

SUMMARY
DNA double-strand breaks (DSBs) occurring within fragile zones of less than 200 base pairs account for the formation of the most common human chromosomal translocations in lymphoid malignancies, yet the mechanism of how breaks occur remains unknown. Here, we have transferred human fragile zones into S. cerevisiae in the context of a genetic assay to understand the mechanism leading to DSBs at these sites. Our findings indicate that a combination of factors is required to sensitize these regions. Foremost, DNA strand separation by transcription or increased torsional stress can expose these DNA regions to damage from either the expression of human AID or increased oxidative stress. This damage causes DNA lesions that, if not repaired quickly, are prone to nuclease cleavage resulting in DSBs. Our results provide mechanistic insight into why human neoplastic translocation fragile DNA sequences are more prone to enzymes or agents that cause longer-lived DNA lesions.
Keywords: human lymphoma chromosomal translocation fragile zones, S. cerevisiae, DNA double-strand break, non-B DNA, transcription, oxidative stress, torsional stress, Artemis, activation-induced deaminase (AID), slipped strand DNA
eTOC Blurb
Pannunzio and Lieber insert mammalian DNA fragile zones into an S. cerevisiae genetic assay and explore how distortion of the DNA duplex combined with ssDNA damaging agents target these zones to create double-strand DNA breaks. This work has implications for formation of the recurrent chromosomal translocations of human lymphoid neoplasms.
INTRODUCTION
Many reciprocal chromosomal translocations that occur in early human B cells involve one DNA double-strand break (DSB) generated by the physiological process of V(D)J recombination at the immunoglobulin heavy chain (IGH) locus, and a second pathological DSB at a non-IGH locus by a different mechanism that has yet to be elucidated (Lieber, 2016). Two non-IGH loci commonly involved in these translocations are CCND1 on chromosome 11 and BCL2 on chromosome 18. Selection is one reason for these oncogenes being involved in recurrent translocations as the dysregulation of each provides cells with a growth advantage. However, after mapping the translocation breakpoints in human patients, it has long been clear that the location of DSBs is not random with many occurring within highly defined zones of less than 200 bp.
DSBs leading to translocations involving CCND1 can occur anywhere within a 344 kb region. Strikingly, 30% of the breaks in patients occur approximately 110 kb away from CCND1 in a 150 bp region known as the BCL1 major translocation cluster (MTC). Similarly, breaks at BCL2 have been mapped to a 31 kb region, but a striking 50% of breaks are located within a 175 bp portion of the 3′ UTR known as the BCL2 major breakpoint region (MBR). This indicates that the MTC and the MBR have a 1000- and 200-fold higher breakage frequency, respectively, relative to the surrounding DNA. We refer to the MTC and the MBR as DNA fragile zones as they appear to be naturally occurring sites of higher frequency DSB formation. Importantly, these regions are distinct from regions termed “common fragile sites” that only become fragile when replication inhibitors (e.g., aphidicolin) are added (Durkin and Glover, 2007).
While a mechanism for DSB formation at fragile zones is still lacking, evidence suggests that the breaks at the MBR and MTC are initiated by the activity of the B cell-specific mutator, activation-induced cytidine deaminase (AID) (Cui et al., 2013; Tsai et al., 2008). Breaks at both fragile zones occur within motifs that are a preferred target for AID (Pham et al., 2003; Yu et al., 2004). Peak AID expression occurs in mature B cells to facilitate somatic hypermutation and class switch recombination, but lower levels of AID expression have been detected in early B cells when V(D)J recombination occurs (Swaminathan et al., 2015). As AID only acts on single-stranded DNA (ssDNA) (Bransteitter et al., 2003), an important question that needs to be addressed is: How does ssDNA form at the fragile zones? A strong possibility is that the MBR and MTC are targeted by AID because of the transient ssDNA character they exhibit, evidenced by the fact that both sequences are sensitive to chemical probing with sodium bisulfite under native (non-denaturing) conditions (Tsai et al., 2009). While we have characterized aspects of the fragile zones in vitro and in human B cells (Cui et al., 2013; Lu et al., 2015; Tsai et al., 2009), transferring these DNA sequences to the genetically tractable S. cerevisiae system is a powerful approach to determine whether this transient ssDNA character is a source of fragility.
We have constructed a yeast genetic assay to examine DSB events in inserted mammalian fragile zones. In addition to testing the sequences themselves, we also examine the effects of transcription, torsional stress, and susceptibility to damage from AID or reactive oxygen species (ROS). Lastly, we test the effect of expression of a constitutively active form of the nuclease Artemis, which has been implicated in DSBs at fragile zones (Cui et al., 2013). We have found that the action of transcription or an increase in torsional stress leads to the formation of transient regions of ssDNA within fragile zones. This ssDNA becomes a target for either AID or ROS, creating DNA lesions that, when left unrepaired, result in DSBs due to active nucleases operating within the cell. This demonstrates that a combination of DNA sequence, ssDNA formation, and long-lived DNA lesions make these human neoplastic translocation zones fragile.
RESULTS
Experimental System for Analysis of DNA Sequence Fragility
Studying the human fragile zones required us to build a yeast genetic assay that could detect DNA DSBs occurring within a defined region of a chromosome. The basic concept for such an assay is shown in Figure 1A and consists of four selectable markers on one non-essential chromosome arm of a haploid yeast cell. Three selectable markers are positioned on the telomeric side of fragile sequences with a fourth marker positioned on the centromeric side. DSBs within fragile zones are detected by selecting for loss of markers 1, 2, and 3 and retention of marker 4. Since these events are the result of a DSB within a defined zone, we refer to them as zonal break events (ZBEs). Based on known mutation rates (Navarro et al., 2007), independent inactivation of markers 1, 2, and 3 would occur in approximately 1 in 1 ×1021 cells per generation. Therefore, each marker increases the sensitivity of the assay by orders of magnitude and ensures that ZBE events are most likely due to loss of the chromosome arm after an unrepaired DSB.
Figure 1. Genetic Assay for Zonal Break Events (ZBEs) Demonstrates Instability Caused by Transcription.

(A) Conceptual design for ZBE assay consisting of four selectable markers. A DSB within the inserted human fragile zone leads to loss of selectable markers 1, 2, and 3 yet retention of selectable marker 4.
(B) The ZBE assay was constructed on chromosome I in S. cerevisiae by inserting the selectable markers KANMX4, URA3, CAN1, and LYS2. The first essential gene on the left arm of chromosome I is PTA1 (several genes have been omitted with the intervening space represented by double slanted lines). Fragile zones are inserted between the CAN1 and LYS2 markers (highlighted in red) where DSBs that result in the loss of the left arm of chromosome I will confer a phenotype of lysine prototrophy (Lys+), 5-fluoroorotic acid resistance (5-FOAR), canavanine resistance (CanR), and G418 sensitivity (G418S). An additional version of the assay contains the GAL1 promoter. The relevant phenotypes for the parental (top) and recombined (bottom) variants and shown in the green and red boxes, respectively. The solid circles and triangles represent the centromere and telomeres, respectively. The dotted line and triangle represent a rebuilt telomere that would form to allow the recombined chromosome to remain mitotically stable.
(C) The ZBE rate with no GAL1 promoter (− transcription) or with the GAL1 promoter (+ transcription). Each rate was calculated from 20 individual trials.
(D) Three mammalian sequences were inserted into the spacer region of the ZBE assay: A 602 bp fragment carrying 12 repeat units from the murine Sγ3 class switch region, the 150 bp human BCL1 major translocation cluster (MTC) fragile site with flanking sequence, and the 175 bp BCL2 major breakpoint region (MBR) fragile site with flanking sequence. Each substrate is inserted downstream of the CAN1 terminator. The size of each fragment is indicated and is consistent throughout. For the MTC and the MBR, the blue box represents the fragile zone and the orange the sequence flanking the fragile zone in human cells. Each rate was calculated from 20 individual trials.
(E) Each substrate is inserted downstream of a GAL1 promoter (black arrow). The p-value indicated compares each substrate to the rate of the GAL1 promoter alone (Figure 1B, + transcription) and was calculated by performing a Mann-Whitney test to compare rates obtained between two data sets. Each rate was calculated from 20 individual trials except for MBR in the forward orientation which used 40 individual trials.
For experiments in (C) to (E), all cells were grown with galactose as the carbon source. The ZBE rate (x10−10) is indicated. Rates were determined by either Luria-Delbrück fluctuation analysis or the Lea and Coulson method of the median. Valves indicated with “<“ are determined by Luria-Delbrück fluctuation analysis and are a theoretical value based on at least one trial within a set yielding ZBEs.
In order to construct the unique ZBE assay (Figure 1B), we have refined the haploid genetic assay from our previous study (Pannunzio and Lieber, 2016). The assay relies on the loss of the URA3, CAN1, and KAN-MX4 markers from the left arm of S. cerevisiae chromosome I, and retention of a LYS2 marker to ensure breaks detected are occurring on the telomeric-side of LYS2 and not in the 25 kb region between LYS2 and the first essential gene on chromosome I, PTA1. Therefore, by screening the Lys+, 5-FOAR, CanR colonies for G418 sensitivity, we can determine the ZBE rate for DNA sequences inserted between the CAN1 and LYS2 genes. The effect of transcription was measured by inserting the GAL1 promoter into this region upstream of the inserted mammalian sequences. We have previously shown that the ectopic promoter leads to stable transcription upon galactose induction (Pannunzio and Lieber, 2016). For consistency, all strains, both those with and without the GAL1 promoter, were grown in YPG (1% yeast extract, 2% peptone, and 2% galactose).
The ZBE Assay Measures the Destabilizing Effect of Transcription on Mammalian Fragile Zones
For the substrate with no GAL1 promoter (i.e., no transcription) and no fragile zone, the ZBE rate was determined to be <0.5 ×10−10 (Figure 1C). This demonstrates the high sensitivity and low background of the ZBE assay and is consistent with previously reported spontaneous recombination rates from various constructs on different chromosomes (Chan and Kolodner, 2011; Meyer and Bailis, 2007; Yadav et al., 2014). Addition of the GAL1 promoter increases the ZBE rate at least 3.6-fold to 1.8 ×10−10 (Figure 1C), confirming previous reports that transcription stimulates recombination (Kim and Jinks-Robertson, 2011; Thomas and Rothstein, 1989).
A known site of DSB formation in the mammalian genome is the IGH locus where, in mature B cells, the DSBs required for class switch recombination (CSR) arise due to the formation of R-loops within transcribed switch sequences where the G-rich, nascent RNA strand anneals to the template DNA strand to create an RNA:DNA hybrid, displacing the non-template DNA strand to create a significant length of ssDNA (Yu et al., 2003). This ssDNA becomes an enzymatic target in a process that results in DSBs (Bransteitter et al., 2003; Shinkura et al., 2003; Yu and Lieber, 2003). Switch sequences are unstable in yeast (Kim and Jinks-Robertson, 2011) and can serve as a positive control in our assay, demonstrating that ssDNA can increase the ZBE rate. The murine Sγ3 region demonstrates particularly strong R-loop formation (Daniels and Lieber, 1995; Roy et al., 2010; Yu et al., 2005). Therefore, we have inserted 12 Sγ3 repeat units into the assay immediately downstream of either the CAN1 terminator or the ectopic GAL1 promoter to test the stability of the sequence with and without transcription.
With no GAL1 promoter present the ZBE rate was again below detection at <0.4 ×10−10 (Figure 1D). In marked contrast, the presence of the GAL1 promoter increased the ZBE rate at least 40-fold to 16 ×10−10 (Figure 1E). This is a synergistic and significant increase of over 8-fold compared to the rate measured with the GAL1 promoter alone (16 ×10−10 vs. 1.8 ×10−10, p=0.006), underscoring the ability of transcription to destabilize switch sequences. More importantly, these results demonstrate that our assay is sensitive enough to detect increases in ZBEs due to mammalian sequences that exhibit a non-B DNA structure.
Next, a 635 bp fragment carrying the 150 bp MTC and a 338 bp fragment carrying the 175 bp MBR were cloned from the human genome and inserted into the ZBE assay again both with and without the GAL1 promoter. As with the 12x Sγ3 substrate with no transcription, the ZBE rate measured for the MTC and the MBR (0.6 ×10−10 and <0.6 ×10−10, respectively, Figure 1D) was not significantly different from the rate with no fragile zone inserted (Figure 1C). The addition of transcription increased the rates for the MTC and the MBR to 2.6 ×10−10 and 1.2 ×10−10, respectively (Figure 1E); though, unlike with the 12x Sγ3 substrate, neither increase was significantly above the rate measured with transcription and no fragile zone (1.8 ×10−10, Figure 1C). Orientation of transcription also did not appear to be a factor, because inverting the MBR sequence failed to significantly change the recombination rate (1.4 ×10−10, p=0.5 compared to GAL1 promoter alone) (Figure 1E).
Since collisions between the transcription and replication machinery can cause genome instability (Kim et al., 2007), we confirmed that local replication origins were not affecting the assay. The insertion of the KAN-MX4 marker of the ZBE assay deletes autonomously replicating sequence 104 (ARS104), the closest active replication origin (Figure S1A) (Siow et al., 2012). A replication bubble originating here would move co-directionally with transcription from the inserted GAL1 promoter. The closest ARS on the centromeric side, ARS105, is nearly twice the distance away (Figure S1A); therefore, loss of ARS104 likely results in more replication forks moving into the assay region from the centromeric side potentiating head-on collisions with transcription from the GAL1 promoter. We found, however, that deletion of ARS104 has no significant effect (Figure S1B). Also, re-orientation of the 12x Sγ3 positive control was only slightly affected by the presence of ARS104 (Figure S1C).
Importantly, as the 12x Sγ3 and MTC regions are similar in size, the increased recombination rate measured for 12x Sγ3 is not simply due to more DNA being available at the break region of the assay and affirms that the sequence of the switch region contributes to instability, likely due to the known ability to form stable R-loops. Since transcription alone does not lead to a significant increase in DSBs for the MBR or the MTC, it does not seem likely that such structures (e.g., R-loops) are forming within these fragile zones. Additional damage to the ssDNA formed at these sequences may be necessary, and this reasoning led us to test various damaging agents known to preferentially target ssDNA.
Expressing Human AID to Target Transient ssDNA
AID deaminates cytosine (C) to uracil (U) and acts only on ssDNA (Bransteitter et al., 2003). Therefore, an effect of AID would also confirm the presence of ssDNA at the assay locus. Following deamination, U is efficiently removed by uracil-DNA glycosylase with the resulting abasic site processed by the major apurinic/apyrimidinic endonuclease, which nicks the phosphate backbone and can result in DSBs when the affected C’s are closely-spaced on opposite strands (DiNoia and Neuberger, 2007; Masani et al., 2013; Popoff et al., 1990).
Upon AID expression in the ZBE construct with no fragile zone but with transcription, the rate was 62 ×10−10 (Figure 2A), a 34-fold increase over the rate measured in Figure 1C (no fragile zone, +transcription, -AID). This reveals that the process of transcription can produce ample ssDNA on which AID can act and that this action of AID can result in DSBs. Next, we tested the effect of inserting the mammalian fragile zones into the assay region. The ssDNA of the Sγ3 R-loop and the non-B character of the fragile zones should make each vulnerable to AID action. The most significant increase caused by AID expression is on the 12x Sγ3 substrate, with the ZBE rate increasing to 143 ×10−10 (Figure 2A). This is 9-fold higher than the rate with 12x Sγ3 and no AID expression (16 ×10−10, Figure 1E) and 2-fold higher than the rate with AID expression and no fragile zone (62 × 10−10, Figure 2A), indicating an additive effect of AID.
Figure 2. Effect of AID Expression and TSA1 loss in ZBE assay.

(A) ZBE rate with expression of galactose inducible human AID from pNP171 with transcription as indicated.
(B) ZBE rate in tsa1Δ mutants with or without transcription through the indicated DNA sequence.
(C) ZBE rate in tsa1Δ mutants with AID expression and with transcription as indicated. For (A) to (C), rates were determined by either Luria-Delbrück fluctuation analysis or the Lea and Coulson method of the median from the indicated number of trials (n). Valves indicated with “<“ are determined by Luria-Delbrück fluctuation analysis and are a theoretical value based on at least 1 trial within a set yielding ZBEs. Error bars represent a 95% confidence interval and can only be calculated from rates determined by the method of the median. In some cases, more accurate p-values were determined by Mann-Whitney tests with comparisons to the “No Fragile Zone” control in each set. Unless explicitly stated: *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
The ZBE rates measured upon transcription through the MBR and the MTC were 70- and 38-fold higher with AID expression than without, respectively (Figure 2A vs Figure 1E). Both rates were increased over the rate measured in the control (no fragile zone, + transcription, +AID, Figure 2A) with the increase in MTC being significant (p=0.012) and the increase with MBR boarder-line significant (p=0.069), indicating that that the fragile zones have a higher sensitivity to AID. Remarkably, in experiments with AID expression but without transcription, the ZBE rate drops below the limit of detection (Figure 2A, Table S2). This clearly indicates that transcription is essential for providing the ssDNA substrate for AID.
Deletion of TSA1 increases the ZBE rate
Bases in duplex DNA should be less prone to attack by ROS (Halliwell and Gutteridge, 2015), but were curious if the transient ssDNA character that the fragile zones exhibit would make them more susceptible to damage upon exposure to reactive oxygen species (ROS). Since we are measuring the rate over several days of culture growth, it was not adequate to use exogenous sources of ROS, such as H2O2, as they decay over time and rate determination requires assay conditions to remain as consistent as possible. Therefore, we increased the endogenous level of ROS genetically.
TSA1 encodes the major peroxiredoxin in S. cerevisiae and TSA1 deletion mutants, while viable, exhibit increased genome instability due to the inability to process ROS (Huang et al., 2003; Iraqui et al., 2009). In tsa1Δ cells with no transcription and no mammalian fragile zone, we measured a ZBE rate of 3.5 ×10−10 (Figure 2B), an increase of at least 7-fold compared to wild-type (WT) cells with no transcription (<0.5 ×10−10 in Figure 1C). Consistent with our previous results, transcription further increased the ZBE rate. The rate measured with an active GAL1 promoter was 20 ×10−10, a significant increase of approximately 11-fold over WT cells with the active GAL1 promoter (compared to 1.8 ×10−10 in Figure 1C, p=0.004) and 5.7-fold higher than the tsa1Δ cells with no promoter (compared to 3.5 ×10−10 in Figure 2B, p=0.006).
Addition of the MBR or the MTC downstream of the GAL1 promoter resulted in ZBE rates of 23 ×10−10 and 13 ×10−10, respectively (Figure 2B). Even though both rates are significantly higher than the rate measured in WT cells with the fragile zones present (see Figure 1E), they are not significantly different than the rate of 20 ×10−10 measured in the control (no fragile zone, +transcription, tsa1Δ, Figure 2B). The significant 3-fold increase conferred by tsa1Δ for the transcribed 12x Sγ3 substrate (49 ×10−10, Figure 2B) does not exceed the 9-fold increase between GAL1 promotor alone versus GAL1 promoter with 12x Sγ3 measured in wild-type cells (1.8 ×10−10 vs. 16 ×10−10 in Figures 1C and 1E).
Interestingly, we found that expressing AID in a tsa1Δ mutant substantially increases the ZBE rate (Figure 2C). With transcription and no fragile zone, the ZBE rate was 3863 ×10−10, a 62 and 193-fold increase over the rate with AID expression or tsa1Δ alone, respectively (Figure 2C vs. Figures 2A and 2B). Significantly higher rates were measured with the 12x Sγ3 (6067 ×10−10, p=0.025) and MTC (7090 ×10−10, p=0.0013) substrates, but not for MBR (4147 ×10−10, p=0.061). Notably, without transcription through the MBR, ZBE levels returned to background levels, a precipitous drop of over 1000-fold. (Figure 2C). This again demonstrates that mutagens appear to require access to vulnerable DNA to cause an increase in the ZBE rate.
Increased Topological Tension Enhances Sensitivity of Fragile Zones to ROS and AID
Transcription is known to increase torsional stress in DNA with the movement of the RNA polymerase introducing positive supercoils ahead of it and negative supercoils behind (Liu and Wang, 1987). The TOP1 gene encodes a type IB topoisomerase that functions by nicking one strand of DNA and relieves both positive and negative supercoiling (Thrash et al., 1985; Wang, 2002). Also, Top1 has been implicated in maintaining genome stability during transcription (Brill and Sternglanz, 1988; Kim and Jinks-Robertson, 2011, 2017; Yadav et al., 2014). Therefore, the ZBE assay was performed in top1Δ mutants to determine if the increased topological tension caused an increase in DSBs at the fragile sequences.
In the control (Figure 3A, no fragile zone, +transcription, top1Δ), there was no significant increase in the ZBE rate in top1Δ cells compared to WT cells (1.7 ×10−10 in Figure 3A vs 1.8 ×10−10 in Figure 1C, p=0.8), suggesting that high transcription alone may not require the participation of Top1 to maintain genome stability. Transcription and the presence of the 12x Sγ3 substrate significantly increased the ZBE rate to 80 ×10−10 (Figure 3A). This is 47-fold higher that the rate measured with the GAL1 promoter alone in a top1Δ background (vs. 1.7 ×10−10 in Figure 3A, p=3 ×10−7) and 5-fold higher than the rate measured in WT cells with a transcribed 12x Sγ3 substrate (vs. 16 ×10−10 in Figure 1E, p=0.002), demonstrating that the combination of transcription and loss of Top1 synergistically increased the ZBE rate at the switch region. No significant increase in ZBE rate was measured in the presence of the MTC or the MBR for a top1Δ single mutant (Figure 3A).
Figure 3. Topoisomerase Dysfunction Sensitizes Fragile Zones to ROS and AID.

(A) ZBE rate in a top1Δ single mutant (red) or top1Δ tsa1Δ double mutants (blue).
(B) ZBE rate with AID expression in a top1Δ mutant (green). Values for the top1Δ single mutant (orange) and AID expression alone (blue) are recapitulated here for comparison. ZBE rates were calculated as described in Figure 2.
Measuring the ZBE rate in top1Δ tsa1Δ double mutants revealed that loss of TOP1 sensitized DNA regions to damage by ROS. The ZBE rate measured in a top1Δ tsa1Δ double mutant with the GAL1 promoter and no fragile zone showed a significant 2.5-fold increase compared to the rate in a tsa1Δ single mutant (50 ×10−10 in Figure 3A vs 20 ×10−10 in Figure 2B, p=0.014). Remarkably, insertion of the MBR sequence downstream of the promotor increased the ZBE rate to 80 ×10−10, a significant increase over promoter alone (p=0.035). This rate is also significantly higher than the rate measured for transcribed MBR in the top1Δ (vs. 0.74 ×10−10 in Figure 3A, p=4 ×10−8) or tsa1Δ (vs. 23 ×10−10 in Figure 2B, p=0.002) single mutants. Again, the ZBE rate measured for the 12x Sγ3 substrate is significantly increased over the rate with no fragile zone, but the MTC insert in the top1Δ tsa1Δ double was not significantly higher than the rate for GAL1 promoter alone in the double mutant (Figure 3A). Loss of transcription in the top1Δ or top1Δ tsa1Δ cells returned the ZBE rate to background levels (Figure 3A).
If increased topological tension results in increased regions of ssDNA, these regions should be more vulnerable to AID, so we tested the effect of expressing AID in a top1Δ background. The ZBE rate for the negative control with no fragile zone was not significantly different from the rate with AID expression alone (Figure 3B 59 ×10−10 vs 62 ×10−10). In contrast, the ZBE rate for the MTC was significantly higher than both AID expression alone and AID expression in a top1Δ background with no fragile zone (Figure 3B, 286 ×10−10 vs 98 ×10−10 and 59 ×10−10, respectively). The 12x Sγ3 substrate showed the highest increase among the fragile zones at 690 ×10−10, highlighting the vulnerability of R-loop forming sequences to both AID and changes in supercoiling (Roy et al., 2008; Yu et al., 2005). Unlike the MTC, the MBR did not display a significant increase in ZBE rate.
Thus, the supercoiling that accumulates upon loss of Top1, is not typically enough to cause a DSB in most sequences of DNA. However, when this excess supercoiling occurs in a sequence that is prone to non-B DNA structures and these structures are exposed to ssDNA damaging agents such as ROS or AID, it can create a situation that favors breakage. Perhaps increased ROS or AID activity helps overcome the loss of TOP1 by relieving torsional stress, but at the cost of increasing DSBs. Interestingly, our data indicate that under these conditions the MBR may be more sensitive to ROS while the MTC is more vulnerable to AID. This is consistent with the MTC sequence showing a significant increase in ZBE rate with AID expression alone (Figure 2A).
Constitutively-Active Artemis Preferentially Cleaves Mammalian Fragile Zones Under Increased ROS Conditions
Previously, our lab implicated the nuclease Artemis as one potential factor that creates DSBs at the MBR fragile zone (Cui et al., 2013). Artemis belongs to the metallo-β-lactamase family of nucleases, has no functional homolog in S. cerevisiae (Chang et al., 2017; Hazrati et al., 2008), and can nick both DNA strands of a symmetrical mismatch created by AID at CpG sites, resulting in a DSB (Chang and Lieber, 2016). The catalytic activity of Artemis resides in the N-terminal β-lactamase and β-CASP domains while the C-terminal tail normally inhibits the catalytic domain until phosphorylated by DNA-PKcs, thus stimulating the endonuclease activity of Artemis (Figure 4A) (Ma et al., 2005; Pannicke et al., 2004; Poinsignon et al., 2004). Truncation mutants that remove the C-terminal tail retain both endonuclease activity in the absence of DNA-PKcs and the intrinsic, DNA-PKcs-independent 5′ exonuclease activity of Artemis (Niewolik et al., 2006). As S. cerevisiae lacks DNA-PKcs, expression of full-length Artemis would likely result in a non-functional protein; therefore, we expressed a galactose-inducible, 413-residue ARM37 truncation to evaluate the ability of Artemis to cleave DNA at the fragile zones (Figure 4A and 4B).
Figure 4. Increased ROS and Expression of Truncated Artemis Synergistically Increases ZBE Rate and Targets Mammalian Fragile Zones.

(A) Comparison of the 692 amino acid full-length Artemis and the ARM37 truncation that ends at residue 413 leaving the N-terminal β-lactamase and β-CASP domains intact, but removing the regulatory C-terminal tail that requires DNA-PKcs for activation. A galactose-inducible, myc-tagged ARM37 cassette was codon optimized for yeast and integrated into the genome at LYS2.
(B) Western blot probed with α-myc comparing cell extracts from a strain with the integrated ARM37 cassette grown with either glucose or galactose as the carbon source. The 50 kDa band expected for ARM37 is detectable only with cells grown in galactose.
(C) Comparison of ZBE rates with ARM37 expression alone (red), with co-expression of AID (blue), or in a tsa1Δ mutants (orange). Conditions and fragile zone substrates are as indicated. ZBE rates were calculated as described in Figure 2.
Expression of ARM37 in assay strains with the 12x Sγ3, MBR, or MTC fragile zones, does not significantly increase the ZBE rate above that measured in wild-type cells with no ARM37 expression (Figure 4C, left vs. Figure 1E). Strikingly, however, when ARM37 is expressed in tsa1Δ cells, a substantial increase in both the ZBE rate and the proportion of total cells containing ZBEs is measured (Figures 4C, right, Figure S2). For transcription alone, the rate increases nearly 100-fold to 281 ×10−10 (compared to 2.9 ×10−10 in Figure 4C, p=1.8 ×10−17). This rate is also 14-fold higher than that measured in tsa1Δ cells without ARM37 (compared to 20 ×10−10 in Figure 2B, p=1.1 ×10−12). Most revealing is that the rates measured in tsa1Δ cells expressing ARM37 are significantly higher for each of the mammalian fragile zones than the rate with no fragile zone, with MBR showing the highest rate at 462 ×10−10 (Figure 4C, right). The rates for 12x Sγ3 and MTC are also significantly higher than with no fragile zone at 443 ×10−10 (p=7 ×10−5) and 411 ×10−10 (p=0.023), respectively. These increases all appear to be dependent on transcription (Figure 4C, Table S2). Furthermore, these results verify the biological activity of the ARM37 protein as the increase in ZBE rate is a direct result of ARM37 expression.
We also tested the effect of AID and ARM37 co-expression (Figure 4C, middle). The ZBE rates, though slightly elevated, were not significantly higher than with AID expression alone for any of the substrates tested (Figure 4C, middle vs. Figure 2A). This tells us something important about the consequences of different types of damage that can be inflicted upon the fragile zones. While AID and ROS can both, in theory, generate lesions that distort the helix and create a substrate for ARM37 cleavage, different types of lesions have different repair kinetics that may influence whether ARM37 has an opportunity to cut.
Deletion of UNG1 Enhances the Effect of AID at Fragile Zones
The U’s generated via cytosine deamination by AID are removed by the highly effective uracil glycosylase encoded by UNG1 in S. cerevisiae. AID expression in the absence of UNG1 should result in an accumulation of mutagenic U’s in the DNA, but a decrease in DSBs since the subsequent cutting of the phosphate backbone by the major apurinc/apyrimidinic endonuclease (Apn1 in yeast, Ape1 in mammals) is now blocked (Poltoratsky et al., 2004). It is also possible that the accumulation of U’s in the genome will lead to a persistent lesion that can be targeted by other nucleases, similar to what we observe with ARM37 expression in tsa1Δ cells.
The ung1Δ deletion alone has no significant effect on the ZBE rate compared to each of the substrates with transcription in WT cells (Figure 5A vs Figures 1C and 1E). Most interesting is the effect of the ung1Δ allele in cells expressing AID (Figure 5A). With transcription, but no fragile zone, loss of UNG1 decreases the ZBE rate 33-fold, compared to AID expression in WT cells (Figure 5A, 1.9 ×10−10 vs 62 ×10−10, p=1.3 ×10−14), clearly demonstrating the UNG1-dependence of DSB formation following AID expression. Intriguingly, this decrease is not uniform among the mammalian fragile zones. For example, the rate measured for the MBR in the ung1Δ + AID background is 23 ×10−10, a significant 12-fold increase over the no fragile zone substrate (p=1.5 ×10−7). This difference between the MBR and no fragile zone is not evident with AID expression alone (Figure 5A, 62 × 10−10 vs 85 ×10−10) and suggests that preventing the efficient removal of excess U’s from the DNA impacts the fragility of these sequences.
Figure 5. AID expression in ung1Δ Mutants Creates a Long-Lived Lesion Prone to DSBs at Fragile zones.

(A) ZBE rate in the indicated background for each of the four DNA substrates. The rates from AID expression alone or AID and ARM37 co-expression are recapitulated here to compare with the rates determined in ung1Δ mutants. ZBE rates were calculated as described in Figure 2.
(C) The % fragility of each substrate normalized to 12x Sγ3. The rate for the 12x Sγ3 substrate under each indicated condition was set at 100% fragility, allowing the fragility of the remaining substrates to be represented as a percentage of the positive control. Red shading, fragility < 50%; yellow shading, fragility between 50 to 90%; green shading, fragility > 90%.
Co-expression of ARM37 and AID in ung1Δ mutants further increases the ZBE rate for some substrates, though not to the levels measured with AID expression alone (Figure 5A). Still, this indicates that the longer-lived lesions can be targeted by ARM37, with the effect on the no-fragile zone and 12x Sγ3 substrates being significantly higher than in the ung1Δ AID background. ARM37 expression does not affect the rate for the MBR, further emphasizing that no single mechanism appears to account for breaks at each of the mammalian fragile zones.
Evaluating the Fragility of Mammalian Sequences
Interestingly, many of the conditions we tested did not greatly affect the transcribed 12x Sγ3 substrate, as the fold increase was typically no greater than that measured in wild-type cells. This suggests that the switch substrate is already at peak fragility in our system under the conditions tested. While this has interesting implications for processing of R-loops in yeast, the major question we were interested in addressing here was if we can generate a condition where the fragile zones exhibit at least as much response in the ZBE assay as the switch region positive control.
If we normalize the ZBE rates for each fragile zone to the 12x Sγ3 substrate (representing 100% fragility), an interesting pattern emerges (Figure 5B). In wild-type, tsa1Δ, ung1Δ or ARM37+ cells, neither the MBR nor the MTC is ever more than 50% as fragile as 12x Sγ3 and is typically much less, exhibiting no more fragility than when no fragile zone is present. AID expression raises the fragility of the MBR and the MTC, but it is still below that of 12x Sγ3 even with co-expression of ARM37. It is only when the tsa1Δ allele and ARM37 expression are combined that both the MBR and the MTC are as fragile as the switch region (Figure 5B). AID expression in tsa1Δ cells increases the fragility of MTC while AID expression in ung1Δ cells seems to more specifically affect the MBR, indicating that different combinations of factors affect the fragility of each zone.
DISCUSSION
Translocations involving BCL2 and CCND1 are present in nearly all cases of follicular lymphoma and mantle cell lymphoma, respectively (Lieber, 2016). A significant number of DSBs at each of these oncogenes occur in tightly focused fragile zones, the MBR of BCL2 and the MTC of CCND1. The advantage of the fragile zones is that we know where the breaks are occurring, allowing us to study what makes them prone to DSBs. While the literature on genome instability is vast, only a very small fraction is devoted to the natural causes of spontaneous DSBs; studies of DNA sequence effects are even rarer. Here, we have evaluated the MBR and the MTC human fragile zones in a S. cerevisiae genetic assay and found that a critical combination of transcription and long-lived lesions account for fragility.
Transcription and the Transient Nature of ssDNA
Transcription had a destabilizing effect, increasing the ZBE rate and having the strongest effect on the 12x Sγ3 substrate. The role of transcription in R-loop formation and DSBs at switch regions in B cells during CSR has well studied, and seeing the phenotype reproduced in yeast confirmed the ability to detect these non-B structures in our system. That transcription alone did not significantly increase the ZBE rate with the MBR and the MTC likely indicates that the mechanism behind DSBs at switch regions and fragile zones is, even though formation of ssDNA is key for both. For switch regions, ssDNA is generated through the transcription-induced formation of R-loops. R-loops are not likely the cause of ssDNA at fragile zones as the single-stranded character detected by chemical probing is still present following pretreatment with RNase H, which digests the RNA in RNA:DNA hybrids, allowing reannealing of the DNA duplex (Tsai et al., 2009). Likely, the ssDNA generated by an R-loop is more extensive and longer-lived than the transient non-B DNA structure the fragile zones can adopt. Therefore, while transcription is necessary to trigger R-loop formation at switch regions, its role for fragile zones may be to disrupt the DNA duplex by separating the DNA strands and increasing torsional stress as the transcription machinery travels along the DNA (Liu and Wang, 1987).
One plausible explanation is that fragility involves the formation of slipped strand DNA (Pearson et al., 1998; Sinden, 1994), where, following strand separation due to transcription, the two strands of the duplex can become misaligned due to the presence of short, closely spaced direct repeats (Figure 6). This creates transient single-stranded loops on each strand of the DNA before the duplex snaps back into register. Indeed, the MBR has an 8 bp sequence that is directly repeated, and the C-strings in both the MBR and the MTC are essentially short direct repeats as well. Furthermore, slipped strand structures may also explain the effect of the TOP1 deletion. Under increased torsional stress, as would occur with topoisomerase dysfunction, a slipped strand DNA structure would relax supercoiling, thus making the transient single-stranded state more stable (Sinden, 1994).
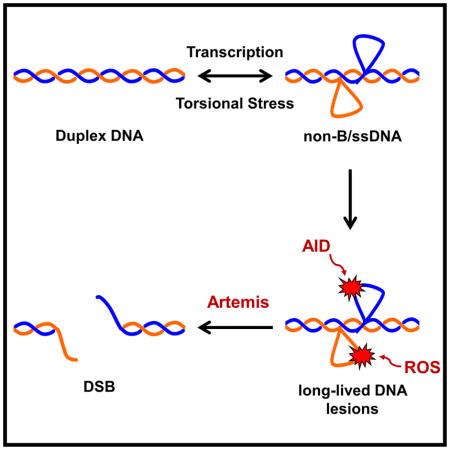
Figure 6. Model for DSB Formation at Human Fragile Zones.

(A) The action of transcription or increased torsional stress can lead to the transient formation of single-strandedness due to slipped strand DNA. Typically, these slipped strand DNA structures are quickly resolved back to B-form DNA. However, while present, the slipped strand structures are prone to damage from AID or ROS. In the presence of AID (blue box), C’s are converted to U’s that are efficiently excised by uracil DNA glycosylase (Ung1). The abasic site is further processed by apurinic/apyrimidinic endonuclease (Apn1), which cleaves the phosphate backbone. When deamination occurs at closely spaced C’s on opposite DNA strands, this process can result in a DSB. Alternatively, in the absence of Ung1, AID activity can result in a persistent lesion that is repaired at a slower rate. Under conditions of increased ROS (orange box), unstacked bases are sensitive to damage, creating oxidized lesions that are also repaired at a slower rate compared to uracil excision, creating a persistent lesion. In both cases the persistent lesion is prone to cleavage by active nucleases, including Artemis (yellow pac-man), that can result in a DSB.
(B) In early human B cells, a DSB forms at the IGH locus on chromosome 14 due to the activity of the RAG complex. Unrepaired DH and JH ends activate Artemis via DNA-PKcs. Concurrently, replication or transcription at fragile zones adjacent to oncogenes lead to DNA strand separation and formation of transient slipped strand DNA structures. meC’s looped out of the duplex are vulnerable to AID, which converts them to T’s. Following re-alignment of the DNA, the T:G mismatches are repaired more slowly than a U:G mismatch and the resulting bubble structure has ss-to-dsDNA boundaries that can be recognized by activated Artemis, cleaving each DNA strand and resulting in a DSB. Depending on repair of the four DNA ends, a reciprocal translocation can form.
Differential Processing of Lesions Generated by AID and ROS
AID and increased ROS were introduced to determine if damage to the ssDNA generated at fragile zones increases DSBs, possibly due to the damage creating cleavable substrates for nucleases such as Artemis. We increased the basal level of ROS by deleting the TSA1 gene encoding the major peroxiredoxin in yeast, since loss of peroxiredoxins is known to increase the DNA damage associated with ROS in yeast and mammals (Iraqui et al., 2009; Neumann et al., 2003; Okazaki et al., 2005). Interestingly, combining ARM37 expression with AID or the tsa1Δ mutant had quite different outcomes. What emerged from our data is that DNA lesions that are long-lived (i.e., having slower repair kinetics) appear to be more vulnerable to ARM37, thus making the human fragile zones more prone to DSBs.
Longevity of the DNA lesions is dictated by the repair processes utilized. The U’s generated by AID are removed by uracil DNA glycosylase (Ung1) at a rate that can be orders of magnitude more efficient than removal of, for example, the oxidized lesion 8-hydroxyguanine (8OHG) by 8OHG glycosylase (Ogg1) (Cravens and Stivers, 2016). Also, Ung1 can act on both ssDNA and dsDNA whereas Ogg1 recognizes the 8OHG:C base pair in duplex DNA (Friedman and Stivers, 2010). Therefore, U’s generated by AID can be removed before the DNA returns to a duplex state.
A critical difference between S. cerevisiae and mammals is that mammals have methylated cytosines (meC) that occur at CpG sites. These CpG’s are also sites of AID action in ssDNA (Bransteitter et al., 2003; Han et al., 2011; Pham et al., 2003). Importantly, while AID deaminates C→U, it deaminates meC→T, with the resulting T:G mismatch being more long-lived than a U:G mismatch (Tsai and Lieber, 2010; Waters and Swann, 1998). Indeed, we have previously demonstrated the importance of meC’s at CpG sites within fragile zones for DSB formation (Cui et al., 2013). Thus, the T:G mismatches generated by meC→T deamination in mammals and the 8OHG:C lesion generated by tsa1Δ in yeast represent long-lived lesions that can create structures recognized by activated Artemis, while U:G mismatches in yeast and mammals are repaired too quickly to be affected by Artemis (Figure 6A).
Like AID, ROS can induce mutations genome-wide, but the fragile zones may be more vulnerable to attack due to the non-B distortion of the helix. Also, the MBR and MTC have a higher GC content (61% and 55%, respectively) than the genome average of S. cerevisiae (38%) and H. sapiens (41%). GC-rich sequences appear especially prone to ROS due to the oxidation of guanine (Madison et al., 2012; Saito et al., 1995) and damage from Cu—H2O2 complexes (Rodriguez et al., 1995). Thus, long-lived oxidized DNA lesions may accumulate at the fragile zones and may explain the large increase in the ZBE rate we observe when ARM37 is expressed in a tsa1Δ mutant.
Using the data collected from our ZBE assay, we can formulate a model for how DSBs are forming at the human fragile zones in yeast and in human B cells (Figure 6). Transcription and torsional stress within the fragile zones leads to strand separation and the presence of strings of C’s potentiates the transient formation of single-stranded slipped strand DNA structures. Typically, slipped strand DNA would rapidly snap back into register, but, while formed, the exposed single-stranded DNA structure would be vulnerable to attack by AID or ROS to generate damage that persists even after snap-back to duplex DNA. The U’s produced by AID deamination are efficiently processed by Ung1 with the subsequent nicking by Apn1 generating a DSB if the deamination sites are closely spaced on opposite strands (Figure 6A, left, blue panel).
Under conditions of high ROS, increased damage to the unstacked bases of the slipped strand DNA may occur (Figure 6A, right, orange panel). The resulting DNA mismatches are repaired more slowly compared to uracil excision, prolonging the ss-to-dsDNA boundaries that are the preferred substrate for Artemis cleavage. Processing of AID-induced lesions by Ung1 occurs too quickly for expression of ARM37 to have an effect, but Ung1-loss creates a persistent U lesion that can either result in mutation or be processed into a DSB by Artemis or another active nuclease (Figure 6A, left, blue panel).
In human B cells, the presence of meC’s within the slipped strand DNA means that AID activity can lead to the longer-lived T:G mismatches that are recognized by activated Artemis to create a DSB (Figure 6B). Translocations involving the fragile zones occur in a very narrow time frame depending on both the failure of the highly efficient NHEJ machinery to join DH and JH ends and simultaneous formation of a DSB at a fragile zone (Jaeger et al., 2000; Welzel et al., 2001). Based on our results, one possible scenario for the latter would involve strand separation leading to slipped strand DNA followed by AID activity converting meC’s to T’s with the T’s in a T:G mismatch removed at a much slower rate compared to U’s (Waters and Swann, 1998). Prior to repair of the T:G mismatch, the lesion would then be susceptible to cleavage by Artemis, which is already activated in pre-B cells for the NHEJ repair required for V(D)J recombination (Figure 6B).
Conclusions
Our data indicate that the sequence of the DNA at the fragile zones may not be the only factor that contributes to DSB formation and that a combination of factors is required. First, separation of the two DNA strands appears critical, likely to expose the fragile zones to damaging agents. Next, we identified that combination of a damage (increased ROS or AID activity) and Artemis contribute in generating DSBs at the fragile zones. A key factor appears to be creating DNA lesions that are repaired at a slower rate, allowing time for factors such as Artemis to act prior to repair of the lesion. The fold difference displayed by the MBR and the MTC, though small, may reflect that once a damage threshold is surpassed, repair efficiency is exceeded, leading to more DSBs in the yeast system as well as in the mammalian cells we are modeling.
Lastly, this study has shown that AID, ARM37, and the tsa1Δ allele are useful tools that allow us to capture and quantitate the transient structures that are quickly resolved in most cells. The ZBE system does not allow us to capture all DSBs (e.g., DSBs that are re-ligated without marker loss); thus, our rates are likely an underestimate of the true number of DSBs. Continued refinement will enhance our ability to detect human fragile zones elsewhere in the genome and to understand why these are markedly more prone to breakage.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information requests may be directed to, and will be fulfilled by, the Lead Contact, Dr. Michael R Lieber (lieber@usc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast Strains
All S. cerevisiae strains are isogenic to the W303 background (Thomas and Rothstein, 1989) except that they carry a wild-type RAD5 allele (Table S1). Standard yeast genetic and molecular techniques were employed (Sambrook et al., 1989; Sherman F., 1986). As before (Pannunzio and Lieber, 2016), the assay relies on the loss of a URA3 and CAN1 cassette from the left arm of S. cerevisiae chromosome I, conferring resistance to 5-fluoroorotic acid (5-FOAR) and canavanine (CanR), respectively. Previously, a DSB anywhere in the 25 kb region between the CAN1 gene and the first essential gene on chromosome I, PTA1, could result in loss of the non-essential chromosome arm. Now we have inserted a LYS2 gene downstream of CAN1, creating a 1.6 kb spacer region between the two genes (Figure 1B). By selecting for the retention of lysine prototrophy (Lys+), we ensure breaks detected are occurring on the telomeric-side of LYS2. Sequence homology from the endogenous URA3, CAN1, and LYS2 loci were deleted from the genome (Table S1). Additionally, the KAN-MX4 cassette, conferring resistance to the antibiotic G418 (G418R), was inserted 2.8 kb telomeric to the URA3 gene.
A second version of the assay has a 492 bp fragment carrying the GAL1 promoter inserted immediately downstream of the CAN1 terminator with transcription directed towards the centromere (Figure 1B), expanding the region between the CAN1 and LYS2 genes to approximately 2.1 kb. Recent GRO-seq data has demonstrated that the transcription rate (mRNA molecules/hour) of the induced GAL1 promoter, while above the median of total transcription rate in yeast, is still approximately 8-fold lower than the strongest yeast promoters (Pelechano et al., 2010) and is within the range of the general transcription rate in mammalian cells (Schwanhausser et al., 2011). This, coupled with the flexibility of induction, makes the GAL1 promoter an ideal choice.
For construction of the assay, as before, the various substrates were first assembled on a plasmid (Pannunzio and Lieber, 2016). A 1059 bp fragment carrying the URA3 CDS plus 177 bp of upstream and 78 bp of downstream sequence was cloned into pUC18, followed by a 2475 bp fragment carrying the CAN1 CDS plus 486 bp of upstream and 216 bp of downstream sequence. Next, a 5′ portion of the BDH2 CDS and promoter and a 707 bp 3′ portion of BDH2 CDS were inserted to target the substrates to the BDH2 locus on chromosome I upon excision from the plasmid and transformation into yeast cells. This plasmid is named pNP121 and was used to create the strain with no promoter and no fragile zone. Insertion of a 492 bp fragment carrying the sequence upstream of the GAL1 CDS and containing three galactose responsive upstream activating sequences was inserted downstream of the CAN1 terminator to create pNP124. The plasmids containing the 12x Sγ3, MTC, and MBR sequences without the GAL1 promoter are pNP146, pNP148, and pNP136, respectively. The plasmids containing the 12x Sγ3, MTC, MBR, and inverted MBR with the GAL1 promoter are pNP122, pNP150, pNP137, and pNP166, respectively.
The substrates were transformed into yeast strains that carried the ars104::KANMX4 and bdh1::LYS2 disruptions on chromosome I, which are also part of the assay. For ars104::KANMX4, oligos NP386 and NP387 (Table S3) were used to generate a 1632 bp fragment from pFA6a-KANMX4 (Bahler et al., 1998) carrying the KANMX4 gene flanked by 60 bp of homology to the ARS104 locus. Fragment insertion results in a 474 bp region containing ARS104 being replaced by the KANMX4 gene. For bdh1::LYS2, the cloned LYS2 gene from pNP106 was amplified using primers NP428 and NP429 to generated a 4571 bp fragment flanked by 60 bp of sequence homologous to BDH1. Fragment insertion replaces a 690 bp fragment of the BDH1 CDS with the LYS2 gene. Additional alleles common to all of the assay strains are ura3::TRP1 and can1::LEU2 generated by fragment insertion (Pannunzio and Lieber, 2016) and the lys2-Δ4443 allele generated by the delitto perfetto method (Storici et al., 2001). All chromosomal insertions and disruptions were confirmed by PCR and meiotic mapping. Strains were backcrossed with wild-type tester strains to remain isogenic.
tsa1Δ
The TSA1 gene on chromosome XIII was disrupted by the HIS3 gene to create the tsa1::HIS3 allele. Oligos NP459 and NP460 were used to generate a 1633 bp fragment carrying the HIS3 gene with upstream and downstream sequence from pUC18-HIS3 (Rothstein, 1991) and flanked by 60 bp of TSA1 sequence. Disruption with this fragment replaces nucleotides 61–531 of the TSA1 CDS with the HIS3 sequence.
ARM37
A DNA fragment carrying the Artemis truncation, ARM37, codon optimized for S. cerevisiae was synthesized by Integrated DNA Technologies (Coralville, IA) and delivered on the plasmid pUCIDT-AMP:ARM37. A 1.2 kb SalI fragment carrying the ARM37 CDS was cloned into the SalI/XhoI site of pESC-TRP (Agilent Technologies, Santa Clara, CA), creating pNP25 and placing ARM37 under the control of a galactose promoter and adding 3′ sequence that encodes a myc epitope. Next, this GAL-ARM37-Myc cassette was inserted into the genome at the LYS2 locus. Since ARM37 insertion offers no selection, the lys2::URA3-KANMX intermediate created during the creation of the lys2-Δ4443 allele by delitto perfetto was utilized. Oligos NP397 and NP398 were used to amplify the ARM37 cassette from pNP25. Oligos NP413 and NP414 were used to amplify a 625 bp fragment upstream of the LYS2 insertion site and oligos NP415 and NP416 where used to amplify a 646 bp fragment downstream of the LYS2 insertion site. All three PCR fragments were combined to generated a 3.6 kb disruption fragment with the ARM37 cassette and the 625 bp and 646 bp flanking homologies, which was transformed into yeast carrying the lys2::URA3-KANMX allele. 5-FOAR and G418S transformants were tested for insertion of ARM37 by PCR. The fidelity of the inserted ARM37 was verified by sequencing. Expression was confirmed by Western blot and the NPT110 strain with the lys2::ARM37 allele was backcrossed three times. Strains used in the ZBE assay were genotyped for the lys2::ARM37 allele by PCR.
AID
The galactose-inducible human AID expression vector codon optimized for yeast was provided on the pESC-LEU-hAIDSc vector (a gift from Dr. Youri Pavlov) (Mayorov et al., 2005). In order to make this vector compatible with the ZBE assay and to maintain selection of the plasmid in YP-Galactose, the LEU2 marker was replaced with the hygromycin B resistance marker hphMX4 from pAG32 (Goldstein and McCusker, 1999). Oligos NP474 and NP475 were used to amplify hphMX a 1.7 kb fragment from pAG32 carrying the hphMX4 gene with upstream and downstream sequence and PflMI restriction endonuclease recognition sites. The fragment replaced the PflMI fragment of pESC-LEU-hAIDSc carrying the LEU2 gene, creating pNP171. pNP171 was highly mitotically unstable, typically with no dissected segregants retaining the plasmid unless selection was maintained up until sporulation. Therefore, when testing strains with pNP171 in the ZBE assay, YPG cultures were supplemented with hygromycin B.
ung1::HIS3
Primers NP493 and NP494 were used to amplify a 1801 bp fragment carrying the HIS3 gene from pUC18-HIS3 with 60 bp of flanking homology to the UNG1 locus. Chromosomal insertion of HIS3 replaces nucleotides 65 to 744 of the UNG1 coding sequence with HIS3 transcribed in the opposite orientation compared to UNG1. Insertion was confirmed by PCR and meiotic mapping and mutant strain was backcrossed three times. Strains bearing the ung1::HIS3 allele are maintained as heterozygous diploids to the mutator effect of ung1Δ.
METHOD DETAILS
Zonal Break Event Assay
The assay was performed in haploid yeast cells as follows. Approximately 50 cells, determined by hemocytometer count, from a single colony growing on uracil-lacking medium were used to inoculate 10 ml of YPG (1% yeast extract, 2% peptone, and 2% galactose). The only exception was with cells carrying the pNP171 vector harboring AID. Then, approximately 50 cells from a single colony growing on YPD (1% yeast extract, 2% peptone, and 2% dextrose) supplemented with 250 μg/ml of hygromycin B were used to inoculate 10 ml of YPG + 250 μg/ml of hygromycin B. The initial inoculum was kept low to ensure an accurate rate determination (Foster, 2006). For each strain, a minimum of 20 cultures were grown with some strains requiring between 60 and 100 cultures for consistent results. Cells were grown to saturation at 30°C (3–4 days). Following one wash in PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.4 mM K2HPO4, pH 7.4), a dilution of the cells was plated to YPD to determine the number of viable cells present in the culture. The remainder of the culture was plated to medium lacking lysine and arginine and containing 5-FOA (750 μg/ml) and canavanine (60 μg/ml). Cells from each culture were distributed over 10, 90mm plates to ensure that the total number of cells per plate did not exceed 108, as this may lead to growth inhibition of selected cells. (Spell and Jinks-Robertson, 2004). Depending on the number of recombinant colonies obtained, either (1) every colony was patched to or (2) a portion of colonies replica plated to YPD supplemented with 200 μg/ml G418 to screen for loss of the KAN-MX4 cassette. The number of G418R colonies was subtracted from the total number of Lys+, 5-FOAR, CanR colonies. This final number was used to determine the ZBE rate (events/cell/generation) either by Luria-Delbrück fluctuation analysis (Luria and Delbruck, 1943) if <50% of the trials yielded colonies or the method of the median (Lea and Coulson, 1949) if ≥50% of the trials yielded colonies. When method of the median was used, a 95% confidence interval could be obtained (Spell and Jinks-Robertson, 2004), otherwise Mann-Whitney tests were used to compare datasets and determine significance.
Yeast Transformation
Linear and circular DNA was transformed into cells by standard lithium acetate (LiAc) transformation (http://www.fhcrc.org/labs/gottschling). Early to mid-log phase cells were harvested from a 50 ml YPD culture growing at 30°C and washed in one half volume of ddH2O. Cells were pelleted and resuspended in 1 ml of 100 mM LiAc, pelleted again, and resuspended in a final volume of 0.5 ml of LiAc. 50 μl of cells were pelleted and resuspended in a solution of 30% PEG-3350, 100 mM LiAc, 100 μg of salmon sperm DNA, plus the DNA to be transformed with the final volume brought to 360 μl with ddH2O. Cells were heat shocked for 40 minutes at 42°C then plated to selective medium.
Western Blot
Single colonies of NPT110, carrying the lys2::ARM37 allele, were used to inoculate 1 ml of YP-Dextrose or YP-Galactose. Following overnight growth at 30°C, 10 μl of each culture was used to inoculate a fresh 1 ml of YP-Dextrose or YP-Galactose and cultures were grown to mid-log phase (approximately 6 hours). Cells were washed 1x in PBS and the pellets were resuspended in 50 μl of suspension buffer (100 mM NaCl, 10 mM Tris-HCl, pH 7.6, 1 mM EDTA, pH 8.0, 1 μg/ml aprotinin, 100 μg/ml PMSF). Next, 50 μl of 2x loading buffer (100 mM Tris-HCl, pH 6.8, 200 mM DTT, 4% SDS, 0.2% bromophenol blue, 20% glycerol) was added and samples were incubated for 10 minutes at 100°C. The insoluble material was pelleted and 20 μl of the supernatant was resolved on a 12% SDS-PAGE gel at 100V using the BioRad mini PROTEAN system. Protein was then transferred to PVDF membrane using the BioRad mini Trans-Blot cell running at 150 mA for 1 hour. The membrane was blocked with a solution of 5% dry milk dissolved in TBST (20 mM Tris-HCl, pH 7.5, 150mM NaCl, 0.05% Tween-20) and blotted with anti-myc antibody. Signal was produced by using the ECL Prime Western Blotting Detection Kit (GE Healthcare) and detected using the BioRad Fluor-S MultiImager.
QUANTIFICATION AND STATISTICAL METHODS
Mann-Whitney tests were performed using the software Past v3.13 (Hammer et al., 2001). All other data was compiled in Microsoft Excel.
Supplementary Material
HIGHLIGHTS.
New insight into transcription as a cause of increased DNA double-strand breaks
Topoisomerase loss sensitizes DNA to AID and to increased oxidative stress
Oxidative stress and AID highlight novel pathways for chromosome breaks
Long-lived DNA lesions in human fragile zones are a critical cause of DSBs
Acknowledgments
The authors thank Y. Pavlov for the pESC-LEU-hAIDSc vector and A. Bailis for select yeast strains. Also, we thank S. Masri and R. Mosteller for helpful comments on the manuscript. The work was supported by NIH grants to M.R.L. Funding for N.R.P. was provided by a generous memorial endowment established by Barbara Knight through the ARCS Foundation, Inc., Los Angeles founder chapter, John H. Richardson Postdoctoral Fellowship.
Footnotes
AUTHOR CONTRIBTIONS
Conceptualization, Methodology and Validation, N.R.P. and M.R.L. Formal Analysis and Investigation, N.R.P. Writing-Original Draft, Review & Editing, N.R.P. and M.R.L. Resources, Supervision, and Project Administration, M.R.L. Funding Acquisition, M.R.L. and N.R.P.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bahler J, Wu JQ, Longtine MS, Shah NG, McKenzie A, 3rd, Steever AB, Wach A, Philippsen P, Pringle JR. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 1998;14:943–951. doi: 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Bransteitter R, Pham P, Scharff MD, Goodman MF. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc Natl Acad Sci U S A. 2003;100:4102–4107. doi: 10.1073/pnas.0730835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brill SJ, Sternglanz R. Transcription-dependent DNA supercoiling in yeast DNA topoisomerase mutants. Cell. 1988;54:403–411. doi: 10.1016/0092-8674(88)90203-6. [DOI] [PubMed] [Google Scholar]
- Chan JE, Kolodner RD. A genetic and structural study of genome rearrangements mediated by high copy repeat Ty1 elements. PLoS Genet. 2011;7:e1002089. doi: 10.1371/journal.pgen.1002089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HH, Lieber MR. Structure-Specific nuclease activities of Artemis and the Artemis: DNA-PKcs complex. Nucleic Acids Res. 2016;44:4991–4997. doi: 10.1093/nar/gkw456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol. 2017 doi: 10.1038/nrm.2017.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravens SL, Stivers JT. Comparative Effects of Ions, Molecular Crowding, and Bulk DNA on the Damage Search Mechanisms of hOGG1 and hUNG. Biochemistry. 2016;55:5230–5242. doi: 10.1021/acs.biochem.6b00482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X, Lu Z, Kurosawa A, Klemm L, Bagshaw A, Tsai AG, Gemmell N, Muschen M, Adachi N, Hsieh CL, et al. Both CpG Methylation and AID are Required for the Fragility of the Human Bcl-2 Major Breakpoint Region: Implications for the Timing of the Breaks in the t(14;18) Mol Cell Biol. 2013;33:947–957. doi: 10.1128/MCB.01436-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels GA, Lieber MR. RNA:DNA complex formation upon transcription of immunoglobulin switch regions: implications for the mechanism and regulation of class switch recombination. Nucl Acids Res. 1995;23:5006–5011. doi: 10.1093/nar/23.24.5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNoia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Ann Rev Biochem. 2007;76:1–22. doi: 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- Durkin SG, Glover TW. Chromosome fragile sites. Annu Rev Genet. 2007;41:169–192. doi: 10.1146/annurev.genet.41.042007.165900. [DOI] [PubMed] [Google Scholar]
- Foster PL. Methods for determining spontaneous mutation rates. Methods Enzymol. 2006;409:195–213. doi: 10.1016/S0076-6879(05)09012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JI, Stivers JT. Detection of damaged DNA bases by DNA glycosylase enzymes. Biochemistry. 2010;49:4957–4967. doi: 10.1021/bi100593a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein AL, McCusker JH. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast. 1999;15:1541–1553. doi: 10.1002/(SICI)1097-0061(199910)15:14<1541::AID-YEA476>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. 5. Oxford University Press; 2015. [Google Scholar]
- Hammer O, Harper DAT, Ryan PD. PAST: Paleontological statistics software package for education and data analysis. Paleontologia Electronica. 2001;4:9. [Google Scholar]
- Han L, Masani S, Yu K. Overlapping activation-induced cytidine deaminase hotspot motifs in Ig class-switch recombination. Proc Natl Acad Sci U S A. 2011;108:11584–11589. doi: 10.1073/pnas.1018726108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazrati A, Ramis-Castelltort M, Sarkar S, Barber LJ, Schofield CJ, Hartley JA, McHugh PJ. Human SNM1A suppresses the DNA repair defects of yeast pso2 mutants. DNA Repair (Amst) 2008;7:230–238. doi: 10.1016/j.dnarep.2007.09.013. [DOI] [PubMed] [Google Scholar]
- Huang ME, Rio AG, Nicolas A, Kolodner RD. A genomewide screen in Saccharomyces cerevisiae for genes that suppress the accumulation of mutations. Proc Natl Acad Sci U S A. 2003;100:11529–11534. doi: 10.1073/pnas.2035018100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iraqui I, Kienda G, Soeur J, Faye G, Baldacci G, Kolodner RD, Huang ME. Peroxiredoxin Tsa1 is the key peroxidase suppressing genome instability and protecting against cell death in Saccharomyces cerevisiae. PLoS Genet. 2009;5:e1000524. doi: 10.1371/journal.pgen.1000524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger U, Bocskor S, Le T, Mitterbauer G, Bolz I, Chott A, Kneba A, Mannhalter C, Nadel B. Follicular lymphomas BCL-2/IgH junctions contain templated nucleotide insertions: novel insights into the mechanism of t(14;18) translocation. Blood. 2000;95:3520–3529. [PubMed] [Google Scholar]
- Kim N, Abdulovic AL, Gealy R, Lippert MJ, Jinks-Robertson S. Transcription-associated mutagenesis in yeast is directly proportional to the level of gene expression and influenced by the direction of DNA replication. DNA Repair (Amst) 2007;6:1285–1296. doi: 10.1016/j.dnarep.2007.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Jinks-Robertson S. Guanine repeat-containing sequences confer transcription-dependent instability in an orientation-specific manner in yeast. DNA Repair (Amst) 2011;10:953–960. doi: 10.1016/j.dnarep.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Jinks-Robertson S. The Top1 paradox: Friend and foe of the eukaryotic genome. DNA Repair (Amst) 2017 doi: 10.1016/j.dnarep.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea DE, Coulson CA. The distribution of the numbers of mutants in bacterial populations. Journal of Genetics. 1949;49:264–285. doi: 10.1007/BF02986080. [DOI] [PubMed] [Google Scholar]
- Lieber MR. Mechanisms of human lymphoid chromosomal translocations. Nat Rev Cancer. 2016;16:387–398. doi: 10.1038/nrc.2016.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu LF, Wang JC. Supercoiling of the DNA template during transcription. Proc Natl Acad Sci USA. 1987;84:7024–7027. doi: 10.1073/pnas.84.20.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Lieber MR, Tsai AG, Pardo CE, Muschen MM, Kladde MP, Hsieh CL. Human Lymphoid Translocation Fragile Zones are Hypomethylated and Have Accessible Chromatin. Mol Cell Biol. 2015;35:1209–1222. doi: 10.1128/MCB.01085-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luria SE, Delbruck M. Mutations of Bacteria from Virus Sensitivity to Virus Resistance. Genetics. 1943;28:491–511. doi: 10.1093/genetics/28.6.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Pannicke U, Lu H, Niewolik D, Schwarz K, Lieber MR. The DNA-PKcs phosphorylation sites of human artemis. J Biol Chem. 2005;280:33839–33846. doi: 10.1074/jbc.M507113200. [DOI] [PubMed] [Google Scholar]
- Madison AL, Perez ZA, To P, Maisonet T, Rios EV, Trejo Y, Ochoa-Paniagua C, Reno A, Stemp ED. Dependence of DNA-protein cross-linking via guanine oxidation upon local DNA sequence as studied by restriction endonuclease inhibition. Biochemistry. 2012;51:362–369. doi: 10.1021/bi201087q. [DOI] [PubMed] [Google Scholar]
- Masani S, Han L, Yu K. Apurinic/apyrimidinic endonuclease 1 is the essential nuclease during immunoglobulin class switch recombination. Mol Cell Biol. 2013;33:1468–1473. doi: 10.1128/MCB.00026-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayorov VI, Rogozin IB, Adkison LR, Frahm C, Kunkel TA, Pavlov YI. Expression of human AID in yeast induces mutations in context similar to the context of somatic hypermutation at G-C pairs in immunoglobulin genes. BMC Immunol. 2005;6:10. doi: 10.1186/1471-2172-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer DH, Bailis AM. Telomere dysfunction drives increased mutation by error-prone polymerases Rev1 and zeta in Saccharomyces cerevisiae. Genetics. 2007;175:1533–1537. doi: 10.1534/genetics.106.068130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro MS, Bi L, Bailis AM. A mutant allele of the transcription factor IIH helicase gene, RAD3, promotes loss of heterozygosity in response to a DNA replication defect in Saccharomyces cerevisiae. Genetics. 2007;176:1391–1402. doi: 10.1534/genetics.107.073056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann CA, Krause DS, Carman CV, Das S, Dubey DP, Abraham JL, Bronson RT, Fujiwara Y, Orkin SH, Van Etten RA. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 2003;424:561–565. doi: 10.1038/nature01819. [DOI] [PubMed] [Google Scholar]
- Niewolik D, Pannicke U, Lu H, Ma Y, Wang LC, Kulesza P, Zandi E, Lieber MR, Schwarz K. DNA-PKcs dependence of artemis endonucleolytic activity: differences between hairpins and 5′ or 3′ overhangs. J Biol Chem. 2006;281:33900–33909. doi: 10.1074/jbc.M606023200. [DOI] [PubMed] [Google Scholar]
- Okazaki S, Naganuma A, Kuge S. Peroxiredoxin-mediated redox regulation of the nuclear localization of Yap1, a transcription factor in budding yeast. Antioxid Redox Signal. 2005;7:327–334. doi: 10.1089/ars.2005.7.327. [DOI] [PubMed] [Google Scholar]
- Pannicke U, Ma Y, Lieber MR, Schwarz K. Functional and biochemical dissection of the structure-specific nuclease Artemis. EMBO J. 2004;23:1987–1997. doi: 10.1038/sj.emboj.7600206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannunzio NR, Lieber MR. Dissecting the Roles of Divergent and Convergent Transcription in Chromosome Instability. Cell Rep. 2016 doi: 10.1016/j.celrep.2015.12.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson CE, Wang YH, Griffith JD, Sinden RR. Structural analysis of slipped-strand DNA (S-DNA) formed in (CTG)n. (CAG)n repeats from the myotonic dystrophy locus. Nucleic Acids Res. 1998;26:816–823. doi: 10.1093/nar/26.3.816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V, Chavez S, Perez-Ortin JE. A complete set of nascent transcription rates for yeast genes. PLoS One. 2010;5:e15442. doi: 10.1371/journal.pone.0015442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham P, Bransteitter R, Petruska J, Goodman MF. Processive AID-catalyzed cytosine deamination on single-stranded DNA stimulates somatic hypermutation. Nature. 2003;424:103–107. doi: 10.1038/nature01760. [DOI] [PubMed] [Google Scholar]
- Poinsignon C, Moshous D, Callebaut I, de Chasseval R, Villey I, de Villartay JP. The metallo-beta-lactamase/beta-CASP domain of Artemis constitutes the catalytic core for V(D)J recombination. J Exp Med. 2004;199:315–321. doi: 10.1084/jem.20031142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poltoratsky VP, Wilson SH, Kunkel TA, Pavlov YI. Recombinogenic phenotype of human activation-induced cytosine deaminase. J Immunol. 2004;172:4308–4313. doi: 10.4049/jimmunol.172.7.4308. [DOI] [PubMed] [Google Scholar]
- Popoff SC, Spira AI, Johnson AW, Demple B. Yeast structural gene (APN1) for the major apurinic endonuclease: homology to Escherichia coli endonuclease IV. Proc Natl Acad Sci U S A. 1990;87:4193–4197. doi: 10.1073/pnas.87.11.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez H, Drouin R, Holmquist GP, O’Connor TR, Boiteux S, Laval J, Doroshow JH, Akman SA. Mapping of copper/hydrogen peroxide-induced DNA damage at nucleotide resolution in human genomic DNA by ligation-mediated polymerase chain reaction. J Biol Chem. 1995;270:17633–17640. doi: 10.1074/jbc.270.29.17633. [DOI] [PubMed] [Google Scholar]
- Rothstein R. Targeting, disruption, replacement, and allele rescue: integrative DNA transformation in yeast. Methods Enzymol. 1991;194:281–301. doi: 10.1016/0076-6879(91)94022-5. [DOI] [PubMed] [Google Scholar]
- Roy D, Yu K, Lieber MR. Mechanism of R-loop formation at immunoglobulin class switch sequences. Mol Cell Biol. 2008;28:50–60. doi: 10.1128/MCB.01251-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy D, Zhang Z, Lu Z, Hsieh CL, Lieber MR. Competition Between the RNA Transcript and the Nontemplate DNA Strand During R-Loop Formation In Vitro: A Nick Can Serve as a Strong R-loop Initiation Site. Mol Cell Biol. 2010;30:146–159. doi: 10.1128/MCB.00897-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito I, Takayama M, Sugiyama H, Nakatani K. Photoinduced DNA Cleavage via Electron Transfer: Demonstration That Guanine Residues Located 5′ to Guanine Are the Most Electron-Donating Sites. J Am Chem Soc. 1995;117:6406–6407. [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning. New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Sherman FFG, Hicks J. Methods in Yeast Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1986. [Google Scholar]
- Shinkura R, Tian M, Khuong C, Chua K, Pinaud E, Alt FW. The influence of transcriptional orientation on endogenous switch region function. Nature Immunol. 2003;4:435–441. doi: 10.1038/ni918. [DOI] [PubMed] [Google Scholar]
- Sinden RR. DNA Structure and Function. San Diego: Academic Press; 1994. [Google Scholar]
- Siow CC, Nieduszynska SR, Muller CA, Nieduszynski CA. OriDB, the DNA replication origin database updated and extended. Nucleic Acids Res. 2012;40:D682–686. doi: 10.1093/nar/gkr1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spell RM, Jinks-Robertson S. Determination of mitotic recombination rates by fluctuation analysis in Saccharomyces cerevisiae. Methods Mol Biol. 2004;262:3–12. doi: 10.1385/1-59259-761-0:003. [DOI] [PubMed] [Google Scholar]
- Storici F, Lewis LK, Resnick MA. In vivo site-directed mutagenesis using oligonucleotides. Nat Biotechnol. 2001;19:773–776. doi: 10.1038/90837. [DOI] [PubMed] [Google Scholar]
- Swaminathan S, Klemm L, Park E, Ford A, Kweon S-M, Trageser D, Hasselfeld B, Henke N, Mooster J, Geng H, et al. Mechanisms of clonal evolution in acute lymphoblastic leukemia. Nature Immunology. 2015 doi: 10.1038/ni.3160. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas BJ, Rothstein R. Elevated recombination rates in transcriptionally active DNA. Cell. 1989;56:619–630. doi: 10.1016/0092-8674(89)90584-9. [DOI] [PubMed] [Google Scholar]
- Thrash C, Bankier AT, Barrell BG, Sternglanz R. Cloning, characterization, and sequence of the yeast DNA topoisomerase I gene. Proc Natl Acad Sci U S A. 1985;82:4374–4378. doi: 10.1073/pnas.82.13.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai AG, Engelhart AE, Hatmal MM, Houston SI, Hud NV, Haworth IS, Lieber MR. Conformational variants of duplex DNA correlated with cytosine-rich chromosomal fragile sites. J Biol Chem. 2009;284:7157–7164. doi: 10.1074/jbc.M806866200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai AG, Lieber MR. Mechanisms of chromosomal rearrangement in the human genome. BMC Genomics. 2010;11(Suppl 1):S1. doi: 10.1186/1471-2164-11-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai AG, Lu H, Raghavan SC, Muschen M, Hsieh CL, Lieber MR. Human chromosomal translocations at CpG sites and a theoretical basis for their lineage and stage specificity. Cell. 2008;135:1130–1142. doi: 10.1016/j.cell.2008.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol. 2002;3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- Waters TR, Swann PF. Kinetics of the action of thymine DNA glycosylase. J Biol Chem. 1998;273:20007–20014. doi: 10.1074/jbc.273.32.20007. [DOI] [PubMed] [Google Scholar]
- Welzel N, TTL, Marculescu R, Mitterbauer G, Chott A, Pott C, Kneba M, Du MQ, Kusec R, Drach J, et al. Templated nucleotide addition and immunoglobulin JH-gene utilization in t(11;14) junctions: implications for the mechanism of translocation and the origin of mantle cell lymphoma. Cancer Res. 2001;61:1629–1636. [PubMed] [Google Scholar]
- Yadav P, Harcy V, Argueso JL, Dominska M, Jinks-Robertson S, Kim N. Topoisomerase I plays a critical role in suppressing genome instability at a highly transcribed G-quadruplex-forming sequence. PLoS Genet. 2014;10:e1004839. doi: 10.1371/journal.pgen.1004839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Chedin F, Hsieh C-L, Wilson TE, Lieber MR. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nature Immunol. 2003;4:442–451. doi: 10.1038/ni919. [DOI] [PubMed] [Google Scholar]
- Yu K, Huang FT, Lieber MR. DNA substrate length and surrounding sequence affect the activation induced deaminase activity at cytidine. J Biol Chem. 2004;279:6496–6500. doi: 10.1074/jbc.M311616200. [DOI] [PubMed] [Google Scholar]
- Yu K, Lieber MR. Nucleic acid structures and enzymes in the immunoglobulin class switch recombination mechanism. DNA Repair. 2003;2:1163–1174. doi: 10.1016/j.dnarep.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Yu K, Roy D, Bayramyan M, Haworth IS, Lieber MR. Fine-structure analysis of activation-induced deaminase accessbility to class switch region R-loops. Mol Cell Biol. 2005;25:1730–1736. doi: 10.1128/MCB.25.5.1730-1736.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.