

Abstract
Rationale:
Primary spinal primitive neuroectodermal tumors (PNETs) are highly malignant tumors, which are extremely rare entities and primary intramedullary PNETs are extremely rare. Till now, only 24 cases of primary intramedullary PNET have been reported.
Patient concerns:
A 26-year-old male presented with progressive low back and lower limb pain for 1 month.
Diagnoses:
Based on MRI and histopathological findings, he was diagnosed with primary intramedullary PNET.
Interventions:
The patient was treated two times with microsurgical resections.
Outcomes:
Follow-up visit at 14 months after the first surgery showed that the patient is neurologically intact and free of disease.
Lessons:
PNETs should be considered in the differential diagnosis of an intramedullary spinal cord tumor manifesting as progressive neurological deterioration.
Keywords: intramedullary, primitive neuroectodermal tumor, spinal tumors
1. Introduction
Primitive neuroectodermal tumors (PNETs) are rare and malignant tumors, first described by Hart and Earle[1] in 1973. According to the 2007 World Health Organization (WHO) classification, PNETs are a group of embryonal tumors composed of undifferentiated or poorly differentiated neuroepithelial cells which can differentiate into neuronal cells, astrocytes, ependymal cells, myocytes, and melanoma cell lines.[2] The diagnosis of the PNETs depends on histopathology and immunohistochemistry.[3] These tumors mostly occur in children and young adults.[4] Primary spinal PNETs are uncommon and can be extradural, intradural extramedullary, and intramedullary, of which primary intramedullary PNETs are extremely rarer.[5,6] Till now, only 24 cases of primary intramedullary PNETs have been reported.[3,5–24] Here we describe an unusual case of primary intramedullary PNET with unique clinical features and poor prognosis.
2. Case presentations
A previously healthy 26-year-old young man presented with progressive pain in the lower back and lower limbs for 1 month. The local hospital considered the diagnosis of lumbar disc herniation. The pain did not relieve after treatment for 3 days. Subsequently, a magnetic resonance imaging (MRI) of the spine showed a mass in the spinal canal from T12 through L1 level, and he was referred to our department for further evaluation.
On physical and neurological examination, movement and sensory findings were normal with no numbness and weakness in both legs. There was no bowel or bladder dysfunction, tone and power in all the limbs were normal, he was conscious and alert, he had no headache or visual field defects, both Achilles tendon reflexes were mildly increased, vital signs were normal, and his family medical history was not notable.
A thoracolumbar MRI revealed a 1.6 × 2.0 × 4.4 cm elliptical-shaped intramedullary mass at the T12-L1 levels, which appeared isointense on T1-weighed imagine and hyperintense on T2-weighed imagine. The lesion showed markedly heterogenous enhancement after gadolinium administration. On the coronal section of enhanced MRI, conus medullaris was pushed to the right side (Fig. 1). We considered the diagnosis of neurogenic tumor and did not exclude ependymoma and astrocytoma.
Figure 1.
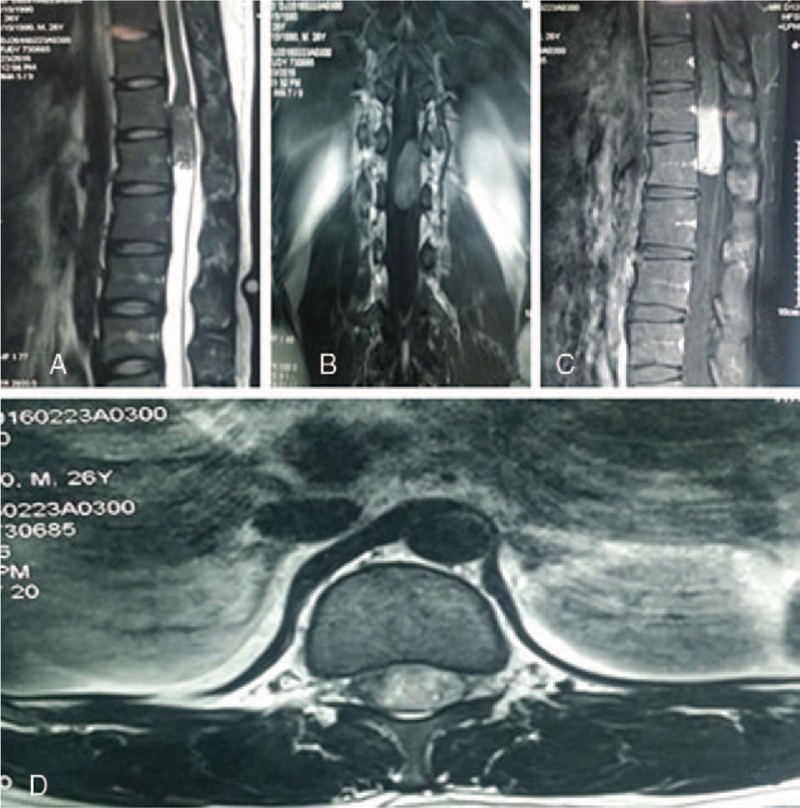
Preoperative MR imaging. (A) Sagittal T2-weighted MR image; (B) coronal T1-weighted MR image; (C) sagittal enhanced T1-weighted MR image; and (D) axial enhanced T1-weighted MR image demonstrate an elliptical-shaped intramedullary mass at T12-L1 levels. The lesion was markedly heterogenous enhancement following gadolinium administration. MR = magnetic resonance.
A gross total excision of the lesion was achieved with T12-L1 laminectomy. After removing the T12-L1 vertebral spinous process and lamina, we can see dura mater was integral with high tension and epidural fat capsule was thin and partially disappeared. The tumor was intramedullary, reddish-gray, soft, hypervascular, adherent to the spinal cord, and extended to cauda equina and was in the left side of the spinal cord. After excision, the compressed spinal cord and cauda equina acquired good decompression. Multiple biopsy specimens were obtained.
Hematoxylin and eosin staining of the tumor specimen revealed a highly cellular tumor, composed of numerous small round cells with hyperchromatic nuclei. Immunohistochemistry revealed a strong cytoplasmic co-expression of CD99 (MIC2) and moderate positivity of S-100, synaptophysin (Syn), Friend leukemia virus integration 1 (FLI-1), and integrase interactor 1 (INI-1). The MIB-1 (Ki-67) proliferation rate was approximately 50%. A few tumor cells expressed desmin. No reactivity was observed by antibodies against CD3, CD20, CD56, terminal deoxynucleotidyl transferase (TdT), cytokeratin (CK), myogenin, and Myo D1 (Fig. 2). The tumor was diagnosed as a primary intramedullary PNET.
Figure 2.
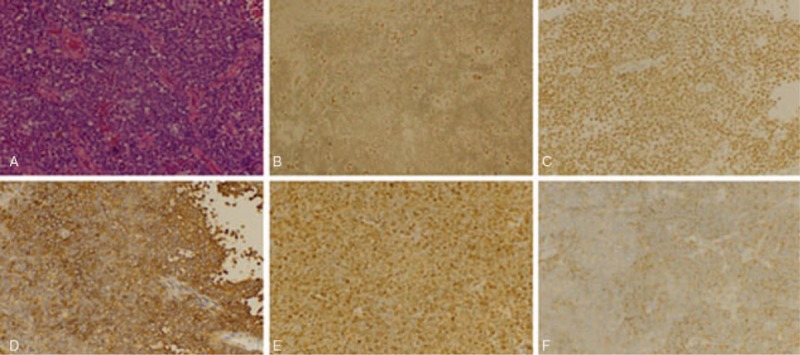
Light microscopy and immunohistochemistry. (A) HE staining: the tumor cells were numerous small round cells with hyperchromatic nuclei (original magnification ×400). Immunohistochemistry showing (B) FLI-I (++), (C) INI-1 (++), (D) CD99 (+++), (E) S-100 (++), (F) Syn (++) (original magnification ×400). HE = hemotoxylin and eosin.
The patient had an uneventful postoperative course. The initial pain of the lower back and lower limbs was significantly relieved and gradually disappeared. Subsequently, a radiation therapy of the neuroaxis and chemotherapy (temozolomide [TMZ]) was carried out. Subsequently, 2 months after the operation, a follow-up MRI did not show any primary lesions elsewhere or a local recurrence of the tumor in the spinal canal (Fig. 3).
Figure 3.
