

Abstract
Purpose
To report spectral domain optical coherence tomography (SD-OCT) and fundus autofluorescence (FAF) documentation of late stage macular findings associated with Sjogren-Larsson Syndrome in three adult siblings.
Methods
Three adult siblings with Sjogren-Larrson Syndrome underwent ophthalmic examination and imaging.
Results
Crystalline maculopathy and subretinal deposits, presumably lipofuscin accumulation, with macular atrophy were present in varying degrees in all three adult siblings.
Discussion
In adults with Sjogren-Larsson Syndrome, crystalline retinopathy can progress to macular atrophy and the appearance of lipofuscin accumulation.
Keywords: Crystalline retinopathy, Fundus autofluorescence, Muller cell, Sjogren-Larsson Syndrome, Spectral domain optical coherence tomography
Introduction
Sjogren-Larsson Syndrome (SLS) is a rare, autosomal recessive disorder with an estimated prevalence of <0.4 per 100,000. This neurocutaneous disorder presents as congenital ichthyosis, spasticity, and mental retardation.1 By age two, ophthalmic changes are generally present and progress with age. SLS is caused by a defect in lipid metabolism, due to a microsomal fatty aldehyde dehydrogenase enzyme (FALDH) deficiency. The consequent accumulation of fatty aldehydes and fatty alcohols disrupts white matter myelin and the barrier functions of the stratum corneum.2–4
Ophthalmic manifestations can involve all retinal layers, including the accumulation of undigested lipid metabolites in retinal ganglion cells, secondary to FALDH deficiency. The formation of foveal and parafoveal crystals is associated with the degeneration of Muller cells in the inner retina, and eventually, retinal pigment epithelium compromise from lipofuscin accumulation, leading to progressive RPE atrophy.5–6 Macular pigment (lutein and zeaxanthin) is reduced in the fovea in SLS and is likely caused by a reduction in carotenoid accumulation and Muller cell dysfunction.7 Patients may complain of photophobia and reduced visual acuity (VA), secondary to Muller cell compromise, retinal thinning, photoreceptor dysfunction, and RPE atrophy.
We present the first reported spectral domain optical coherence tomography and fundus autofluorescence documentation of lipofuscin accumulation and RPE atrophy, which are advanced macular findings associated with Sjogren-Larsson Syndrome, in three non-consanguineous Caucasian adult siblings.
Case Report
Three siblings (35, 38, and 42- years-old) presented with congenital ichthyosis, spasticity, and mental retardation. The diagnosis of Sjogren-Larsson Syndrome (SLS) had been made at prior institutions during childhood and included confirmatory genetic testing. A detailed ophthalmologic examination included assessment of best-corrected visual acuity (BCVA), slit-lamp examination, fundoscopy, and extensive fundus imaging using standard color fundus photography, auto-fluorescence (FAF) and near-infrared reflectance imaging (Heidelberg HRA2), and spectral domain optical coherence tomography (SD-OCT; Heidelberg Spectralis OCT).
Patient 1
A 35-year-old woman presented with best-corrected visual acuity (BCVA) of 20/50 OU with mild myopia. On adnexal exam scaling of her upper lids was noted. Anterior segment exam was normal as was her peripheral retinal exam. Her optic nerves appeared healthy. On macular evaluation, intraretinal crystals were present OU along with retinal pigmentary changes and atrophy. These changes were symmetrical between the two eyes, except for more prominent lipofuscin accumulation in the right macula (Figure 1A). Fundus autofluorescence imaging revealed scattered areas of hyperautofluorescence, corresponding to pigmented areas of the macula and signifying areas of increased lipofuscin. Additional areas of hypoautofluorescence signified regions with RPE atrophy (Figure 1B). Spectral domain OCT showed crystals in the outer plexiform layer (OPL), inner plexiform layer (IPL), and nerve fiber layer OU, and pseudocystic atrophy in the right eye. Areas of ellipsoid zone disruption and retinal pigment epithelium (RPE) disruption are noted along with subretinal deposits, presumably lipofuscin accumulation OU (Figure 1C–D). Decreased central retinal thickness (CRT) was also appreciated OU.
Figure 1.
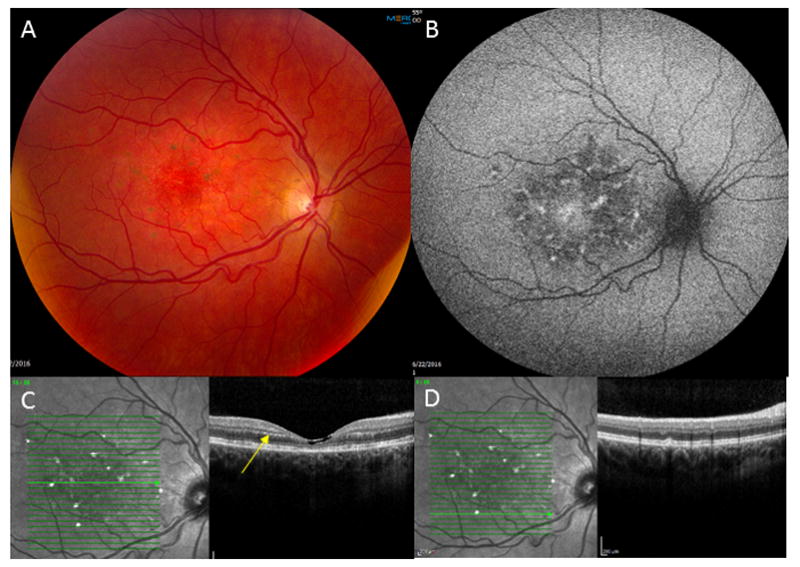
A—Fundus photo OD showing intraretinal crystals in the macular region along with retinal pigmentary changes and atrophy. Several hyperpigmented spots of greater lipofuscin accumulation can be seen. B—Fundus Autoflouresence OD showing scattered hyperflouresence consistent with pigmented regions of the macula and areas of increased lipofuscin accumulation, and patchy hypoflouresence indicative of RPE atrophy. C— SDOCT with crystals (yellow arrow) noted in the OPL, IPL, and nerve fiber layer OD. Additional regions of EZ and RPE disruption with subretinal lipofuscin accumulation is appreciated. Foveal pseudocystic atrophy is also appreciated. D—Subretinal (presumably lipofuscin) deposit.
Patient 2
The 38-year-old man presented with BCVA of 20/70 OD and 20/60 OS and mild hyperopia. Scaling was noted on bilateral upper lids. Anterior segment, optic nerve, and peripheral retinal examination were all normal. Macular evaluation revealed intra-retinal crystals and RPE changes in the fovea and para-foveal zones. One pigmented nidus of lipofuscin accumulation was noted in each macula superotemporal to the fovea (Figure 2A). Hyperautofluorescence was noted in the fovea and in the few areas of lipofuscin accumulation OU on FAF imaging (Figure 2B). Crystals were noted in the OPL and IPL OU and pseudocystic atrophy was present OU along with significant retinal thinning, ellipsoid zone (EZ) disruption and a subretinal deposit OU on SD-OCT (Figure 2C). Central retinal thickness was reduced OU. Atrophic changes and lipofuscin deposits were less prominent compared to Patient 1.
Figure 2.

A—Fundus photo OD with milder crystalline deposition and RPE atrophy than Patient 1. A single nidus of dark pigmentation secondary to lipofuscin accumulation is noted in the superotemporal region of the macula (arrow). B—FAF with hyperflouresence from lipofuscin accumulation. C—SDOCT OD shows crystalline maculopathy and pseudocystic atrophy with significant retinal thinning.
Patient 3
The 42-year-old woman presented with BCVA of 20/70 OD and 20/80 OS with mild myopia. No lid scaling was present. Anterior segment, optic nerve, and peripheral retinal examination were all normal. Macular evaluation revealed fewer intraretinal crystals compared to her younger siblings and more prominent pigmentary changes and atrophy OU. General fundus pigmentation was lighter than her siblings as well (Figure 3A). FAF showed foveal hyperautofluorescence surrounded by a ring of hypoautofluorescence punctuated by areas of increased lipofuscin and hyperautofluorescence (Figure 3B). Fewer crystals were present relative to her siblings on SD-OCT, but EZ and RPE disruption were more prominent, as were areas of subretinal deposits, presumably lipofuscin accumulation (Figure 3C).
Figure 3.

A—Fundus photo OD with prominent pigmentary changes and retinal atrophy, and diffuse fundus hypopigmentation. B—FAF shows foveal hyperflouresence and a ring of hypoflouresence. C—SDOCT reveals fewer crystals relative to the patient’s siblings. There is prominent EZ and RPE disruption, and subretinal (presumably) lipofuscin accumulation.
Discussion
This report demonstrates advanced SLS maculopathy in three adult siblings in the second largest family with genetically confirmed SLS, and the first from non-consanguineous origins. All three patients developed diplegic spasticity requiring walkers. The two oldest siblings were first discussed in a dermato-pathology paper in 1982.8 Considering no prior ophthalmic findings were recorded, a longitudinal comparison was not possible.
A crystalline maculopathy of varying severity was present in these three adult siblings. Crystals can be appreciated in any retinal layer but are usually most prominent in the outer and inner plexiform layers of the retina in patients with SLS. Considering that the Muller cell body is located in the inner nuclear layer with processes extending into the outer limiting membrane and inner limiting membrane of the inner retina, crystals found in this distribution could be considered a marker of Muller cell damage.9 Crystal formation has been documented in SLS patients as young as 3 years old and seems to be a relatively early step in pathogenesis.10
Muller cells function as support cells for other cells in the retina as they phagocytose outer segments shed from cones and defend against oxidative stress.9 They also act as optical fibers within the retina, without which photophobia could be exacerbated, as occurs in SLS patients. Muller cell dysfunction can also contribute to a loss of macular pigment, which is a feature of SLS maculopathy. Loss of macular pigment can lead to an increase in oxidative stress as well.
A consequence of the fatty aldehyde dehydrogenase enzyme (FALDH) deficiency in SLS is an accumulation of fatty aldehydes and fatty alcohols in several body tissues, including the retina. This accumulation of lipid and/or the secondary oxidative byproducts from lipid metabolism may result in Muller cell dysfunction. Muller cell damage could also further compromise the retina’s ability to metabolize and remove excess lipid and minimize the production of oxidative byproducts. Muller cell compromise can lead to neuronal apoptosis and photoreceptor damage, clinically demonstrated as pseudocystic atrophy, retinal thinning, increased lipofuscin, and RPE atrophy. These macular findings are the end-stage changes in SLS maculopathy that are present in varying degrees in our three adult patients.
Retinal crystals in this particular distribution and the atrophic retinal changes (IS/OS and RPE disruption) appear similar to those found in type 2 idiopathic macular telangiectasia and tamoxifen retinopathy, meaning that all three conditions may involve Muller cell dysfunction as an early step in a pathway which culminates in retinal atrophy. Unlike type 2 idiopathic macular telangiectasia and tamoxifen retinopathy, SLS maculopathy features subretinal deposits (presumably lipofuscin accumulation) late in the disease progression.
To the best of our knowledge we offer the first documentation of spectral domain optical coherence tomography and fundus autofluorescence findings of lipofuscin accumulation and RPE atrophy in advanced stage Sjogren-Larsson Syndrome maculopathy in three non-consanguineous Caucasian adult siblings. The maculopathy in this condition likely involves Muller cell dysfunction and has similar features to type 2 idiopathic macular telangiectasia and tamoxifen retinopathy, suggesting a common pathway that culminates in retinal atrophy. Reclassification of these conditions and potentially others based on Muller cell dysfunction rather than the presence of retinal crystals may be indicated.
Supplementary Material
Summary.
We present the first reported spectral domain optical coherence tomography (SD-OCT) and fundus autofluorescence (FAF) documentation of lipofuscin accumulation and RPE atrophy, which are advanced macular findings associated with Sjogren-Larsson Syndrome, in three non-consanguineous Caucasian adult siblings.
Acknowledgments
Funding was provided by NIH center grant P30-EY014801 and by an unrestricted grant to the University of Miami from Research to Prevent Blindness, New York, NY. The sponsor or funding organization had no role in the design or conduct of this research
Footnotes
No conflicting relationship exists for any author. This article was completed at Bascom Palmer Eye Institute Miller School of Medicine at the University of Miami.
References
- 1.Sjogren T, Larsson T. Oligophrenia in combination with congenital ichthyosis and spastic disorder; a clinical and genetic study. Acta Psychiatr Neurol Scand Suppl. 1957;113:1–112. [PubMed] [Google Scholar]
- 2.Willemsen MAAP, Johannes RMC, Rotteveel JJ, et al. Juvenile macular dystrophy associated with deficient activity of fatty aldehyde dehydrogenase in Sjögren-Larsson syndrome. American Journal of Ophthalmology. 2000;130(6):782–89. doi: 10.1016/s0002-9394(00)00576-6. [DOI] [PubMed] [Google Scholar]
- 3.De Laurenzi V, Rogers GR, Hamrock DJ, et al. Sjögren-Larsson syndrome is caused by mutations in the fatty aldehyde dehydrogenase gene. Nat Genet. 1996;12:52–7. doi: 10.1038/ng0196-52. [DOI] [PubMed] [Google Scholar]
- 4.Lossos A, Khoury M, Rizzo WB, et al. Phenotypic variability among adult siblings with Sjögren-Larsson syndrome. Archives of Neurology. 2006;63(2):278–280. doi: 10.1001/archneur.63.2.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Newman E, Reichenbach A. The Müller cell: a functional element of the retina. Trends Neurosci. 1996;19:307–12. doi: 10.1016/0166-2236(96)10040-0. [DOI] [PubMed] [Google Scholar]
- 6.Nilsson SE, Jagell S. Lipofuscin and melanin content of the retinal pigment epithelium in a case of Sjögren-Larsson syndrome. British journal of ophthalmology. 1987;71(3):224–26. doi: 10.1136/bjo.71.3.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van der Veen RL, Fuijkschot J, Willemsen MA, et al. Patients with Sjögren-Larsson syndrome lack macular pigment. Ophthalmology. 2010;117(5):966–971. doi: 10.1016/j.ophtha.2009.10.019. [DOI] [PubMed] [Google Scholar]
- 8.Matsuoka LY, Kousseff BG, Hashimoto K. Studies of the skin in Sjogren-Larsson syndrome by electron microscopy. The American Journal of Dermatopathology. 1982;4(4):295–302. doi: 10.1097/00000372-198208000-00002. [DOI] [PubMed] [Google Scholar]
- 9.Reichenbach A, Bringmann A. New functions of Müller cells. Glia. 2013;61(5):651–78. doi: 10.1002/glia.22477. [DOI] [PubMed] [Google Scholar]
- 10.Fuijkschot J, