

Abstract
Rationale:
Charcot–Marie–Tooth disease (CMT) is typically an autosomal dominant, inherited neuropathy, although there is a rare male X-linked CMT. Such patients show central nervous system (CNS) involvement in addition to peripheral neuropathy. Recently, we encountered a patient who presented with acute disseminated encephalomyelitis (ADEM)-like symptoms, but was later diagnosed as having X-linked CMT (CMTX) due to a mutation.
Patient concerns:
A previously healthy 11-year-old boy was admitted for a sudden transient weakness of his left side extremities.
Diagnoses:
The patient was diagnosed with left side hemiparesis. Brain magnetic resonance imaging (MRI) showed ADEM-like demyelinating lesions on both centrum semiovale. A diagnosis of probable ADEM was made, and the patient soon recovered. After 4 months, a second MRI showed complete resolution of the brain lesions. However, the symptoms recurred 2 years later. A third MRI revealed white matter abnormalities, and a physical examination demonstrated pes cavus deformities and peripheral muscle wasting of both lower extremities.
Interventions:
On the basis of the brain MRI lesions and physical findings, we suspected CMTX. Genotyping confirmed a mutation in the GJB1 gene.
Outcomes:
When the symptoms recurred 2 years later, dysarthria and demyelinating MRI lesions were present. We could not identify any triggering factors.
Lessons:
Differential diagnosis of recurrent ADEM-like lesions in the cerebral white matter and peripheral neuropathy should include the possibility of CMTX disease.
Keywords: acute disseminated encephalomyelitis, CNS symptoms, GJB1 mutation, X-linked CMT
1. Introduction
Charcot–Marie–Tooth disease (CMT) is a hereditary peripheral sensory and motor neuropathy. CMT is associated with a change in the structure of several proteins, including myelin protein zero, peripheral myelin protein 22, early growth response factor 2, connexin 32, myotubularin related protein 2, periaxin, neurofilament light chain, kinesin 1B, and N-myc downstream regulated gene 1 product.[1] CMT affects approximately 1 in 2500 individuals. CMT is a common hereditary peripheral neuropathy.[2] CMT can be categorized as type 1 (demyelinating) and type 2 (axonal), depending on where the damage appears in the peripheral nerves.[3] Histopathological and electrophysiological analyses can distinguish 2 types of CMT disease, that is, primary peripheral demyelinating (CMT1) and primary peripheral axonal neuropathies (CMT2).[1,4] The common phenotype of CMT includes bone deformities such as pes caves, twirling of the ankle, painful foot callosities, and sparse bony appearances termed pes planus. In addition, 10% of CMT disease cases involve scoliosis.[5,6] CMT disease progresses gradually, usually in the second or third decade of life.[7] CMT may also appear later, but mostly with a less threatening clinical course. CMT can be autosomal dominant, autosomal recessive, or X-linked.[8] X-linked CMT (CMTX) is the second most common form next to CMT type 1A.
The majority of patients with CMT have the autosomal dominant inherited form that mainly involves the peripheral nervous system. However, central nervous system (CNS) symptoms and signs, as well as peripheral neuropathy, can present in male patients with CMTX. Here, we report our experience with a patient with X-linked CMT presenting as acute disseminated encephalomyelitis (ADEM)-like illness, diagnosis of which was confirmed by identifying a GJB1 mutation.
2. Case report
A previously healthy 11-year-old boy was referred to our hospital because of a sudden transient weakness of his left side extremities. One day before being admitted to our hospital, the patient experienced a brief and recurrent motor weakness on the left side of the body and walked unstably. He was the second child born to healthy parents. Pregnancy and birth at term were uneventful. He was delivered by normal spontaneous vaginal delivery. The birth weight was 3.1 kg (50th percentile) and his development was normal. Upon a physical examination in the emergency room, pes cavus deformities were detected. The patient had no significant neurodevelopmental family history and no family members had pes cavus deformities. A neurological examination revealed a mild left side hemiparesis, but no other neurological symptoms, such as nystagmus, neck stiffness, or headache were observed. A laboratory examination that included cerebrospinal fluid analysis showed no abnormal findings. Transaxial diffusion-weighted images showed relatively symmetric high signal intensities on both centrum semiovale and on the splenium of the corpus callosum (Fig. 1A). A diagnosis of probable ADEM was made and he was admitted and treated using high-dose intravenous methylprednisolone (20 mg/kg/day) for 3 days. The motor grade recovered to grade level 5 on both sides. He was discharged without neurological symptoms and sequelae and was followed up at the outpatient department. The steroid treatment was tapered to extinction stepwise for 1 month. At 4 months after onset, a second MRI was conducted in the outpatient department and showed that the brain lesion had completely resolved.
Figure 1.
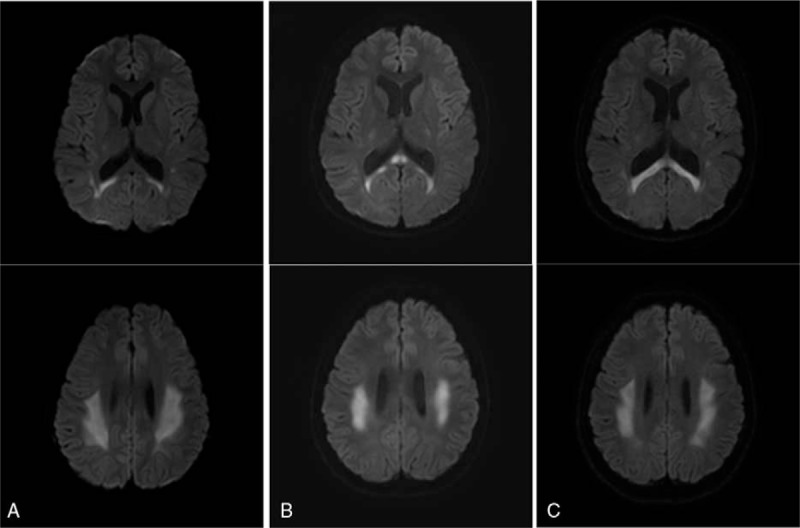
(A–C) Transaxial diffusion-weighted images (TR: 3069.5, TE: 81.0, b = 1000) showed relatively symmetric high signal intensities on both centrum semiovale and the splenium of the corpus callosum (A. Brain MRI scan was taken at the time of the first episode, B. The second episode, C. The third episode.).
Two years later (at age 13), the patient's symptoms recurred, with left side hemiparesis for 60 minutes. A third MRI showed white matter abnormalities in the internal capsule, corpus callosum, and periventricular areas (Fig. 1B). Upon physical examination, the patient showed typical pes cavus deformities and peripheral muscle wasting on both lower extremities. A neurological examination revealed grade 4 motor weakness in the left extremities, no sensory loss, and an intact deep tendon reflex. On the basis of the lesions seen on the brain MRI images and physical findings, we suspected that the patient had X-linked CMT. We identified a G283A mutation in GJB1, resulting in the substitution of valine to methionine (CMTX, GJB1: c.283G > A; Fig. 2). Neurological symptoms were relieved using high-dose intravenous methylprednisolone for 3 days, which was slowly tapered to extinction over 9 weeks.
Figure 2.
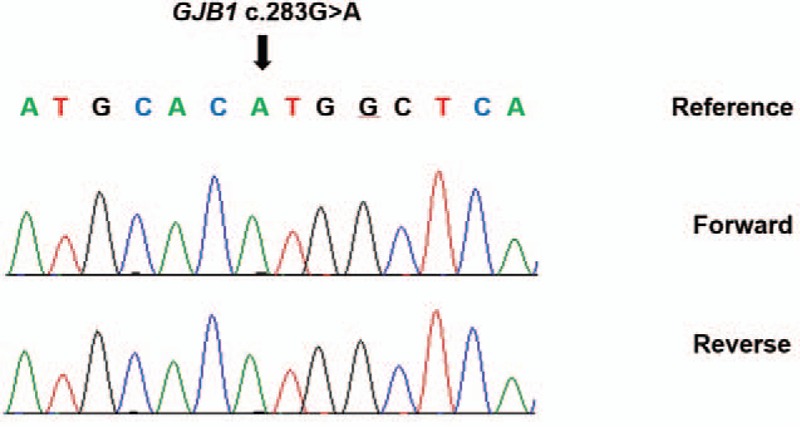
DNA sequencing chromatographs of GJB1 gene showed the c.283G > A variation.
Four years later (age 15), the neurological symptoms recurred. Dysarthria and similar MRI lesions were present (Fig. 1C), but we could not identify any triggering factors. This study was ethically approved by the Institutional review board of Chonbuk National University Hospital.