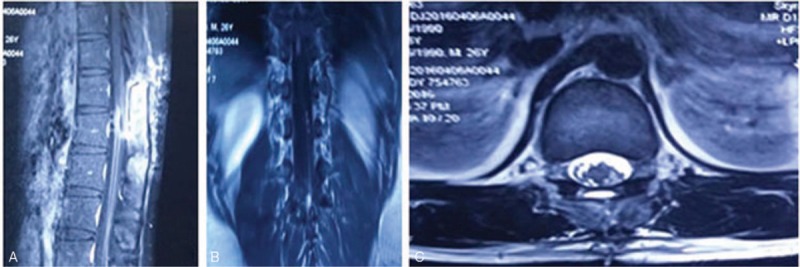
A follow-up MR imaging (2 months after the initial operation). (A) Sagittal enhanced T1-weighted MR image. (B) Coronal enhanced T1-weighted MR image. (C) Axial enhanced T2-weighted MR image. MRI showing no primary lesions elsewhere and local recurrence. MRI = magnetic resonance imaging.
Nine months after the initial operation, the patient was again hospitalized because of intermittent dysuria with low back and lower limbs pain. He was admitted to the Department of Urology of our hospital. Doppler ultrasonography of kidney, ureter, bladder, and prostate revealed no abnormality. The urodynamic examination found a acontractile detrusor. The patient received suprapubic cystostomy due to the acute urinary retention. Postoperatively, the low back and lower limbs pain still existed. The patient was referred to our department again due to the suspicion of tumor recurrence by a urologist.
According to MRI of spinal column, the patient developed recurrence at T12-L1 levels and multiple seeding metastases at L3-L5 levels (Fig. 4), and another microsurgical resection was performed. The spinal cord and nerve roots were infiltrated by the tumors and no distinct planes were evident between the mass and the cord. Considering the high malignancy of the tumor and avoiding impairing the spinal cord and nerve roots, a partial excision was performed. After the excision, the spinal cord was decompressed. The patient endured the operation well. Postoperative pathological findings and immunohistochemistry were compatible with PNET. After operation, there was no neurologic deterioration with significant relief of low back and lower limbs pain. The patient received postoperative radiotherapy and chemotherapy (TMZ). Follow-up visit at 14 months after the first surgery showed that the patient is neurologically intact and free of disease.
Figure 4.
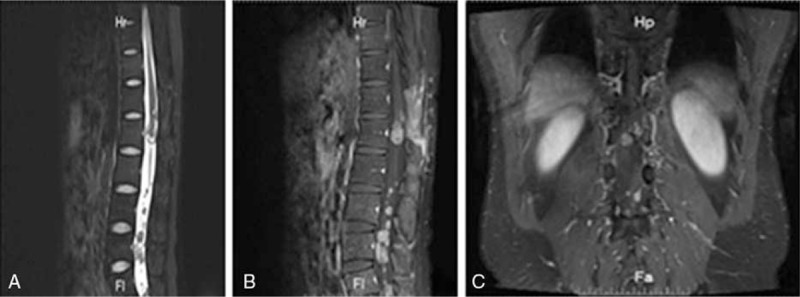
A MR imaging before the second operation (9 months after the initial operation). MR imaging showing recurrence at T12-L1 levels and multiple seeding metastases at L3-L5 levels. (A) Sagittal T2-weighted MR image; (B) sagittal T1-weighted MR image; (C) coronal T1-weighted MR image. MR = magnetic resonance.
Written informed consent was obtained from the patient to use the content and imaging material for publication.
3. Discussion
Primitive neuroectodermal tumor is rare and malignant tumor with poor prognosis; majority of PNETs are described as isolate cases by rare case reports in previous neurosurgical literatures. Here, a young man with rare primary intramedullary PNET has been reported, and the comprehensive descriptions about this disease are as following.
3.1. PNET classification
According to the 2007 WHO classification, PNETs are a group of embryonal tumors composed of undifferentiated or poorly differentiated neuroepithelial cells, which can differentiate into neuronal cells, astrocytes, ependymal cells, myocytes, and melanoma cell lines.[2] However, in the 2016 WHO classification of tumors of the central nervous system (CNS), the term primitive neuroectodermal tumor or PNET was removed from the diagnostic lexicon. According to whether there is the C19MC amplification or not, the corresponding diagnoses are embryonal tumor with multilayered rosettes (ETMR), C19MC-altered or ETMR, not otherwise specified (NOS), and a tumor with histological features of medulloepithelioma should be diagnosed as medulloepithelioma.[25] Our case occurred before the 2016 CNS WHO classification, and our pathological diagnostic used the previous criteria, so we still use the term PNET to describe this disease.
3.2. Clinical manifestation
These tumors predominantly occur in children and young adults, and can be extradural or intradural extramedullary or intramedullary.[4,5] The clinical presentation of primary spinal PNETs varied according to tumor locations, the degree of tumor invasion, and involved structures.[26,27] Muscle weakness is the most common symptom. Sensory symptoms, local pain, and radiculopathy are also common presentations.[4]
3.3. Pathological features
As for diagnosis of PNET, histopathological examination and immunohistochemistry analysis are both required.[3] Microscopically, these tumors consisted of mainly small round undifferentiated cells with hyperchromatic nuclei.[26] The tumors display high mitotic activity and necrosis.[4] Immunohistochemically, the tumor cells are usually positive for neuron-specific enolase (NSE), S-100, nestin, vimentin, or microfilaments. Syn or glial fibrillary acidic protein (GFAP) may be positive resting with the tumor cell differentiation.[5] In terms of subtyping, negativity for CD99 support the diagnosis of cPNET. Conversely, strong positivity for CD99 support the diagnosis of pPNET,[3] and the membrane protein FLI-1 is also commonly expressed in pPNETs.[4]
3.4. MRI characteristics
In MRI, primary spinal PNETs may occur at all levels of the spine like extradural or extramedullary, or intradural or intramedullary spinal location, but they mainly occur in the thoracolumbar and extramedullary intradural region.[28] In our case, the lesion occurred in the thoracolumbar and intramedullary region. Although MRI characteristics are not specific, it is important to investigate the condition of patients. MRI provides high-resolution imaging for soft tissue within the spinal column, and any intramedullary lesions can be found easily.[29] On T1-weighted images, the lesion shows hypointense signal on T1 and hyperintense signal on T2-weighted images, with a markedly heterogenous enhancement after gadolinium administration.[28] Our patient is consistent with these characteristics. Due to rarity of this disease, intramedullary tumor, astrocytoma, and ependymoma should be considered in the differential diagnosis; in addition, intramedullary metastasis needs to be included in the differential diagnosis.
3.5. Treatment and prognosis
There is no standard therapy for primary spinal PNETs.[4] At present, the therapy of PNETs mainly includes surgery, radiotherapy, and chemotherapy, which are crucial for the prognosis of this tumor.[29,30] Primary spinal PNETs are highly malignant tumors, with characteristics of infiltrative growth. In this patient, although, combined treatments of surgery, radiotherapy, and chemotherapy were given, and he was free of tumor progression for only 9 months.
Moreover, the tumors do not have an explicit plane of cleavage between tumor and neural structures. Therefore, gross total resection is usually impossible.[24] Furthermore, due to the small number of primary spinal PNET cases, it is unclear if the radiotherapy and chemotherapy plays an effective role in the prognosis.[16] However, from the existing studies, radiotherapy and chemotherapy seems an effective treatment, which may delay progression and improve prognosis of this disease.[29] Unfortunately, although undergoing surgery, radiotherapy, and chemotherapy, primary spinal PNETs still have a poor prognosis.[4,30]
3.6. Literature review
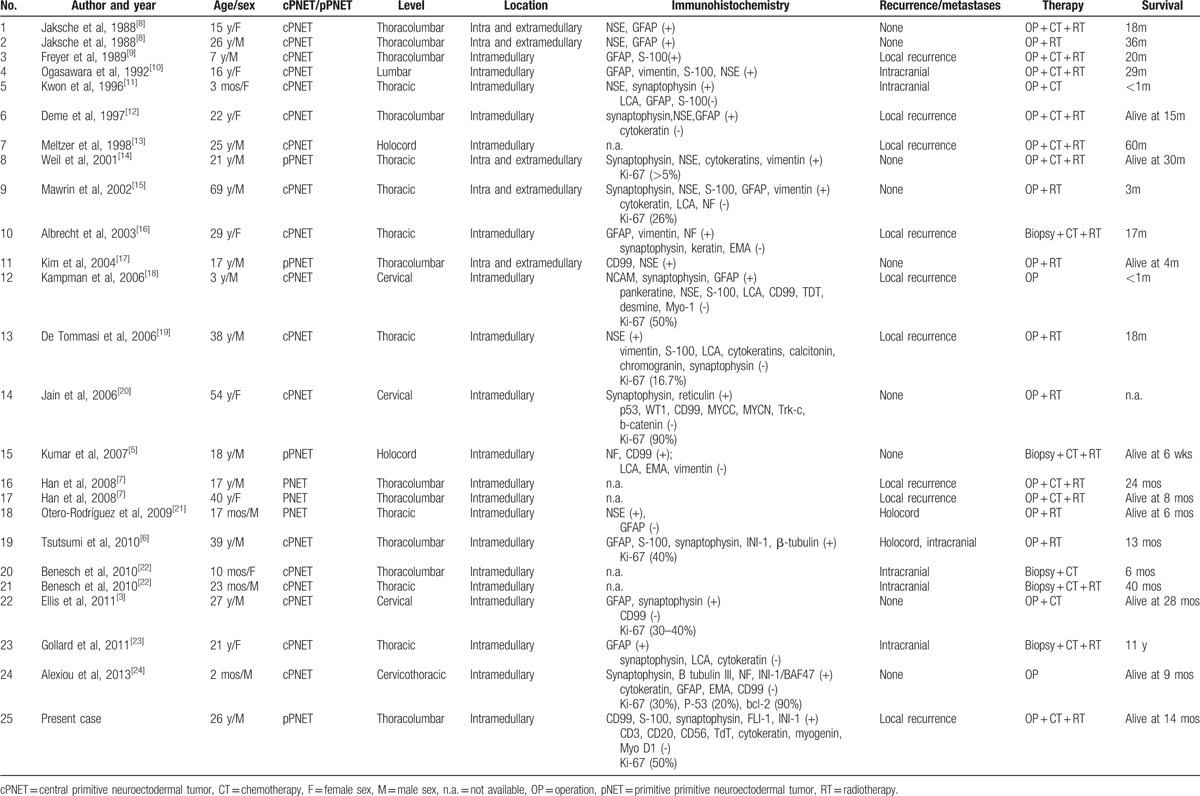
There are only 24 patients with primary intramedullary PNETs reported so far.[3,5–24] Also, a summary of these patients and our present case is shown in Table 1. There are 9 female patients and 16 male patients, with ages that ranged from 2 months to 69 years, and median age is 21 years. The male-to-female ratio is 1.78:1. These tumors mostly occur in children and young adults, and a male preponderance was observed. The tumor involved the cervical spine in 3 patients (12%), the cervicothoracic spine in 1 patient (4%), the thoracic spine in 8 patients (32%), the thoracolumbar spine in 10 patients (40%), the lumbar spine in 1 patient (4%), and the holocord in 2 patients (8%). We can find that this tumor can occur at any level of the spine; however, it mainly occurred in thoracolumbar cord. Of the 25 cases, 22 cases had been classed to cPNET or pPNET, and 18 cases were cPENT and 4 cases were pPNET. In the primary intramedullary PNET, cPNET is more common than pPNET. There are 16 cases (64%) suffering from recurrence or metastases. Considering some cases did not conduct comprehensive follow-up examinations or some cases died early, cases with recurrence or metastases may not have been recorded. Therefore, the rate of recurrence or metastases was above 64%. After operation and biopsy, 13 patients (52%) were treated with chemoradiation, 3 (12%) had chemotherapy only, 7 (28%) had radiotherapy, and 2 (8%) did not receive adjuvant therapy. Gollard et al[23] reported a case with 11 years of survival, which was the only one surviving above 5 years in our reviews. Other cases progressed rapidly and died within 5 years. Long-term outcomes were reported for 24 patients. Only 9 were reported to be alive at the time of publication (mean of 12.8 months since diagnosis, median of 9 months). The other 15 died on average at 27.9 months (median of 18.0 months) from presentation.
Table 1.
Summary of patients with primary intramedullary PNETs.
4. Conclusions
In conclusion, PNETs are rare and malignant tumors, and primary intramedullary PNETs occur even more rarely. Histopathological examination and immunohistochemistry analysis are both required for the diagnosis of PNETs. The therapy of these tumors mainly includes surgery, radiotherapy, and chemotherapy; however, the prognosis of these tumors is poor. The striking features of PNETs include: young patient, short course, rapid progress, long T1 and long T2 signal intensity with a markedly heterogenous enhancement after gadolinium administration in MRI at thoracolumbar cord. PNETs should be considered in the differential diagnosis of an intramedullary spinal cord tumor manifesting as progressive neurological deterioration, especially symptom is consistent with the above features.
Footnotes
Abbreviations: CK = cytokeratin, CNS = central nervous system, cPNET = central PNET, ETMR = embryonal tumor with multilayered rosettes, FLI-1 = Friend leukemia virus integration 1, GFAP = glial fibrillary acidic protein, INI-1 = integrase interactor 1, MRI = magnetic resonance imaging, NOS = not otherwise specified, NSE = neuron-specific enolase, PNET(s) = primitive neuroectodermal tumor(s), pPNET = peripheral PNET, Syn = synaptophysin, TdT = terminal deoxynucleotidyl transferase, TMZ = temozolomide, WHO = World Health Organization.
The authors report no conflicts of interest.
References
- [1].Hart MN, Earle KM. Primitive neuroectodermal tumors of the brain in children. Cancer 1973;32:890–7. [DOI] [PubMed] [Google Scholar]
- [2].Patnaik A, Mishra S, Mishra S, et al. Primary spinal primitive neuroectodermal tumour: report of two cases mimicking neurofibroma and review of the literature. Neurol Neurochir Pol 2012;46:480–8. [DOI] [PubMed] [Google Scholar]
- [3].Ellis JA, Rothrock RJ, Moise G, et al. Primitive neuroectodermal tumors of the spine: a comprehensive review with illustrative clinical cases. Neurosurg Focus 2011;30:E1. [DOI] [PubMed] [Google Scholar]
- [4].Tong X, Deng X, Yang T, et al. Clinical presentation and long-term outcome of primary spinal peripheral primitive neuroectodermal tumors. J Neurooncol 2015;124:455–63. [DOI] [PubMed] [Google Scholar]
- [5].Kumar R, Reddy SJ, Wani AA, et al. Primary spinal primitive neuroectodermal tumor: case series and review of the literature. Pediatr Neurosurg 2007;43:1–6. [DOI] [PubMed] [Google Scholar]
- [6].Tsutsumi S, Nonaka Y, Abe Y, et al. Intramedullary primitive neuroectodermal tumor presenting with rapidly-progressive cauda equina syndrome. Neurol Med Chir 2010;50:1031–5. [DOI] [PubMed] [Google Scholar]
- [7].Han IH, Kuh SU, Chin DK, et al. Surgical treatment of primary spinal tumors in the conus medullaris. J Korean Neurosurg Soc 2008;44:72–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jaksche H, Wöckel W, Wernert N. Primary spinal medulloblastomas? Neurosurg Rev 1988;11:259–65. [DOI] [PubMed] [Google Scholar]
- [9].Freyer DR, Hutchinson RJ, McKeever PE. Primary primitive neuroectodermal tumor of the spinal cord associated with neural tube defect. Pediatr Neurosci 1989;15:181–7. [DOI] [PubMed] [Google Scholar]
- [10].Ogasawara H, Kiya K, Kurisu K, et al. Intracranial metastasis from a spinal cord primitive neuroectodermal tumor: case report. Surg Neurol 1992;37:307–12. [DOI] [PubMed] [Google Scholar]
- [11].Kwon OK, Wang KC, Kim CJ, et al. Primary intramedullary spinal cord primitive neuroectodermal tumor with intracranial seeding in an infant. Childs Nerv Syst 1996;12:633–6. [DOI] [PubMed] [Google Scholar]
- [12].Deme S, Ang LC, Skaf G, et al. Primary intramedullary primitive neuroectodermal tumor of the spinal cord: case report and review of the literature. Neurosurgery 1997;41:1417–20. [DOI] [PubMed] [Google Scholar]
- [13].Meltzer CC, Townsend DW, Kottapally S, et al. FDG imaging of spinal cord primitive neuroectodermal tumor. J Nucl Med 1998;39:1207–9. [PubMed] [Google Scholar]
- [14].Weil RJ, Zhuang Z, Pack S, et al. Intramedullary Ewing sarcoma of the spinal cord: consequences of molecular diagnostics. Case report. J Neurosurg 2001;(suppl 2):270–5. [DOI] [PubMed] [Google Scholar]
- [15].Mawrin C, Synowitz HJ, Kirches E, et al. Primary primitive neuroectodermal tumor of the spinal cord: case report and review of the literature. Clin Neurol Neurosurg 2002;104:36–40. [DOI] [PubMed] [Google Scholar]
- [16].Albrecht CF, Weiss E, Schulz-Schaeffer WJ, et al. Primary intraspinal neurectodermal tumor: report of two cases and review of literature. J Neurooncol 2003;61:113–20. [DOI] [PubMed] [Google Scholar]
- [17].Kim YW, Jin BH, Kim TS, et al. Primary intraspinal primitive neuroectodermal tumor at conus medullaris. Yonsei Med J 2004;45:533–8. [DOI] [PubMed] [Google Scholar]
- [18].Kampman WA, Kros JM, De Jong TH, et al. Primitive neuroectodermal tumours (PNETs) located in the spinal canal; the relevance of classification as central or peripheral PNET: case report of a primary spinal PNET occurrence with a critical literature review. J Neurooncol 2006;77:65–72. [DOI] [PubMed] [Google Scholar]
- [19].De Tommasi A, De Tommasi C, Occhiogrosso G, et al. Primary intramedullary primitive neuroectodermal tumor (PNET): case report and review of the literature. Eur J Neurol 2006;13:240–3. [DOI] [PubMed] [Google Scholar]
- [20].Jain A, Jalali R, Nadkarni TD, et al. Primary intramedullary primitive neuroectodermal tumor of the cervical spinal cord. Case report. J Neurosurg Spine 2006;4:497–502. [DOI] [PubMed] [Google Scholar]
- [21].Otero-Rodríguez A, Hinojosa J, Esparza J, et al. Purely intramedullary spinal cord primitive neuroectodermal tumor: case report and review of the literature. Neurocirugia (Astur) 2009;20:381–7. [DOI] [PubMed] [Google Scholar]
- [22].Benesch M, Sperl D, von Bueren AO, et al. Primary cental nervous system primitive neurectodermal tumors (PNETs) of the spinal cord in children: Four cases from the German HIT database with a critical review of the literature. J Neurooncol 2010;104:279–86. [DOI] [PubMed] [Google Scholar]
- [23].Gollard RP, Rosen L, Anson J, et al. Intramedullary PNET of the spine: long-term survival after combined modality therapy and subsequent relapse. J Paediatr Hamatol Oncol 2011;33:107–12. [DOI] [PubMed] [Google Scholar]
- [24].Alexiou GA, Siozos G, Stefanaki K, et al. Intramedullary spinal cord primitive neuroectodermal tumor presenting with hydrocephalus. J Child Neurol 2013;28:246–50. [DOI] [PubMed] [Google Scholar]
- [25].Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 2016;131:803–20. [DOI] [PubMed] [Google Scholar]
- [26].Khmou M, Malihy A, Lamalmi N, et al. Peripheral primitive neuroectodermal tumors of the spine: a case report and review of the literature. BMC Res Notes 2016;9:438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cabral GA, Nunes CF, Melo JO, Jr, et al. Peripheral primitive neuroectodermal tumor of the cervical spine. Surg Neurol Int 2012;3:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wu G, Ghimire P, Zhu L, et al. Magnetic resonance imaging characteristics of primary intraspinal peripheral primitive neuroectodermal tumour. Can Assoc Radiol J 2013;64:240–5. [DOI] [PubMed] [Google Scholar]
- [29].Venkataraman S, Pandian C, Kumar SA. Primary spinal primitive neuroectodermal tumour: a case report. Ann Neurosci 2013;20:80–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Meng XT, He SS. Primitive neuroectodermal tumor in the spinal canal: a case report. Oncol Lett 2015;9:1934–6. [DOI] [PMC free article] [PubMed] [Google Scholar]