

Abstract
Background:
Genetic factors in the pathogenesis of cardiomyopathies have received a lot of attention during the past 2 decades. Some studies have reported that angiotensin-converting enzyme (ACE) gene has been associated with hypertrophic cardiomyopathy (HCM). However, there have been inconsonant results among different studies. To clarify the influence of ACE on HCM, a systemic review and meta-analysis of case–control studies were performed.
Methods:
The following databases were searched to indentify related studies: PubMed database, the Embase database, the Cochrane Central Register of Controlled Trials database, China National Knowledge Information database, and Chinese Scientific and Technological Journal database. Search terms included “hypertrophic cardiomyopathy,” “angiotensin converting enzyme” or “ACE,” and “polymorphism or mutation.”
Results:
Fifteen separate studies were suitable for the inclusion criterion. The selected studies contained 2972 participants, including 1047 in HCM group and 1925 controls. Pooled odds ratios (ORs) were calculated to assess the association between ACE insertion/deletion (I/D) polymorphism and HCM. Our case–control data indicated that D allele carrier is a risk allele in all genetic models: allele contrast (D vs I: OR = 1.35, 95% confidence interval [CI]: 1.10–1.65, P = .004), homozygous comparison (DD vs II: OR = 1.69; 95% CI: 1.12–2.54; P = .01), dominant model (DD + ID vs II: OR = 1.52, 95% CI: 1.15–2.02, P = .003), and recessive model (DD vs ID + II: OR = 1.34, 95% CI: 0.99–1.81, P = .03).
Conclusion:
In summary, the current meta-analysis provided solid evidence suggesting that ACE gene I/D polymorphism was probably a genetic risk factor for HCM.
Keywords: ACE, hypertrophic cardiomyopathy, meta-analysis, polymorphism
1. Introduction
Left-ventricular hypertrophy (LVH) is a physiological adaptation of the heart to increased workload. LVH is frequently secondary to clinical conditions such as hypertension, valvular disease, and myocardial infarction.[1,2] However, some patients develop the cardiac hypertrophy in the absence of these conditions that impose overwork to the heart. This primary/essential form of LVH is frequently familial and caused by mutations in sarcomeric genes, and is designated as hypertrophic cardiomyopathy (HCM).[3] HCM, the most common hereditary cardiac disease, affects 1 in every 500 people in the general population and represents a major cause of sudden cardiac death in adolescent athletes.[4]
HCM is frequently caused by mutations in genes encoding sarcomeric proteins.[5–7] It is reported that several gene polymorphisms, including those encoding the components of the renin–angiotensin system (RAS), have been associated with the risk of developing LVH, and could also modify the clinical phenotype in HCM patients.[8,9] Previous studies suggested that RAS acted on cellular hypertrophy and cell proliferation,[10] and therefore played a regulatory role in cardiac function, blood pressure, and electrolyte homeostasis.[11] In the end, it can affect both left cardiac ventricle (LV) hypertrophy and remodeling.[12] It has been demonstrated that components of the RAS such as angiotensinogen, renin, angiotensin-converting enzyme (ACE), and angiotensin II receptors exist within the heart and may function independently from the circulating RAS.[13] ACE, through conversion of angiotensin I to angiotensin II, the latter as trophic as well as mitogenic hormone, acts as a growth factor for cardiac myocytes and induces cardiac hypertrophy independent of hemodynamic or neurohumoral effects.[14]ACE is 21 kb length, including 26 exons, located on long arm of chromosome 17 (17q23.3) locus of the human genome. It will be inherited independently of the diseased sarcomeric genes which are located on different chromosomes. The restriction fragment length polymorphism, a 287 base pair (bp) insertion/deletion (I/D), is located inside intron 16 of the ACE gene and corresponds to an Alu repetitive sequence. DD genotype subjects have a higher level of ACE and angiotensin II and, consequently, an increase in hypertrophy and fibrosis.[12,14–16] That is, the ACE levels in the human heart are in part determined by the so-called I/D polymorphism.[16] Therefore, the angiotensin II levels increase and then also affect the phenotypic expression in HCM.[15]
In spite of the above-mentioned reports associating RAS and HCM, the studies from different populations have been conflicting and the role of the RAS system in modifying the phenotype in HCM remains controversial. As meta-analysis is a reliable way to combine information from many studies and thus may provide more conclusive answers, we decide to evaluate the influence of ACE polymorphisms on the HCM phenotype.
2. Methods
This study was approved by the ethics committees of the First Hospital of Jilin University and conformed to the principles of the Declaration of Helsinki. Written informed consent was obtained from each participant before entry into the study, and all of the procedures were in accordance with institutional guidelines.
2.1. Search strategy
The following database was searched to identify related studies: PubMed database, the Embase database, the Cochrane Central Register of Controlled Trials database, China National Knowledge Information data base, and the Wanfang databases. For the association of ACE I/D and HCM, the following search terms were used in searching the PubMed database: “hypertrophic cardiomyopathy”, “angiotensin converting enzyme” or “ACE” and “polymorphism or mutation”. The full texts of the retrieved articles were scrutinized to inspect whether data on the topic of interest were included. We systematically searched eligible studies reported before Nov 2016. The references of all retrieved articles were also screened.
2.2. Inclusion/exclusion criteria
The studies included in the meta-analysis must meet all the following three criteria: evaluating the association of ACE I/D polymorphism with HCM; using case–control design; containing genotype data of II, ID, and DD, and comprehensive statistical indicators directly or indirectly: odds ratio (OR) values and 95% confidence interval (CI); using similar themes and methods; and satisfying Hardy–Weinberg equilibrium (HWE) among the controls. However, all the patients were excluded for potential stimulus such as hypertension, ischemic heart disease, valvular heart disease, congenital malformations of the heart or vessels, and intrinsic pulmonary disease for HCM.
2.3. Data extraction
Two investigators independently reviewed all studies and extracted the data using a standard information extraction and reached consensus on all items. If there was discrepancy between them, it was settled by discussion until a consensus was reached. The data extracted from the studies included such details as the first author, publication year, country, ethnicity, age, genotype distribution in cases and controls, source of controls, diagnostic criteria, and HWE test. The excluded literatures were comprised of studies of poor research quality, providing little or insufficient data, violating the inclusion criteria, or repeated publications. If the same research result appeared in different articles, the result was only adopted once in the present meta-analysis.
2.4. Statistical methods
The pooled ORs and corresponding 95% CIs of different studies were calculated to compare dichotomous statistics between studies. The pooled ORs were calculated using 4 models: allele model (D vs I), homozygous comparison (DD vs II), dominant model (DD + ID vs II), and recessive model (DD vs II + ID). To assess heterogeneity across the studies, Cochrane Q test[17] and I2 statistic[18,19] were calculated. If the studies were shown to be homogeneous with P ≥ .10 and I2 < 50%, the fixed-effects model (the Mantel–Haenszel method) was selected. Otherwise, the random-effects model (the DerSimonian and Laird method) was applied.
If the outcomes were heterogeneous, prespecified subgroup comparisons were conducted to detect the influence of the following factors on the ACE gene I/D polymorphism-HCM correlation.
In addition, a sensitivity analysis was performed to assess the stability of the results. The significance of the pooled ORs was determined by the Z-test, and a P < .05 was considered significantly. Sensitivity analyses were conducted by deleting a single study each time involved in the meta-analysis to identify the potential influence of the individual data set on the pooled ORs. Egger test and the visual symmetry of funnel plot were assessed to examine publication bias of the related studies. This meta-analysis was performed using the software STATA version 13.0 (Stata Corporation, TX) and Review Manager 5.3 (Cochrane Collaboration, Oxford, UK). All P-values were based on 2-sided tests.
3. Results
3.1. Characteristics of eligible studies
Initially, 378 studies were identified as potentially eligible candidates from the electronic and manual searches. After screening the titles and abstracts, 322 studies were excluded because of not case–control trials or irrelevant studies or no desired polymorphism (including other review papers). Full texts of 56 papers were retrieved and most were excluded because they were duplicate publications or focused on ACE gene other polymorphisms or included no sufficient data. Additionally, studies no satisfying HWE were excluded. Finally, 15 articles met the selection criteria. Therefore, a total of 15 eligible original reports were included in the final meta-analysis. A flow diagram of the study selection is shown in Fig. 1.
Figure 1.
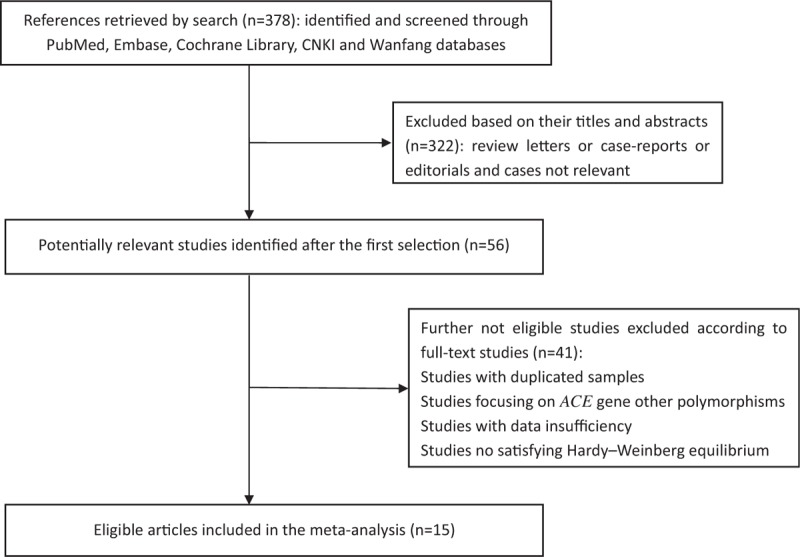
Flow chart of study selection.
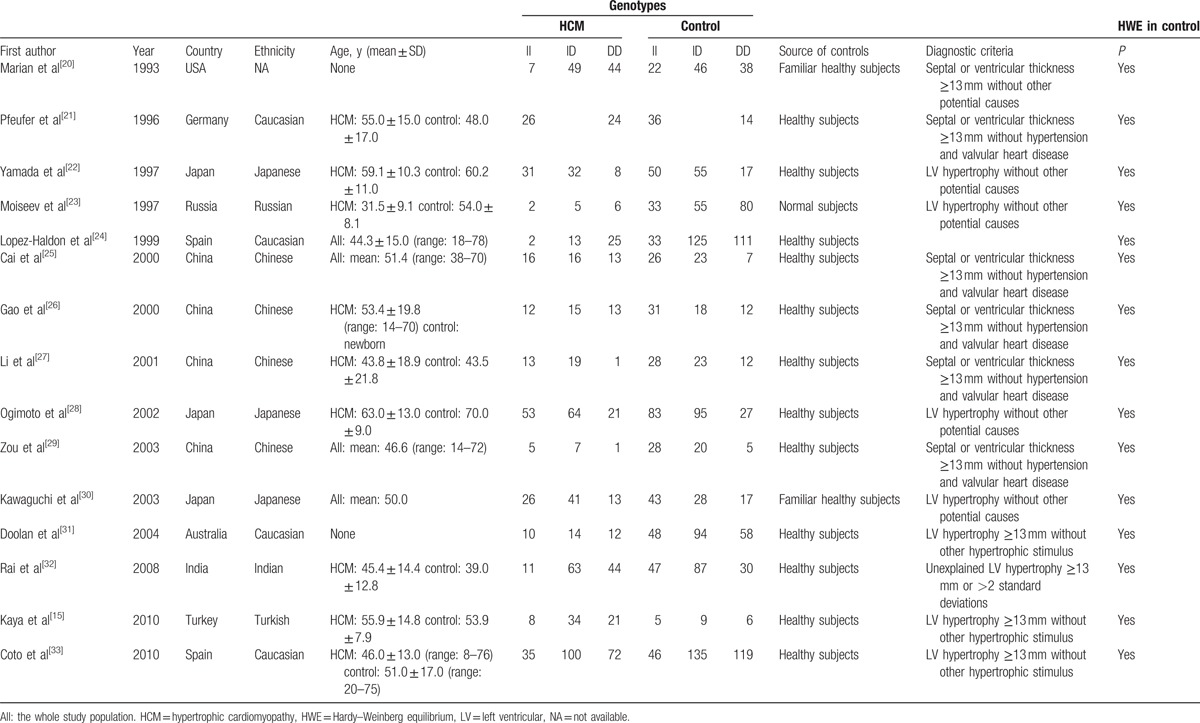
Table 1 describes the characteristics of selected studies included in the meta-analysis. Briefly, the meta-analysis in the present study was performed with 15 articles.[15,20–33] A total of 8 studies included Asian populations,[24,26–31,33] and 6 studies included Caucasian populations.[19,23,25,32,34,35] All studies were case–control studies. The distributions of the genotypes in the control populations were consistent with HWE in all of the studies.
Table 1.
Characteristic of eligible studies in the meta-analysis.
3.2. Association of ACE I/D polymorphisms and HCM susceptibility
For the genetic variant ACE I/D in the 15 studies, including 1047 cases and 1925 controls, D allele frequency was significantly higher in HCM group (75.5%) than in control group (71.0%), P = .009.
The potential heterogeneity was found in all comparisons (all P < .10), so random model was used in the meta-analysis (Table 2). For ACE I/D variant, the summary OR for allele D versus I is shown in Fig. 2 and Table 2, and the summary OR was 1.35 (allel model, D vs I: 95% CI: 1.10–1.65; P = .004). Overall comparison of DD genotype with II genotype showed significant association of this variant with HCM risk (homozygous model, DD vs II: OR = 1.69; 95% CI: 1.12–2.54; P = .01; Fig. 3). In the current meta-analysis, the association between ACE I/D polymorphism and the risk of HCM was also investigated under both the dominant genetic model and recessive genetic model. Under dominant genetic model, the association was also detected between the ACE I/D variant and HCM risk and the pooled OR was 1.52 (DD + ID vs II: 95% CI: 1.15–2.02; P = .003; Fig. 4). Under recessive genetic model, the ACE DD genotype was significantly associated with HCM risk compared with the wild-type I allele, and the pooled OR was 1.34 (DD vs ID + II: 95% CI: 0.99–1.81; P = .03; Table 2). In total, results in across different ethnic populations strongly indicated that D allele and DD genotype of the ACE gene I/D polymorphism were probably the genetic risk factor of HCM.
Table 2.
Odds ratio and heterogeneity tests for ACE I/D polymorphism and HCM in different models.

Figure 2.
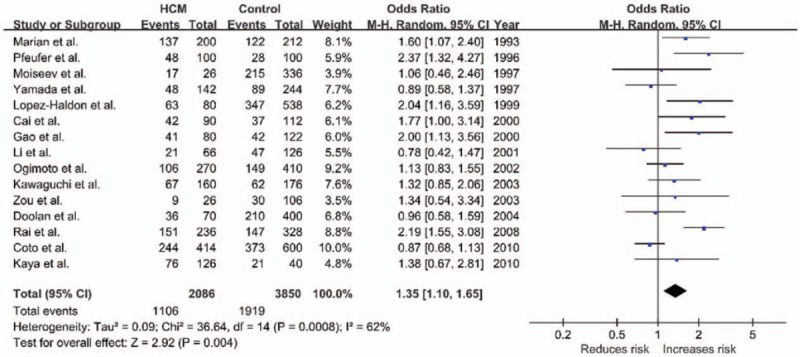
Forest plot of HCM and ACE I/D in an allel model (D vs I), the horizontal lines correspond to the study-specific OR and 95% CI, respectively. The area of the squares reflects the study-specific weight. The diamond represents the pooled results of OR and 95% CI. ACE = angiotensin-converting enzyme, CI = confidence interval, HCM = hypertrophic cardiomyopathy, I/D = insertion/deletion, OR = odds ratio.
Figure 3.
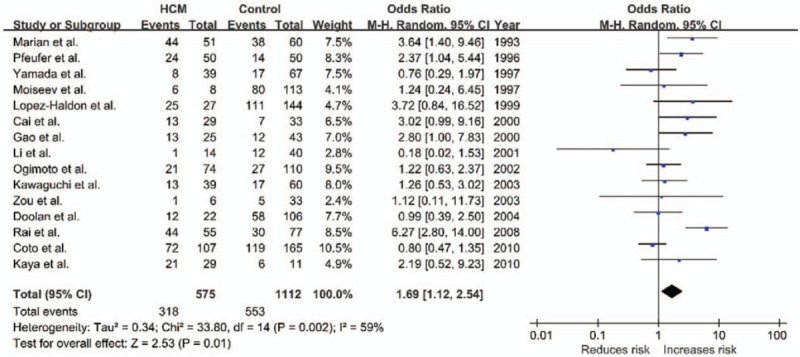
Forest plot of HCM and ACE I/D in a homozygous model (DD vs II), the horizontal lines correspond to the study-specific OR and 95% CI, respectively. The area of the squares reflects the study-specific weight. The diamond represents the pooled results of OR and 95% CI. ACE = angiotensin-converting enzyme, CI = confidence interval, HCM = hypertrophic cardiomyopathy, I/D = insertion/deletion, OR = odds ratio.
Figure 4.
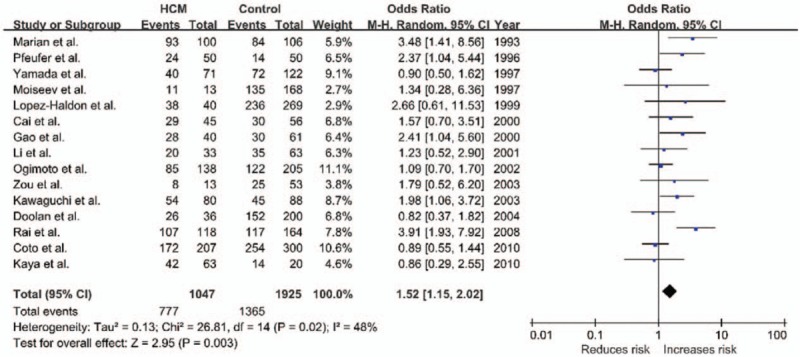
Forest plot of HCM and ACE I/D in a dominant model (DD + ID vs II), the horizontal lines correspond to the study-specific OR and 95% CI, respectively. The area of the squares reflects the study-specific weight. The diamond represents the pooled results of OR and 95% CI. ACE = angiotensin-converting enzyme, CI = confidence interval, HCM = hypertrophic cardiomyopathy, I/D = insertion/deletion, OR = odds ratio.
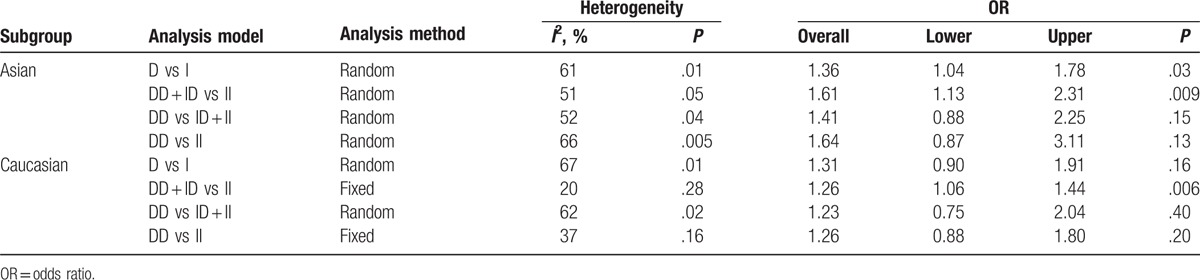
We performed a meta-analysis in 2 subgroups based on ethnicity: Asian population and Caucasian population. Results of the analysis in the Asian subgroup and the Caucasian subgroup are presented in Table 3. From our subgroup analyses, we found that Asian and Caucasian subgroups with the DD genotype of ACE showed a higher risk of HCM; however, between-study heterogeneity was not eliminated apart from Caucasian subgroup (DD + ID vs II: Pheterogeneity = .28, I2 = 20%; DD vs II: Pheterogeneity = .16, I2 = 37%).
Table 3.
Subgroup analysis of different genetic models by ethnicity.
3.3. Publication bias
Funnel plots of all the studies above were listed in Fig. 5. No publication bias was observed, as the shape of the funnel plots seemed to show no evident asymmetry in each meta-analysis. It is further validated by the Egger test (P > . 05).
Figure 5.
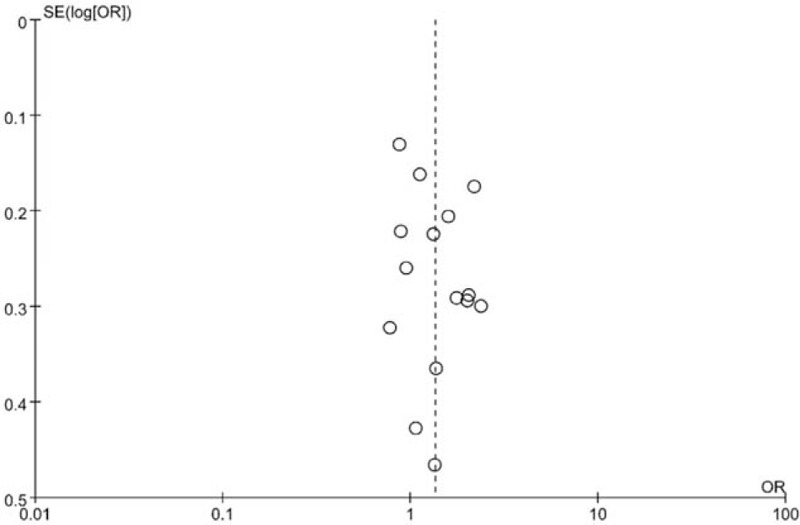
Begg funnel plot for publication bias tests. Each point represents a separate study for the indicated association. Log[OR] represents natural logarithm of odds ratio (OR). Vertical line represents the mean effects size.
3.4. Sensitivity analysis
Deletion of 1 single study from the overall pooled analysis each time to check the influence of the removed dataset to the overall ORs to assess the sensitivity analysis did not alter or impact the overall ORs. This indicated that the results of the meta-analysis about ACE gene I/D polymorphism and risk of HCM were relatively stable and reliable (Fig. 6).
Figure 6.
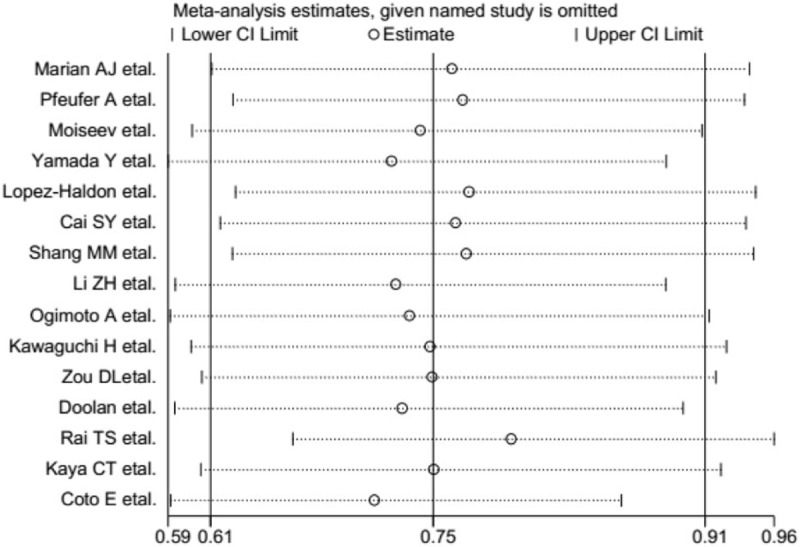
Potential outliers (ie, data points that are far outside the norm) were identified by a sensitivity analysis.
4. Discussion
In the present study, we systematically reviewed all available published studies and performed a meta-analysis to evaluate the association of ACE gene I/D polymorphisms with HCM. Fifteen studies were included in this meta-analysis. Pooled ORs showed a significant association between ACE I/D polymorphism and HCM susceptibility in the genetic models (allele, dominant, and recessive). Sensitivity analysis further showed that the association was stable, and Begg and Egger tests indicated a lack of publication bias. We conducted a comprehensive meta-analysis on 15 published studies with 1047 cases and 1925 controls relating the variant of the ACE I/D to the risk of HCM, which provided better ability to detect smaller effect sizes. Its strength was based on the accumulation of published data, giving greater information to detect significant differences.
The angiotensin I converting enzyme enhances the synthesis of angiotensin II (Ang II), which induces cell proliferation, migration, and hypertrophy, and enhances the proinflammatory cytokines and matrix metalloproteinases. Thus, overexpression of Ang II plays a powerful role in cardiomyopathy. Previous studies have found that ACE I/D polymorphisms are related with plasma Ang II levels. ACE I/D polymorphisms have been extensively examined for a variety of clinical endpoints, such as hypertension, coronary artery disease,[34] cough,[35] and pulmonary complications following esophagectomy.[36] The ACE I/D polymorphisms also modulate the phenotype in patients with HCM. However, studies from different populations have demonstrated conflicting data. Rai et al[32] found that D allele of ACE I/D polymorphism significantly influences the HCM phenotypes. In contrast, Yamada et al[22] reported that the ACE I/D polymorphisms are not related to HCM in a Japanese population. Analyses assuming additive, dominant, or recessive effects of the D allele failed to show any association with HCM. Moreover, our current meta-analysis found that compared with ACE II genotype, patients with D allele showed a significantly increased risk of HCM, suggesting that ACE I/D polymorphisms might attribute to HCM risk.
As ACE gene may modify the phenotypic expression of the HCM, the administration of angiotensin-converting enzyme inhibitor (ACEI) or the angiotensin II type 1 receptor (AT1-R) antagonist remains interesting in HCM. It is now known that in presence of HCM, patients expressing D/D genotype for ACE gene show an increased level of serum ACE, have an increased risk of sudden death, and present an increased severity of hypertrophy.[20,37–39] Angiotensin II has trophic effects on the heart and plays an important role in the development of myocardial hypertrophy.[40] This knowledge has generated interest in ACEI or AT1-R antagonist as a potential therapeutic tool to prevent or reduce myocardial fibrosis, perhaps reduce the risk of arrhythmias and sudden death, and reduce the progression of diastolic dysfunction in HCM.[41–43] A double-blind, placebo-controlled, randomized study showed that the long-term administration of the AT1-R antagonist candesartan in patients with HCM was associated with the significant regression of LVH, improvement of left ventricular function, and exercise tolerance.[43] Thus, AT1-R antagonist has the potential to attenuate myocardial hypertrophy and may, therefore, provide a new treatment option to prevent sudden cardiac death in patients with HCM.
The results of this meta-analysis should be interpreted with some degree of caution, because there were several limitations in our analysis. First, we failed to subgroup the familial HCM and sporadic HCM in HCM patients due to the relatively insufficient studies. Second, heterogeneity among the included studies may affect the interpretation of the results of the meta-analysis. Third, most of the sample sizes of the referenced studies are relatively small, which might weaken the meta-analysis results. Furthermore, we could not investigate various ethnic distributions because most of the studies included only the Caucasian or Asian populations. It may be a result of various factors such as differences in study designs, environmental backgrounds, genetic constitution, or sample selection between studies. Taken together, all of these limitations may have affected the results of the present study.
5. Conclusions
The present meta-analysis finds an association between HCM and ACE I/D polymorphism. The findings of the current study may add benefit to risk stratification strategies in patients with HCM and may encourage further study focusing on the effect of ACE I/D polymorphisms on HCM risk. These results also suggest a potential treatment approach by regulating RAS in HCM patients. Prospective and more genome-wide association studies are needed to clarify the real role of the ACE gene in determining susceptibility to HCM.
Footnotes
Abbreviations: ACE = angiotensin-converting enzyme, CI = confidence interval, HCM = hypertrophic cardiomyopathy, HWE = Hardy–Weinberg equilibrium, I/D = insertion/deletion, LVH = left-ventricular hypertrophy, OR = odds ratio, RAS = renin–angiotensin system, SHCM = sporadic hypertrophic cardiomyopathy.
The authors have no funding and conflicts of interest to disclose.
References
- [1].Sadoshima J, Izumo S. The cellular and molecular response of cardiac myocytes to mechanical stress. Annu Rev Physiol 1997;59:551–71. [DOI] [PubMed] [Google Scholar]
- [2].Lorell BH, Carabello BA. Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation 2000;102:470–9. [DOI] [PubMed] [Google Scholar]
- [3].Bos JM, Towbin JA, Ackerman MJ. Diagnostic prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J Am Coll Cardiol 2009;54:201–11. [DOI] [PubMed] [Google Scholar]
- [4].Orenes-Pinero E, Hernandez-Romero D, Jover E, et al. Impact of polymorphisms in the renin-angiotensin-aldosterone system on hypertrophic cardiomyopathy. J Renin Angiotensin Aldosterone Syst 2011;12:521–30. [DOI] [PubMed] [Google Scholar]
- [5].Marian AJ, Roberts R. Recent advances in the molecular genetics of hypertrophic cardiomyopathy. Circulation 1995;92:1336–47. [DOI] [PubMed] [Google Scholar]
- [6].Niimura H, Bachinski LL, Sangwatanaroj S, et al. Mutations in the gene for cardiac myosin-binding protein C and lateonset familial hypertrophic cardiomyopathy. N Engl J Med 1998;338:1248–57. [DOI] [PubMed] [Google Scholar]
- [7].Spirito P, Seidman CE, McKenna WJ, et al. The management of hypertrophic cardiomyopathy. N Engl J Med 1997;336:775–85. [DOI] [PubMed] [Google Scholar]
- [8].Bleumink GS, Schut AF, Sturkenboom MC, et al. Genetic polymorphisms and heart failure. Genet Med 2004;6:465–74. [DOI] [PubMed] [Google Scholar]
- [9].Keren A, Syrris P, McKenna WJ. Hypertrophic cardiomyopathy: the genetic determinants of clinical disease expression. Nat Clin Pract Cardiovasc Med 2008;5:158–68. [DOI] [PubMed] [Google Scholar]
- [10].Sadoshima J, Izumo S. Molecular characterization of angiotensin II-induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ Res 1993;73:413–23. [DOI] [PubMed] [Google Scholar]
- [11].Griendling KK, Murphy TJ, Alexander RW. Molecular biology of the renin-angiotensin system. Circulation 1993;87:1816–28. [DOI] [PubMed] [Google Scholar]
- [12].Wang JG, Staessen JA. Genetic polymorphisms in the renin-angiotensin system: relevance for susceptibility to cardiovascular disease. Eur J Pharmacol 2000;410:289–302. [DOI] [PubMed] [Google Scholar]
- [13].Lindpainter K, Ganfen D. The cardiac renin-angiotensin system: an appraisal of present experimental and clinical evidence. Circ Res 1991;68:905–21. [DOI] [PubMed] [Google Scholar]
- [14].Perkins MJ, Van Driest SL, Ellsworth EG, et al. Gene-specific modifying effects of pro-LVH polymorphisms involving the reninangiotensin-aldosterone system among 389 unrelated patients with hypertrophic cardiomyopathy. Eur Heart J 2005;26:2457–62. [DOI] [PubMed] [Google Scholar]
- [15].Kaya CT, Gurlek A, Altin T, et al. The relationship between angiotensin converting enzyme gene I/D polymorphism and QT dispersion in patients with hypertrophic cardiomyopathy. J Renin Angiotensin Aldosterone Syst 2010;11:192–7. [DOI] [PubMed] [Google Scholar]
- [16].Danser AH, Schalekamp MA, Bax WA, et al. Angiotensin-converting enzyme in the human heart. Effect of the deletion/insertion polymorphism. Circulation 1995;92:1387–8. [DOI] [PubMed] [Google Scholar]
- [17].DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials 1986;7:177–88. [DOI] [PubMed] [Google Scholar]
- [18].Higgins JP, Thompson SG, Deeks JJ, et al. Measuring inconsistency in meta-analyses. BMJ 2003;327:557–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].DerSimonian R. Meta-analysis in the design and monitoring of clinical trials. Stat Med 1996;15:1237–48. discussion 49–52. [DOI] [PubMed] [Google Scholar]
- [20].Marian AJ, Yu QT, Workman R, et al. Angiotensinconverting enzyme polymorphism in hypertrophic cardiomyopathy and sudden cardiac death. Lancet 1993;342:1085–6. [DOI] [PubMed] [Google Scholar]
- [21].Pfeufer A, Osterziel KJ, Urata H, et al. Angiotensin-converting enzyme and heart chymase gene polymorphisms in hypertrophic cardiomyopathy. Am J Cardiol 1996;78:362–4. [DOI] [PubMed] [Google Scholar]
- [22].Yamada Y, Ichihara S, Fujimura T. Lack of association of polymorphisms of the angiotensin converting enzyme and angiotensinogen genes with nonfamilial hypertrophic or dilated cardiomyopathy. Am J Hypertens 1997;10:921–8. [DOI] [PubMed] [Google Scholar]
- [23].Moiseev VS, Demurov LM, Kobalava Zh D, et al. The polymorphism of the angiotensin- converting enzyme gene in patients with hypertension, left ventricular hypertrophy and the development of a myocardial infarct at a young age. Preliminary report. Ter Arkh 1997;69:18–23. [PubMed] [Google Scholar]
- [24].Lopez-Haldon J, Garcia-Lozano JR, Martinez Martinez A, et al. The effect of polymorphisms of the angiotensinconverting enzyme and angiotensinogen genes on the phenotypic expression of Spanish patients with hypertrophic cardiomyopathy. Med Clin (Barc) 1999;113:161–3. [PubMed] [Google Scholar]
- [25].Cai SY, Wu X. The relationship between angiotensin-converting enzyme gene polymorphism in patients with hypertrophic cardiomyopathy and left ventricular hypertrophy. Zhejiang Med J 2000;9:521–3. [Google Scholar]
- [26].Gao MM, Xiao B, Hu DY, et al. Angiotensin-converting enzyme genotype distribution in patients with hypertrophic cardiomyopathy. J Capital Univ Med Sci 2000;2:112–4. [Google Scholar]
- [27].Li ZH, Ma AQ, Wu GR, et al. The relationship between angiotensin- converting enzyme gene I/D polymorphism and hypertrophic cardiomyopathy. J Clin Cardiol (China) 2001;3:231–4. [Google Scholar]
- [28].Ogimoto A, Hamada M, Nakura J, et al. Relation between angiotensin-converting enzyme II genotype and atrial fibrillation in Japanese patients with hypertrophic cardiomyopathy. J Hum Genet 2002;47:184–9. [DOI] [PubMed] [Google Scholar]
- [29].Zou DL, Yuan FX, Guo JJ, et al. Relationship between hypertrophic cardiomyopathy and dilated cardiomyopathy and angiotensin-converting enzyme gene polymorphism. J China Med Univ 2003;162–4. [Google Scholar]
- [30].Kawaguchi H. Angiotensin-converting enzyme and angiotensinogen gene polymorphism in hypertrophic cardiomyopathy. Exp Clin Cardiol 2003;8:155–9. [PMC free article] [PubMed] [Google Scholar]
- [31].Doolan G, Nguyen L, Chung J, et al. Progression of left ventricular hypertrophy and the angiotensin-converting enzyme gene polymorphism in hypertrophic cardiomyopathy. Int J Cardiol 2004;96:157–63. [DOI] [PubMed] [Google Scholar]
- [32].Rai TS, Dhandapany PS, Ahluwalia TS, et al. ACE I/D polymorphism in Indian patients with hypertrophic cardiomyopathy and dilated cardiomyopathy. Mol Cell Biochem 2008;311:67–72. [DOI] [PubMed] [Google Scholar]
- [33].Coto E, Palacin M, Martin M, et al. Functional polymorphisms in genes of the angiotensin and serotonin systems and risk of hypertrophic cardiomyopathy: AT1R as a potential modifier. J Transl Med 2010;8:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kato N, Tatara Y, Ohishi M, et al. Angiotensinconverting enzyme single nucleotide polymorphism is a genetic risk factor for cardiovascular disease: a cohort study of hypertensive patients. Hypertens Res 2011;34:728–34. [DOI] [PubMed] [Google Scholar]
- [35].Nishio K, Kashiki S, Tachibana H, et al. Angiotensin-converting enzyme and bradykinin gene polymorphisms and cough: a meta-analysis. World J Cardiol 2011;3:329–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lee JM, Lo AC, Yang SY, et al. Association of angiotensin-converting enzyme insertion/deletion polymorphism with serum level and development of pulmonary complications following esophagectomy. Ann Surg 2005;241:659–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lechin M, Quinones MA, Omran A, et al. Angiotensin-I converting enzyme genotypes and left ventricular hypertrophy in patients with hypertrophic cardiomyopathy. Circulation 1995;92:1808–12. [DOI] [PubMed] [Google Scholar]
- [38].Rigat B, Hubert C, Alhenc-Gelas F, et al. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of the serum enzyme level. J Clin Invest 1990;86:1343–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Jan Danser AH, Schalekamp M, Bax WA, et al. Angiotensin-converting enzyme in human heart. Effect of the deletion/insertion polymorphism. Circulation 1995;92:1387–8. [DOI] [PubMed] [Google Scholar]
- [40].Kawano H, Do YS, Kawano Y, et al. Angiotensin II has multiple profibrotic effects in human cardiac fibroblasts. Circulation 2000;101:1130–7. [DOI] [PubMed] [Google Scholar]
- [41].Amberger CN, Glardon O, Glaus T, et al. Effects of benazepril in the treatment of feline hypertrophic cardiomyopathy. Results of a prospective, open-label, multicenter clinical trial. J Vet Cardiol 1999;1:19–26. [DOI] [PubMed] [Google Scholar]
- [42].Rush JE, Freeman LM, Brown DJ, et al. The use of enalapril in the treatment of feline hypertrophic cardiomyopathy. J Am Anim Hosp Assoc 1998;34:38–41. [DOI] [PubMed] [Google Scholar]
- [43].Martin P, Pavel G, Roman K, et al. The effects of candesartan on left ventricular hypertrophy and function in nonobstructive hypertrophic cardiomyopathy a pilot, randomized study. J Mol Diagn 2009;11:35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]