

Abstract
Rationale:
Chronic progressive external ophthalmoplegia (CPEO) is a classical mitochondrial ocular disorder characterized by bilateral progressive ptosis and ophthalmoplegia. Kearns -Sayre syndrome (KSS) is a multisystem disorder with PEO, cardiac conduction block, and pigmentary retinopathy. A few individuals with CPEO have other manifestations of KSS, but do not meet all the clinical diagnosis criteria, and this is called “CPEO plus.”
Patient concerns:
We report a 48-year-old woman exhibiting limb weakness, ptosis, ophthalmoparesis, and cerebellar dysfunctions.
Diagnoses:
The patient was diagnosed as exhibiting CPEO plus syndrome.
Interventions:
The patient underwent clinical, genetic, histological, and histochemical analysis. She was treated orally with CoQ10, vitamin Bs, L-carnitine, and vitamin E.
Outcomes:
The patient's serum creatine kinase levels, electrocardiography, and nerve conduction study results were normal; an electromyogram revealed myopathic findings. Magnetic resonance imaging showed global brain atrophy, particularly in the brainstem and cerebellum areas. A muscle biopsy showed the presence of abundant ragged red fibers. Sequencing of the mitochondrial DNA from the skeletal muscle biopsy revealed C960del mutation in 12S rRNA and homozygous mutation C2835T in 16S rRNA. She took medicines on schedule, the clinical features were similar as 2 years ago.
Lessons:
This is the first report of 2 rRNA mutations in a patient with MRI findings showing global brain atrophy, particularly in brainstem and cerebellum areas. Early recognition and appropriate treatment is crucial. This case highlights the cerebellar ataxia can occur in CPEO plus.
Keywords: 12S rRNA, 16S rRNA, C2835T, C960del, chronic progressive external ophthalmoplegia plus, mitochondrial, MRI
1. Introduction
Mitochondrial diseases, comprising a heterogeneous group of disorders characterized by primary defects in mitochondrial function, can be related to mutations of genes encoding either nuclear DNA (nDNA) or mitochondrial DNA (mtDNA).[1] Organs with high demand for aerobic energy, such as the brain, skeletal muscle, and heart, appear to be most commonly affected by mitochondrial dysfunction, although any, and indeed all tissues can be involved.[2] What is more, same mutation can lead to different phenotypes, largely depending on the classical “rules” of mitochondrial medicine: dual genetic control mtDNA and nDNA, tissue energy demand, levels of heteroplasmy, mitotic segregation, and maternal inheritance.[3]
Pathogenic mtDNA mutations may be classified to point mutations, mtDNA depletion, and large-scale rearrangements (duplications, single deletions, or multiple deletions).[4] Single deletions or duplications are responsible for 3 major phenotypes: progressive external ophthalmoplegia (PEO), Kearns–Sayre syndrome (KSS), and Pearson syndrome.[5] PEO is a mitochondrial myopathy with ptosis, ophthalmoplegia, and variably severe proximal limb weakness. KSS is a multisystem disorder with PEO, cardiac conduction block, and pigmentary retinopathy. Pearson syndrome is featured by sideroblastic anemia and exocrine pancreas dysfunction, and is usually fatal in infancy.[5] When PEO occurs in isolation, it is often referred to as chronic progressive external ophthalmoplegia (CPEO).[6] A few individuals with CPEO have other manifestations of KSS, but do not meet all the clinical diagnosis criteria, and is called “KSS minus” or “CPEO plus.”[7] Horga et al[8] established that the most common genetic defect associated with CPEO is single mitochondrial DNA deletion. However, the genotype–phenotype correlations are extremely variable.
C960del mutation in 12S rRNA and point mutation C2835T in 16S rRNA have been reported before. Mitochondrial 12S rRNA was associated with nonsyndromic and aminoglycoside-induced hearing loss.[9] Among 66 patients with bilateral vestibulopathy, C960del mutation in 12S rRNA was found in 1 patient.[9] Point mutation C2835T in 16S rRNA has been found in the family of Leber hereditary optic neuropathy (LHON) [10] to cause Rett syndrome,[11] which is a childhood neurodevelopmental disorder, with an approximate prevalence of 1 in 10,000 to 15,000 live female births. The syndrome is characterized by normal early development, followed by a loss of purposeful use of hands, distinctive hand movements, slowed brain and head growth, mental retardation, gait abnormalities, and seizures.[12]
Here, 2 mutations including C960del in 12S rRNA and a homozygous point mutation C2835T in 16S rRNA have been found in 1 patient whose clinical features manifested with CPEO plus syndrome. The patient's brain magnetic resonance imaging (MRI) revealed whole brain atrophy with the greatest severity in the cerebellar and brainstem areas.
2. Case presentation
This study was conducted according to the guidelines of the Ethical Committees of the Affiliated Hospital of Jining Medical University, China. Written informed consent was obtained from all individuals.
A 48-year-old Chinese Han nationality woman was referred to Affiliated Hospital of Jining Medical University, China, in 2015. She complained of mild limb weakness, especially when exercising, difficulty in opening her eyes, and blurred vision without diplopia for 7 years. At first, she could walk by herself, but was prone to fall to the right-hand side, the symptom worsened progressively, she stood unstably, and could not walk by herself. She showed dysarthria without dysphagia for about 3 years, and she never went to the hospital and did not take any medication. She never suffered stroke-like episodes, migraine, and epileptic seizure. Ten days before going to the outpatient clinic of our hospital, she suffered from a common cold and felt that the above symptoms worsened, her speech could not be understood by family members.
The neurological examination of the patient presented the normal mental status was good cooperated. The visual acuities were 0.3 for the right eye and 0.5 for the left eye because of cataract, and eye fundus examination showed no signs of pigmentary retinopathy. The pupillary sizes were equal and the light reflexes were normal. Bilateral incomplete ptosis and limitation of eye movements in every direction, and a bilateral gaze-evoked, horizontal, gross nystagmus were found. The bilateral audition, pharyngeal reflex, and functions of other cranial nerves were normal. Cerebellar dysarthria was observed. The muscle strength of the neck and proximal of the 4 extremities was 4 and of the distal muscles of the 4 extremities was 5−. The tendon reflexes were (+) in the 4 extremities. The bilateral diadochokinesis, finger-to-nose test, and heel-knee-shin maneuver were all positive, which revealed cerebellar ataxia. The patient displayed a remarkably broad-based, drunken gait while walking helped by others. Romberg test showed that she was unable to keep balance of both opening and closing the eyes. The sensory and autonomic nervous systems were unremarkable.
In Simpson test, fatigue was observed on sustained lid and eye elevation was negative. Intramuscular neostigmine 1 mg showed no improvement in symptoms. The 2 tests were used to differentiate myasthenia gravis. The resting serum lactate in the blood was 2.2 mmol/L (normal 0.7–2.1 mmol/L); no aerobic exercise tests were performed as she could not stand by herself. Serum creatine kinase levels and electrocardiography results were normal. Electroretinogram was not done. The needle electromyogram findings were myopathic. Right anterior tibial muscle, right caput mediale musculi gastrocnemii, right deltoid muscle, and right common extensor muscle of digits were examined. All results showed the normal duration of insertion potential, but with fibrillation and positive sharp waves. With slight contraction, the duration, amplitude of motor unit potentials (MUPs), and the polyphasic MUPs were within normal range. Right caput mediale musculi gastrocnemii and right deltoid muscle showed a pathological interference pattern. Nerve conduction study results and repetitive nerve stimulation tests were normal. MRI revealed whole brain atrophy, with the greatest severity in the cerebellar and brainstem areas (Fig. 1).
Figure 1.
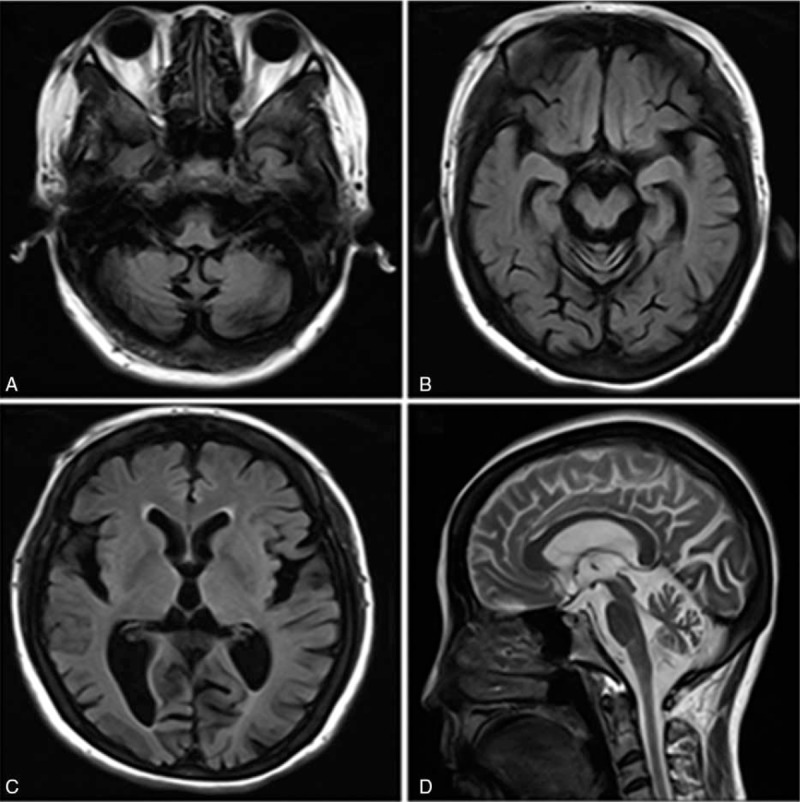
Brain magnetic resonance imaging revealing whole brain atrophy with greatest severity in the cerebellar and brainstem areas. T1W1 (A and B), T2 flair (C), and T2W2 (D).
The patient's parents were not consanguineous; her mother died of stroke at the age of 68 and showed no similar symptoms during her life time. The patient had a full sibling sister who died at her 30s, with a similar clinical presentation, but without going to a hospital, so the concrete symptoms, signs, and diagnosis were not known. All of the patient's children (2 daughters and 1 son, separately aged at 26, 23, and 17 years old) exhibited no symptoms and signs.
2.1. Muscle histology and histochemistry analysis
A biopsy of the right biceps femoris muscle was performed according to standard procedures. For histological and histochemical analysis, 10 mm frozen sections were stained with hematoxylin and eosin, modified Gomori trichrome, cytochrome c oxidase (COX), and succinate dehydrogenase (SDH). Muscle histology showed fiber size variation, and the modified Gomori trichrome staining and SDH showed ragged red fibers, accounting for 12% of whole fibers. The proportion of muscle fibers exhibiting a mitochondrial oxidative phosphorylation defect could be accurately quantified by normal COX-positive fibers staining of brown; COX-negative fibers presented blue as a result of impaired complex IV activity (Fig. 2).
Figure 2.
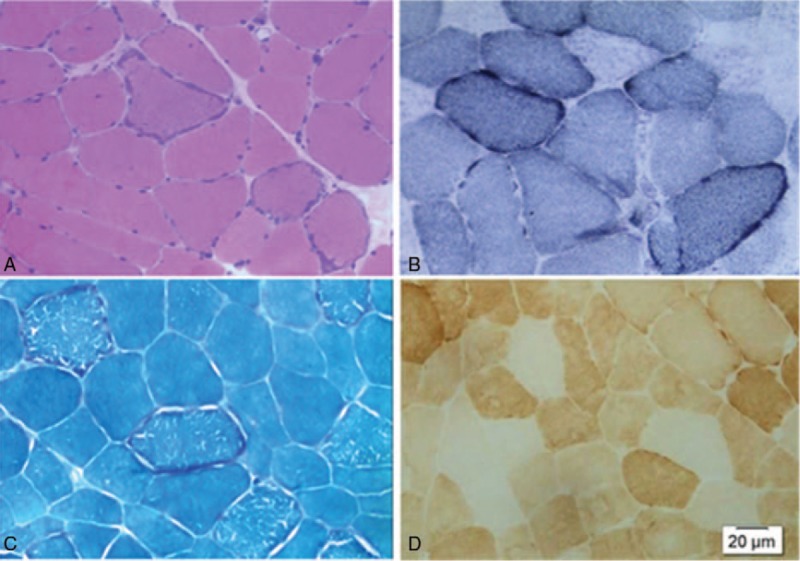
Skeletal muscle histological and histochemical analysis. (A) Frozen sections stained with haematoxylin and eosine (100×). (B) Frozen sections stained with succinate dehydrogenase (SDH) (100×). (C) Frozen sections stained with modified Gomori trichrome (100×). (D) Frozen sections stained with cytochrome c oxidase (100×).
2.2. Mutation screening
Total DNA was extracted from 10 mL aliquots of ethylenediaminetetraacetic acid blood using a salting-out method, and from muscle biopsy specimens using the QiaAmp DNA Mini Kit (#51306, Qiagen). Muscle DNA from 20 Chinese individuals without any signs of mitochondrial diseases was also analyzed as control. MLPA technology was used, and no large-scale rearrangements or multiple DNA deletions were found. For direct sequence analysis of the entire mitochondrial genome, 42 overlapping PCR fragments were amplified from skeletal muscle DNA and sequenced by a commercial sequencing service (KingMed diagnostic, China). The coding regions of 42 nuclear genes including NDUFS1, NDUFS4, NDUFS7, NDUFS8, NDUFV1, SDHA, SDHAF1, NDUFS2, SDHA, UQCRB, SURF1, LRPPRC, SCO1, SCO2, COX10, COX15, BCS1L, ATPAF2, GFM1, MRPS16, PUS1, TUFM, TACO1, POLG, POLG2, C10orf2, SLC25A4, TYMP, TK2, DGUOK, SUCLA2, RRM2B, AIF1, TRMU, GFER, COQ2, COQ9, CABC1, ETFDH, TAZ, SLC25A3, and MPV17 were sequenced. The mutation screening revealed C960del mutation in 12S rRNA and homozygous mutation C2835T in 16S rRNA (Figure 3). The primers and the PCR conditions are available in request. Her children were not detected because of their disagreement.
Figure 3.
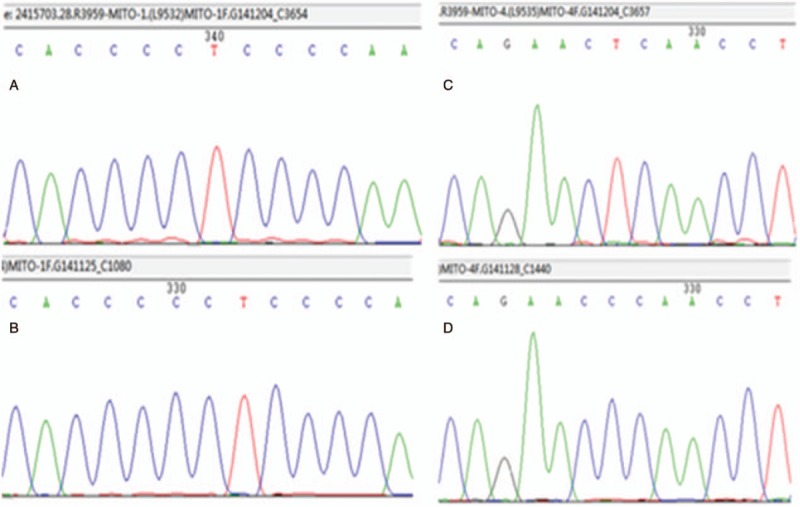
Mutation screening. Mutation screening reveals C960del mutation in 12S rRNA (A) and homozygous mutation C2835T in 16S rRNA (C). B and D represent controls.
2.3. Treatment and follow-up
According to the clinical features, muscle pathology, and genetic analysis, the patient was diagnosed to be with CPEO plus syndrome, and she was treated orally with CoQ10, vitamin Bs, L-carnitine, and vitamin E. During the 2 years, she took medicines on schedule, the clinical features were similar as 2 years ago, and she could walk with help. All of the patient's children were not affected until now.
3. Discussion
Diagnostic work-up for the suspected mitochondrial disease is a stepwise procedure.[13,14] The first step comprises a comprehensive individual and family history and clinical investigations by neurologist, ophthalmologist, aurist, cardiologists, dermatologist, and so on. We reported a patient with a subtle onset of clinical features of a slowly progressive neuromuscular condition, ptosis, eye movement abnormalities, cerebellar dysarthria, and cerebellar ataxia. The MRI showed the whole brain atrophy with the greatest severity in the cerebellar and brainstem areas. A muscle biopsy showed the presence of abundant ragged red fibers; thus the mitochondrial disease was suspected. The second step is to define the phenotype and determine whether it is sporadic or inherited, the patient here was diagnosed to be with CPEO plus syndrome, but regrettably the other members of her family were untested, so the trait of inheritance was uncertain. The genetic test is the third step depending on steps 1 and 2. After the entire mitochondrial genome and the coding region of nuclear genes sequencing, C960del mutation in 12S rRNA and homozygous mutation C2835T in 16S rRNA were discovered. Currently, the management of mitochondrial disease is largely supportive, and various treatments such as co-enzyme Q10, dichloroacetate, carnitine, creatine, and vitamin “cocktails” have been showed to have no meaningful clinical efficacy.[15] Although there are still no effective treatments for the majority of patients, the disease of this patient did not develop rapidly.
There are 3 types of the polymorphic rRNA variants: deleterious, neutral, and adaptive, and the 2 variants found in this report were reported to be neutral.[16] The location in 12S rRNA and 16s rRNA could be found in MITOMAP (www.mitomap.org). The phenotypes of the 2 mutations had been reported before. Elstner et al[9] reported a man with C960 del mutation in 12S rRNA had concurrent hearing loss after gentamycin therapy. The C2835T in 16S rRNA was reported in the family of LHON,[10] which was rapid and painless, and showed bilateral loss of central vision. C2835T has also been reported to cause Rett syndrome,[11] which is a pediatric neurological disorder, defined by the presence of severe neurodevelopment decline, acquired microcephaly, movement abnormalities, dementia, autistic behavior, and seizures in young female children. Breathing irregularities, hand stereotypies, nocturnal unrest, and anxiety or inappropriate fears, and low mood have also been commonly reported among patients with Rett syndrome.[17,18] However, our patient was of normal intelligence, and did not exhibit hearing and vision loss; so the clinical features were different from the previously reported cases.
As the genotype-phenotype may have correlations, we speculate the correlation between the mutation and the clinical features. It was reported that rearrangements (deletions and duplications) of mitochondrial DNA may cause CPEO,[19] so we speculate that the clinical features of ptosis, eye movement abnormalities may be caused by C960 del. One of the most prevalent clinical manifestations of mitochondrial disorders is ataxia,[20] many syndromes include cerebellar ataxia, such as CPEO plus syndrome, KSS, myoclonus epilepsy with ragged-red fibers (MERRF), mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS), neurogenic weakness, ataxia, retinitis pigmentosa (NARP), May–White syndrome, and so on.[21–24]This patient manifested with the cerebellar dysfunctions, and MRI revealed a larger extent of brain shrinkage with the greatest severity in the cerebellar and brainstem areas, which was not rare and in consistent with the previous studies.[25] It was reported that the mean atrophy score in CPEO patients due to a single mitochondrial gene mutation is significantly higher than that in control patients.[26] Of 10 CPEO patients, 1 patient had severe and 4 had moderate cerebellar atrophy.[27] Patients with CPEO plus features had significantly reduced total gray matter and cerebellar volumes compared with controls.[28] Thus, we speculate that the cerebellar dysfunction may be caused by C960 del.
Our reports had some limitations. First, the patient's children refused to have mitochondrial DNA sequencing and they were young; thus they may not achieve the age of onset, so we need to follow up for a period of time. Second, because of limited references reported, the phenotype separately caused by C960 del and C2835T in this patient was unknown; and the last is that the underlying mechanism of the 2 mutations causing the phenotype was uncovered.
4. Conclusions
We first reported a case clinically manifested with CPEO plus syndrome, the brain MRI revealed whole brain atrophy with the greatest severity in the cerebellar and brainstem areas, C960del mutation in 12S rRNA and homozygous mutation C2835T in 16S rRNA were found after DNA sequencing, which is the first report of 2 rRNA mutations in 1 patient. Until now, the mechanism caused by the 2 mutations was still unknown, which needs further study. In conclusion, both the patient's clinical manifestation and the 2 rRNA mutations broaden the spectrum of CPEO plus syndrome.
Acknowledgment
We would like to thank the patient for her participation in this study.
Footnotes
Abbreviations: COX = cytochrome c oxidase, CPEO = chronic progressive external ophthalmoplegia, KSS = Kearns–Sayre syndrome, LHON = Leber hereditary optic neuropathy, MELAS = mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes, MERRF = myoclonus epilepsy with ragged-red fibers, MRI = magnetic resonance imaging, mtDNA = mitochondrial DNA, MUPs = motor unit potentials, NARP = neurogenic weakness, ataxia, retinitis pigmentosa, nDNA = nuclear DNA, PCR = polymerase chain reaction, PEO = progressive external ophthalmoplegia, SDH = succinate dehydrogenase.
Funding: This work was supported by grant 81401064 from the National Natural Science Foundation of China (to Dr Zhanyun Lv), grant 81271398 from the National Natural Science Foundation of China (to Dr Yanlei Hao), and 2013-ZD-003 from the MiaoPu project of the Affiliated Hospital of Jining Medical College (to Dr Zhanyun Lv, 2013).
The authors report no conflicts of interest.
References
- [1].Filosto M, Mancuso M. Mitochondrial diseases: a nosological update. Acta Neurol Scand 2007;115:211–21. [DOI] [PubMed] [Google Scholar]
- [2].Meyers DE, Basha HI, Koenig MK. Mitochondrial cardiomyopathy: pathophysiology, diagnosis, and management. Tex Heart Inst J 2013;40:385–94. [PMC free article] [PubMed] [Google Scholar]
- [3].DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med 2003;348:2656–68. [DOI] [PubMed] [Google Scholar]
- [4].Davis RL, Sue CM. The genetics of mitochondrial disease. Semin Neurol 2011;31:519–30. [DOI] [PubMed] [Google Scholar]
- [5].Moraes CT, DiMauro S, Zeviani M, et al. Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns-Sayre syndrome. N Engl J Med 1989;320:1293–9. [DOI] [PubMed] [Google Scholar]
- [6].McFarland R, Taylor RW, Turnbull DM. A neurological perspective on mitochondrial disease. Lancet Neurol 2010;9:829–40. [DOI] [PubMed] [Google Scholar]
- [7].Salvatore D, Michio H. Mitochondrial DNA Deletion Syndromes. GeneReviews® [Internet]. In: Pagon RA, Adam MP, Ardinger HH, editors. Seattle, WA: University of Washington; 1993–2017. [PubMed]
- [8].Horga A, Pitceathly RD, Blake JC, et al. Peripheral neuropathy predicts nuclear gene defect in patients with mitochondrial ophthalmoplegia. Brain 2014;137(Pt 12):3200–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Elstner M, Schmidt C, Zingler VC, et al. Mitochondrial 12S rRNA susceptibility mutations in aminoglycoside-associated and idiopathic bilateral vestibulopathy. Biochem Biophys Res Commun 2008;377:379–83. [DOI] [PubMed] [Google Scholar]
- [10].Qu J, Zhou X, Zhang J, et al. Extremely low penetrance of Leber's hereditary optic neuropathy in 8 Han Chinese families carrying the ND4 G11778A mutation. Ophthalmology 2009;116:558–64. e553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tang J, Qi Y, Bao XH, et al. Mutational analysis of mitochondrial DNA of children with Rett syndrome. Pediatr Neurol 1997;17:327–30. [DOI] [PubMed] [Google Scholar]
- [12].Lyst MJ, Bird A. Rett syndrome: a complex disorder with simple roots. Nat Rev Genet 2015;16:261–75. [DOI] [PubMed] [Google Scholar]
- [13].Parikh S, Goldstein A, Koenig MK, et al. Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med 2015;17:689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Finsterer J, Harbo HF, Baets J, et al. EFNS guidelines on the molecular diagnosis of mitochondrial disorders. Eur J Neurol 2009;16:1255–64. [DOI] [PubMed] [Google Scholar]
- [15].Pfeffer G, Majamaa K, Turnbull DM, et al. Treatment for mitochondrial disorders. Cochrane Database Syst Rev 2012;CD004426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ruiz-Pesini E, Wallace DC. Evidence for adaptive selection acting on the tRNA and rRNA genes of human mitochondrial DNA. Hum Mutat 2006;27:1072–81. [DOI] [PubMed] [Google Scholar]
- [17].Mount RH, Charman T, Hastings RP, et al. The Rett Syndrome Behaviour Questionnaire (RSBQ): refining the behavioural phenotype of Rett syndrome. J Child Psychol Psychiatry 2002;43:1099–110. [DOI] [PubMed] [Google Scholar]
- [18].Cianfaglione R, Clarke A, Kerr M, et al. A national survey of Rett syndrome: behavioural characteristics. J Neurodev Disord 2015;7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zeviani M, Di Donato S. Mitochondrial disorders. Brain 2004;127:2153–72. [DOI] [PubMed] [Google Scholar]
- [20].Finsterer J. Mitochondrial ataxias. Can J Neurol Sci 2009;36:543–53. [DOI] [PubMed] [Google Scholar]
- [21].Zeviani M, Simonati A, Bindoff LA. Ataxia in mitochondrial disorders. Handb Clin Neurol 2012;103:359–72. [DOI] [PubMed] [Google Scholar]
- [22].Kawai H, Akaike M, Yokoi K, et al. Mitochondrial encephalomyopathy with autosomal dominant inheritance: a clinical and genetic entity of mitochondrial diseases. Muscle Nerve 1995;18:753–60. [DOI] [PubMed] [Google Scholar]
- [23].Mancuso M, Filosto M, Bellan M, et al. POLG mutations causing ophthalmoplegia, sensorimotor polyneuropathy, ataxia, and deafness. Neurology 2004;62:316–8. [DOI] [PubMed] [Google Scholar]
- [24].Mancuso M, Orsucci D, Angelini C, et al. Redefining phenotypes associated with mitochondrial DNA single deletion. J Neurol 2015;262:1301–9. [DOI] [PubMed] [Google Scholar]
- [25].Bindu PS, Arvinda H, Taly AB, et al. Magnetic resonance imaging correlates of genetically characterized patients with mitochondrial disorders: a study from south India. Mitochondrion 2015;25:6–16. [DOI] [PubMed] [Google Scholar]
- [26].Pitceathly RD, Morrow JM, Sinclair CD, et al. Extra-ocular muscle MRI in genetically-defined mitochondrial disease. Eur Radiol 2016;26:130–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Heidenreich JO, Klopstock T, Schirmer T, et al. Chronic progressive external ophthalmoplegia: MR spectroscopy and MR diffusion studies in the brain. AJR Am J Roentgenol 2006;187:820–4. [DOI] [PubMed] [Google Scholar]
- [28].Yu-Wai-Man C, Smith FE, Firbank MJ, et al. Extraocular muscle atrophy and central nervous system involvement in chronic progressive external ophthalmoplegia. PLoS One 2013;8:e75048. [DOI] [PMC free article] [PubMed] [Google Scholar]