3. Discussion
It should be noted that in the present case, neither parent had symptoms or a family history of CMT disease. Although the initial clinical features of the patient were indicative of a peripheral neuropathy, they were also similar to those of ADEM, which is a CNS disease. Notably, ataxia, a high signal lesion load on MRI, and involvement of the deep gray matter are most commonly observed in ADEM. The diagnosis was able to be differentiated when the symptoms recurred and the patient showed wasting and weakness of both legs, as well as definite pes cavus deformities. Given the presence of these clinical features, we eliminated ADEM and suspected X-linked CMT.
The CNS lesions in CMTX are similar to those in ADEM when seen on MRI. ADEM is a multifocal multiple lesion with a relatively asymmetric distribution in both hemispheres, with subcortical U-fiber invasion. On the contrary, CMTX in this case was relatively symmetrically distributed, and invaded the centrum semiovale and/or the splenium of the corpus callosum; notably, subcortical U-fibers were generally spared. ADEM usually consists of a single episode or attack of widespread demyelinating damage. CMTX tends to be recurrent, though involvement of connexin 32 embedded in the myelin structure allows damage to both peripheral and central neurons to occur.[9] In CMTX, the CNS is rarely involved symptomatically, although occasional transient, severe CNS involvement characterized by ataxia and dysarthria has been described. However, patients may have mild asymptomatic CNS involvement in CMTX. Patients with 1 of 14 different mutations demonstrate central slowing of brainstem auditory-evoked responses, as well as subclinical central conduction-related slowing of visual-evoked potentials and central motor-evoked potentials.[10] CMTX, being X-linked, primarily affects male patients. It occurs in more than 90% of GJB1 mutations. The known mutations are predicted to cause a loss of function, and hence, virtually all men with GJB1 mutations have similar age-related phenotypes.[11] However, specific mutations causing CNS involvement have yet to be reported.
We report that the patient with the GJB1 mutation had CMTX with recurrent ADEM-like lesions in the cerebral white matter, pes cavus deformities, and muscle wasting of both the lower extremities. In conclusion, CMTX should be included in the differential diagnosis in children with transient CNS impairment and any signs of peripheral neuropathy.
Footnotes
Abbreviations: ADEM = acute disseminated encephalomyelitis, CMT = Charcot–Marie–Tooth disease, CNS = central nervous system, MRI = magnetic resonance imaging.
The authors report no conflicts of interest.
References
- [1].Boerkoel CF, Takashima H. Charcot-Marie-Tooth disease and related neuropathies: mutation distribution and genotype-phenotype correlation. Ann Neurol 2002;51:190–201. [DOI] [PubMed] [Google Scholar]
- [2].Antonellis A1, Ellsworth RE, Sambuughin N. Glycol-tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet 2003;72:1293–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mersiyanova IV. A new variant of Charcot-Marie-Tooth disease type 2 is probably the result of a mutation in the neurofilament-light gene. Am J Hum Genet 2000;67:137–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pareyson D, Marches C. Diagnosis, natural history, and management of Charcot–Marie–Tooth disease. Lancet Neurol 2009;8:654–67. [DOI] [PubMed] [Google Scholar]
- [5].Holmes JR, Hansen ST. Foot and ankle manifestations of Charcot Marie Tooth disease. Foot Ankle 1993;14:476–86. [DOI] [PubMed] [Google Scholar]
- [6].Banchs I, Casasnovas C, Albertí A, et al. Diagnosis of Charcot-Marie-Tooth disease. J Biomed Biotechnol 2009;2009:985415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Piazza S, Ricci G, Ienco EC, et al. Pes cavus and hereditary neuropathies: when a relationship should be suspected. J Orthop Traumatol 2010;11:195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lupski JR, Reid JG. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med 2010;362:1181–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nakagawa M, Takashima H, Umehara F, et al. Clinical phenotype in X-linked Charcot-Marie-Tooth disease with an entire deletion of the connexin 32 coding sequence. J Neurol Sci 2001;185:31–7. [DOI] [PubMed] [Google Scholar]
- [10].Taylor RA, Simon EM, Marks HG, et al. The CNS phenotype of X-linked Charcot-Marie-Tooth disease. Neurology 2003;61:1475–8. [DOI] [PubMed] [Google Scholar]
- [11].Shy ME, Siskind C, Swan ER, et al. CMT1X phenotypes represent loss of GJB1 gene function. Neurology 2007;68:849–55. [DOI] [PubMed] [Google Scholar]