dehydrogenation of D-glycals by palladium supported on activated charcoal under ethylene atmosphere: synthesis of 1,5-anhydrohex-1-en-3-uloses. Synthesis. 1999:1869–1871. [Google Scholar]
- 28.Fieser LF, Rajagopalan S. Selective oxidation with N-bromosuccinimide. I. cholic acid. J Am Chem Soc. 1949;71:3935–3938. [Google Scholar]
- 29.Welankiwar SS, Murphy WS. Stereoselective oxidation of fusidic acid derivatives. J Chem Soc Perkin Trans 1. 1976:710–712. [Google Scholar]
- 30.Chung K, et al. Chemoselective Pd-catalyzed oxidation of polyols: synthetic scope and mechanistic studies. J Am Chem Soc. 2013;135:7593–7602. doi: 10.1021/ja4008694. [DOI] [PubMed] [Google Scholar]
- 31.Yi CS, He Z, Guzei IA. Transfer hydrogenation of carbonyl compounds catalyzed by a ruthenium-acetamido complex: evidence for a stepwise hydrogen transfer mechanism. Organometallics. 2001;20:3641–3643. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data reported in this paper are available in the Supplementary Materials. Crystallographic data for the structures reported in this paper are deposited at the Cambridge Crystallographic Data Centre (CCDC) under the deposition numbers CCDC 1556838 (Ru-3-Cl), CCDC 1556837 (Ru-2-DABIII-alkoxide), CCDC 1556836 (Ru-2), CCDC 1556835 (2e), CCDC 1556834 (2d), CCDC 1556833 (2c) and CCDC 1556613 (1l). Copies of these data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif.