

Abstract
Rationale:
Sporadic Creutzfeldt-Jakob disease (sCJD) mainly occurs in the elderly, with the peak age of onset ranging from 55 to 75 years. The symptoms of sCJD are not unique, and laboratory tests such as magnetic resonance imaging (MRI), electroencephalogram (EEG) and cerebrospinal fluid (CSF)14-3-3 protein have low sensitivity or specificity. Therefore, excluding treatable diseases and establishing a diagnosis could be difficult in young patients with suspected sCJD. Recently, real-time quaking-induced conversion (RT-QuIC) has been used in the diagnosis of sCJD, with more than 95% sensitivity and 100% specificity.
Patient concerns:
We report the case of an 18-year-old woman presented with cerebellar ataxia, blurred vision, rapidly progressive dementia, tremor and involuntary movements, urinary incontinence, mutism, and eventually myoclonus for 16 weeks. Brain MRI scans were unremarkable at the 4th and 8th week after initial symptom presentation, but showed hyperintensity in bilateral basal ganglia and cortical ribboning at the 16th week. Typical periodic bilateral triphasic sharp wave complexes on EEG did not appear until the 16th week after initial symptom presentation.
Diagnoses:
Due to the young age of the patient and the originally unremarkable MRI and EEG findings, we first considered treatable diseases such as autoimmune encephalitis, infections, organic acidemias and toxication. However, extensive tests ruled out these diseases. When she was finally diagnosed with probable sCJD, we were unable to perform a brain biopsy. We confirmed the diagnosis by detecting the scrapie form of prion protein in the CSF using RT-QuIC.
Interventions:
Experimental treatments with corticosteroids, intravenous immunoglobulin and ganciclovir were given.
Outcomes:
Experimental treatments were ineffective. The patient's parents discharged her from our clinic.
Lessons:
We present a case of probable sCJD with an early onset and a complex clinical picture confirmed by RT-QuIC. This case report suggests that RT-QuIC has great value for the diagnosis of atypical cases.
Keywords: age at onset, real-time quaking-induced conversion, sporadic Creutzfeldt–Jakob disease
1. Introduction
Sporadic Creutzfeldt–Jakob disease (sCJD) is a progressive, fatal disease with a mean survival of about 6 months.[1] This disease is relatively common compared with other forms of CJD (genetic CJD and acquired CJD), and accounts for 85% of all cases of human prion diseases.[2] sCJD mainly occurs in the elderly, with a peak age of onset of 55 to 75 years, and a mean age of onset of 64 years.[1] The current Centers for Disease Control (CDC) diagnostic criteria for probable sCJD consist of typical neurological symptoms (rapidly progressive dementia, with at least 2 of the 4 following symptoms: myoclonus, visual or cerebellar signs, pyramidal/extrapyramidal signs, and akinetic mutism), positive laboratory tests (cerebrospinal fluid [CSF] 14-3-3 protein, typical magnetic resonance imaging [MRI], or electroencephalogram [EEG] abnormalities), and the exclusion of alternative diagnoses.[3] The diagnosis of definite sCJD requires a brain biopsy or autopsy.
However, without an invasive brain biopsy, the premortem diagnosis of sCJD is difficult. In younger patients, excluding treatable diseases and establishing a diagnosis can be especially challenging. Many nonprion diseases cause progressive dementia,[4] and the appearance of the 4 other major symptoms varies considerably.[5] In the methionine (M)/valine (V)2, VV2 and MM1 subtypes in which basal ganglia hyperintensity is most common, the frequency of MRI abnormality varies from 70% to 79%.[6] Typical EEG abnormalities of periodic bilateral triphasic sharp wave complexes (PSWC) appear later in the disease course, with a low sensitivity of 44%.[7] The test sensitivity of CSF 14-3-3 protein detection is 86% but the specificity is 68%.[7]
Recently, real-time quaking-induced conversion (RT-QuIC) has been used to diagnose prion diseases. This novel approach uses recombinant prion protein (PrP) as a substrate to amplify femtograms of scrapie form of PrP (PrPSc) seed in CSF to detectable levels in CJD patients.[2,8] The reported sensitivity for both sCJD and genetic CJD is >80%,[2,8,9] and the specificity is >98%.[2]
In this article, we report a case of probable sCJD with an unusually early onset in an 18-year-old woman. Because of her age, a great effort was made to identify treatable diseases. Repeated brain MRI with negative results and nonspecific EEG changes at the early stage of the disease complicated the clinical picture. When the diagnosis was made, it was confirmed by detecting PrPSc in the CSF using RT-QuIC.
2. Case report
An 18-year-old woman presented with gait imbalance after coughing for a week. She then developed blurred vision, diplopia, dementia, dysarthria, and tremor of her head and limbs during the following 8 weeks. She was otherwise healthy, with no history of alcohol ingestion or drug use. Her CSF pressure, cytology and biochemistry results were normal. Brain MRI findings at the 4th week after initial symptom presentation were unremarkable. Visual evoked potentials showed prolonged latency of bilateral P100 waves. She was treated empirically with intravenous pulse steroid therapy followed by high-dose corticosteroids for 4 weeks, intravenous immunoglobulin (IVIg) for 5 days, and ganciclovir for 2 weeks. However, her symptoms continued to deteriorate. She was then referred to our clinic 8 weeks after initial symptom presentation.
Upon admission, examinations of orientation, short-term memory, calculation, concentration, and comprehension revealed a global loss of cognitive function. Physical examination showed a prominent cerebellar syndrome (truncal and limb ataxia, intention tremor, dysarthria). Babinski signs were positive bilaterally. Both static and action tremors of her head and limbs were elicited. In addition, suspicious nuchal rigidity was detected.
We first considered autoimmune encephalitis, central nervous system (CNS) infections, heavy metal poisoning, and organic acidemias. However, chest, abdomen, and pelvic computed tomography scans were unremarkable. In addition, an extensive workup for autoimmunity provided negative results (including serum and CSF anti-N-methyl-d-aspartate receptor, antivoltage-gated potassium channel-complex, anti-γ-aminobutyric acid receptor, anti-α-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor, anticontactin-associated protein 2 antibodies, and classical paraneoplastic antibodies, as well as serum antiglutamic acid decarboxylase antibody, antinuclear, antidouble stranded, antiendomysial, antineutrophil cytoplasmic, and thyroid peroxidase antibodies). Therefore, autoimmune encephalitis was unlikely. Blood and urine toxicity tests including heavy metal and narcotics screening were also negative. Tests for organic acidemias (including blood amino acids, fatty acyl carnitine, and chromatographic analysis of urine metabolic components) and vitamin B12 level were normal. Therefore, we excluded metabolic diseases such as methylmalonic aciduria, 4-hydroxybutyric aciduria, and vitamin B12 insufficiency. We performed a second lumber puncture. CSF biochemistry test showed a normal glucose level, a raised protein level (0.61 g/L), and no leukocytes. Microbiologic examination of the CSF did not detect any viral or bacterial causative agents, including Lyme and measles antibodies. In addition, serum syphilis and human immunodeficiency virus tests were negative. Therefore, CNS infections were unlikely. At the 8th week after initial symptom presentation, repeat brain MRI scans were still unremarkable (Fig. 1A and B), and EEG findings showed diffused θ and δ activities (Fig. 2A). On the 14th week, a second EEG showed further slowing of the background and paroxysmal triphasic waves of bilateral frontal dominancy (Fig. 2B).
Figure 1.
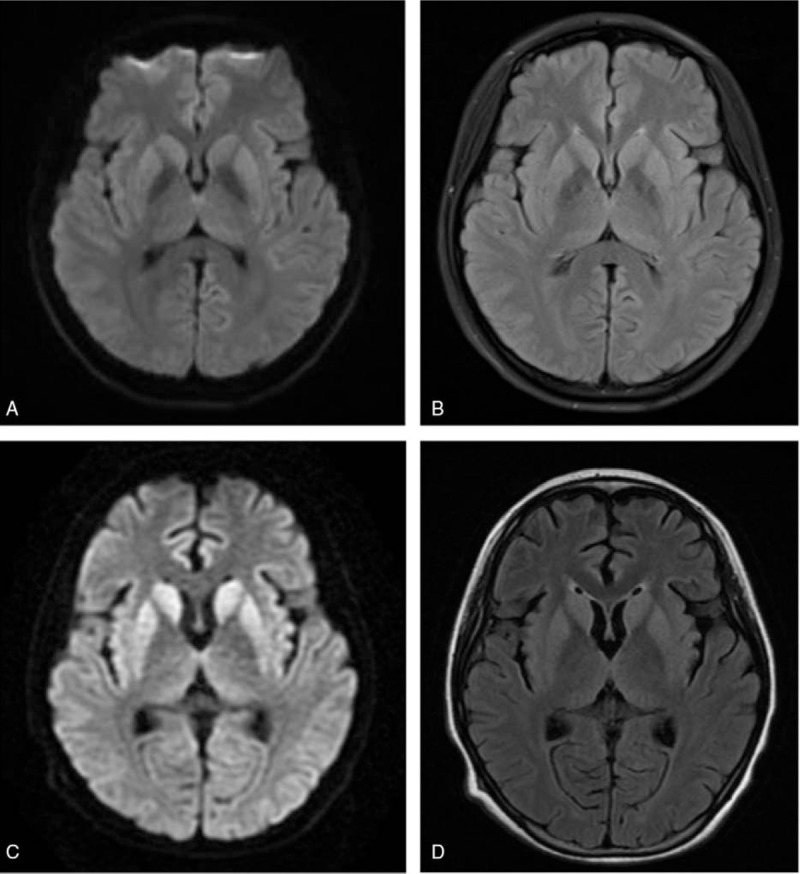
Brain magnetic resonance imaging. (A, B) Diffusion-weighted imaging (DWI) and T2 fluid attenuated inversion recovery sequences showed unremarkable results at the 8th week after initial symptom presentation. (C, D) DWI and T2 flair sequences showed hyperintensity of bilateral putamen and head of caudate nucleus at the 16th week after initial symptom presentation. Widespread cortical ribboning also appeared in DWI.
Figure 2.
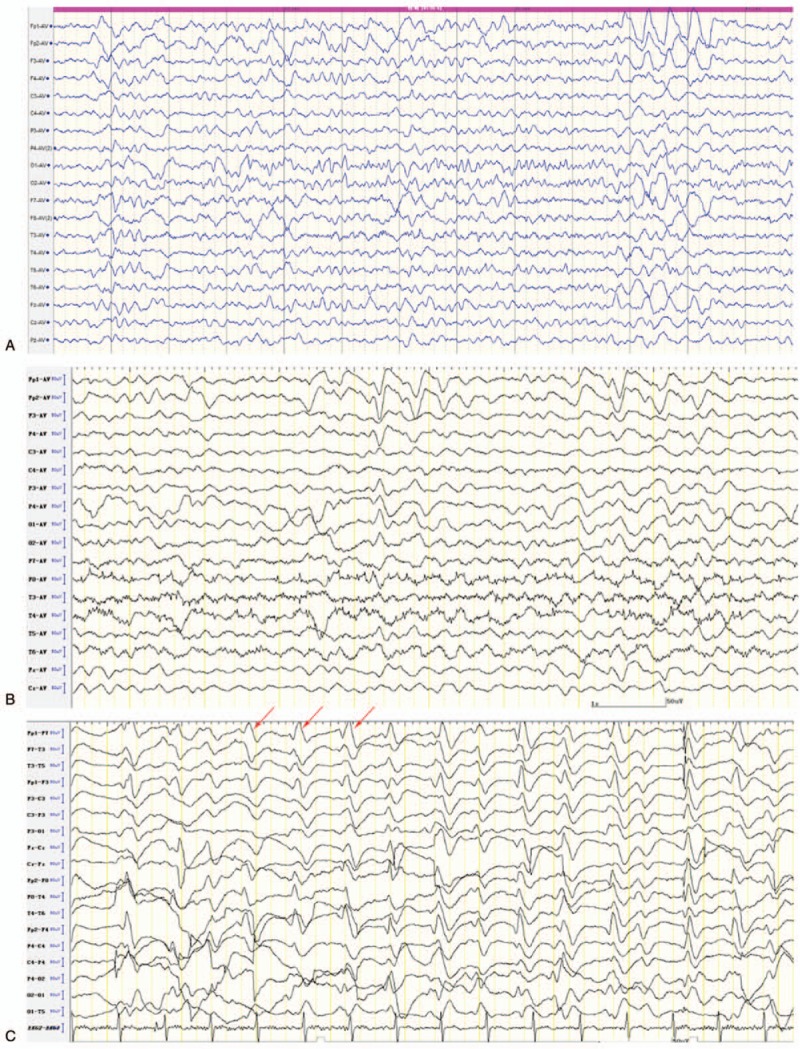
Interictal EEG. (A) At the 8th week after initial symptom presentation, the EEG showed diffused θ and δ activities. (B) At the 14th week after initial symptom presentation, the EEG showed further slowing of the background, and paroxysmal triphasic waves of bilateral frontal dominancy. (C) At the 16th week after initial symptom presentation, the EEG showed periodic bilateral triphasic sharp wave complexes lasting 300 ms that occurred every 0.5 to 1 s (arrows). EEG = electroencephalogram.
Despite 2 more sessions of IVIg and continued oral prednisone treatment, her condition deteriorated to include mutism, urinary incontinence, hyperhidrosis, limb rigidity, and orofacial and head involuntary movement, and eventually myoclonus by the 16th week. Then, re-examination of brain MRI showed increase in bilateral putamen and head of caudate nucleus on diffusion-weighted imaging (DWI) and T2 fluid attenuated inversion recovery (FLAIR) signals (Fig. 1C and D). Cortical ribboning in bilateral occipital, temporal, insular, and frontal lobes also appeared in DWI. A third EEG examination showed PSWC lasting 300 ms that occurred every 0.5 to 1 s (Fig. 2C). Bursts of generalized polyspike complexes were associated with myoclonus of the limbs. Based on the combination of clinical symptoms, the evolution of MRI abnormalities and EEG patterns, CJD was considered. A subsequent lumbar puncture showed further elevated protein level (0.9 g/L), a normal glucose level, and no leukocytes. A test for 14-3-3 protein was negative. The CSF tau level was not determined. PrP gene (PRNP) amplification and sequencing of the whole gene revealed no mutations, thus precluding genetic CJD. The genotype of PRNP codon 129 was MM, which is the genotype of variant CJD. However, the early predominant features of variant CJD include sensory disturbances and psychiatric symptoms.[10] These symptoms did not fit our patient's clinical profile. The absence of previous history in receiving blood transfusion, hormone injection, corneal transplant, or neurosurgical procedures excluded the possibility of iatrogenic CJD. According to the CDC's diagnostic criteria in 2010,[3] she was diagnosed with probable sCJD. We did not measure the molecular mass of PrPSc. Therefore, we could not further classify the case into a subtype. MM1 was the most likely based on the clinical symptoms and the MRI and EEG findings. Because she was only 18 years old, the diagnosis was made with caution. To further confirm our diagnosis, we performed RT-QuIC to detect PrPSc in the CSF, and the result was positive. Corticosteroids were then discontinued. After learning the prognosis of the disease, her parents refused a brain biopsy and discharged her from our clinic.
3. Discussion
This is the youngest case of probable sCJD reported to our knowledge. In China, the peak incidence of sCJD is at 60 to 69 years of age,[5] which is similar to other populations.[1] Based on the current diagnostic criteria, the youngest probable sCJD case reported in China was 21 years, and the youngest possible sCJD case was 18 years.[5] Among different subtypes of sCJD, VV1 is characterized by early onset.[11] However, according to the genotype of codon 129, our patient should be classified into MM subtypes rather than VV1.
The first diagnoses that we considered include diseases that could occur in young adults, such as subacute sclerosing panencephalitis (SSPE), organic acidemias, autoimmune encephalitis, or poisoning. She presented with cerebellar ataxia, followed by visual symptoms, rapidly progressive dementia, extrapyramidal features, autonomic symptoms, mutism, and eventually myoclonus. Although the symptoms of our patient were quite typical from the beginning, this combination is not unique. It could appear in other diseases such as autoimmune encephalitis, SSPE or progressive myoclonus epilepsy.
Blurred vision and diplopia were among her earliest complaints. Prolonged latency of bilateral P100 waves suggested impairment of her optic nerves. Although her brain MRI showed no abnormalities in the occipital lobes when blurred vision appeared, we could not exclude MRI negative lesions causing cortical blindness. Previous research has shown that blurred vision is a presenting feature in about 9% of CJD cases.[12] Visual acuities could be impaired by occipital lobe disease such as the Heidenhain variant of sCJD.[13] Blurred vision could also be the result of optic nerve damage, since PrPSc has been discovered in optic nerves of sCJD patients.[14] Diplopia is also commonly reported, possibly caused by cortical perceptual disturbances.[12]
The diagnosis was further obscured by the unremarkable brain MRI findings and the nonspecific changes in the EEG of our patient at the early stage of disease. As the disease progressed, the patient's MRI showed hyperintensity in bilateral putamen and caudate nucleus, as well as widespread cerebral cortex involvement. These MRI changes were consistent with those seen in the MM1 subtype.[6] The typical presentation of MRI has been described in the MRI-CJD Consortium criteria in 2009 and CDC's diagnostic criteria in 2010. Both criteria require high signal abnormalities in basal ganglia,[3,7] or in at least 2 cortical regions[7] on either DWI or T2 FLAIR. Therefore, re-examination of MRI is strongly recommended in patients with initial normal results. However, the time-course over which MRI changes occur in sCJD are poorly characterized. By contrast, it is widely accepted that typical EEG abnormalities do not appear until approximately 12 weeks.
Given the complexity of clinical symptoms and brain MRI and EEG abnormalities, sCJD can mimic treatable conditions such as immune-mediated encephalitis, infections, and metabolic disorders. The combined specificity of clinical symptoms, EEG, MRI, and 14-3-3 protein in diagnosing sCJD is only 70.8%, although the sensitivity is 98%.[7] According to the present diagnostic criteria, a definitive diagnosis relies on the presence of protease-resistant PrP and/or the presence of scrapie-associated fibrils in the CNS tissue.[3] However, patients may refuse invasive brain biopsy. In addition, the results are often inconclusive because not all areas of the brain will show the classic histological changes in CJD.[15] This issue could lead to a conundrum as to whether experimental treatments such as corticosteroids or anti-infective agents should be discontinued, especially in younger patients.
In this case, we cannot make a definitive diagnosis in the absence of a brain biopsy. Instead, we applied a much less invasive test, RT-QuIC, to further confirm the diagnosis. RT-QuIC was first used on CJD patients in 2011, and this technique showed >80% sensitivity and 100% specificity with CSF specimen.[16] In recent large-cohort prospective studies using second-generation RT-QuIC in CSF, the sensitivity for the diagnosis of sCJD was increased to 95%, with 100% specificity.[2,8] RT-QuIC using olfactory mucosa sampling is even less invasive compared with lumbar puncture, and showed a similar sensitivity of 90% to 97% and a specificity of 100%.[8,17] Furthermore, the combined results of RT-QuIC assays on CSF and olfactory mucosa samples reportedly allows an antemortem diagnosis of sCJD with 100% specificity and sensitivity.[8] According to these reports, a positive result should indicate definite sCJD. Given the convenience and the specificity of this test, it could provide tremendous help in atypical cases such as that described herein. Whether the timing of sampling affects the sensitivity has not been studied. Therefore, it remains unknown whether RT-QuIC is applicable for the diagnosis of sCJD before the manifestation of typical clinical symptoms, MRI lesions, or EEG abnormalities.
4. Conclusion
We report a case of early-onset probable sCJD confirmed by RT-QuIC. To our knowledge, this is the youngest reported case of probable sCJD in the literature. The case suggests that sCJD should not be ruled out simply due to young age. It also suggests that brain MRI should be repeated when the result is normal at the early stage of the disease. In atypical cases, RT-QuIC should be considered when a brain biopsy is not available.
Footnotes
Abbreviations: CDC = Centers for Disease Control, CNS = central nervous system, CSF = cerebrospinal fluid, DWI = diffusion-weighted imaging, EEG = electroencephalogram, FLAIR = fluid attenuated inversion recovery, IVIg = intravenous immunoglobulin, M = methionine, MRI = magnetic resonance imaging, PRNP = PrP gene, PrP = prion protein, PrPSc = scrapie form of PrP, PSWC = periodic bilateral triphasic sharp wave complexes, RT-QuIC = real-time quaking-induced conversion, sCJD = sporadic Creutzfeldt–Jakob disease, SSPE = subacute sclerosing panencephalitis, V = valine.
The procurement of clinical data and samples for the laboratory tests from hospitalized patients has been approved by the Peking Union Medical College Hospital ethics committee, and written informed consent was obtained from the guardian of the patient when she was admitted to our hospital.
Written informed consent for publication of the clinical details and clinical images was obtained from the father of the patient. A copy of the consent form is available for review by the Editor of this journal when required.
All data analyzed during this study are included in this article. The original clinical data are included in an additional file.
YY acquired and analyzed the data and drafted and revised the manuscript; XD performed RT-QuIC and revised the manuscript; HG participated in the acquisition of data and revised the manuscript; QL participated in the analysis of the data and critically revised the manuscript for intellectual content. All authors read and approved the final manuscript.
The authors have no funding and conflicts of interest to disclose.
References
- [1].Geschwind MD. Prion diseases. Continuum (Minneap Minn) 2015;21:1612–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Foutz A, Appleby BS, Hamlin C, et al. Diagnostic and prognostic value of human prion detection in cerebrospinal fluid. Ann Neurol 2017;81:79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].CDC's Diagnostic Criteria for Creutzfeldt–Jakob Disease (CJD), 2010. Available from: https://www.cdc.gov/prions/cjd/diagnostic-criteria.html. Accessed December 19, 2016. [Google Scholar]
- [4].Chitravas N, Jung RS, Kofskey DM, et al. Treatable neurological disorders misdiagnosed as Creutzfeldt–Jakob disease. Ann Neurol 2011;70:437–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gao C, Shi Q, Tian C, et al. The epidemiological, clinical, and laboratory features of sporadic Creutzfeldt–Jakob disease patients in China: surveillance data from 2006 to 2010. PLoS ONE 2011;6:e24231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Meissner B, Kallenberg K, Sanchez-Juan P, et al. MRI lesion profiles in sporadic Creutzfeldt–Jakob disease. Neurology 2009;72:1994–2001. [DOI] [PubMed] [Google Scholar]
- [7].Zerr I, Kallenberg K, Summers DM, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt–Jakob disease. Brain 2009;132:2659–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bongianni M, Orrù C, Groveman BR, et al. Diagnosis of human prion disease using real-time quaking-induced conversion testing of olfactory mucosa and cerebrospinal fluid samples. JAMA Neurol 2017;74:155–62. [DOI] [PubMed] [Google Scholar]
- [9].Sano K, Satoh K, Atarashi R, et al. Early detection of abnormal prion protein in genetic human prion diseases now possible using real-time QUIC assay. PLoS ONE 2013;8:e54915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zeidler M, Stewart GE, Barraclough CR, et al. New variant Creutzfeldt–Jakob disease: neurological features and diagnostic tests. Lancet 1997;350:903–7. [DOI] [PubMed] [Google Scholar]
- [11].Cali I, Castellani R, Yuan J, et al. Classification of sporadic Creutzfeldt–Jakob disease revisited. Brain 2006;129:2266–77. [DOI] [PubMed] [Google Scholar]
- [12].Armstrong RA. Creutzfeldt–Jakob disease and vision. Clin Exp Optom 2006;89:3–9. [DOI] [PubMed] [Google Scholar]
- [13].Wong A, Matheos K, Danesh-Meyer HV. Visual symptoms in the presentation of Creutzfeldt–Jakob disease. J Clin Neurosci 2015;22:1688–9. [DOI] [PubMed] [Google Scholar]
- [14].Head MW, Northcott V, Rennison K, et al. Prion protein accumulation in eyes of patients with sporadic and variant Creutzfeldt–Jakob disease. Invest Ophthalmol Vis Sci 2003;44:342–6. [DOI] [PubMed] [Google Scholar]
- [15].Manix M, Kalakoti P, Henry M, et al. Creutzfeldt–Jakob disease: updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy. Neurosurg Focus 2015;39:E2. [DOI] [PubMed] [Google Scholar]
- [16].Atarashi R, Satoh K, Sano K, et al. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat Med 2011;17:175–8. [DOI] [PubMed] [Google Scholar]
- [17].Orrú CD, Bongianni M, Tonoli G, et al. A test for Creutzfeldt–Jakob disease using nasal brushings. N Engl J Med 2014;371:519–29. [DOI] [PMC free article] [PubMed] [Google Scholar]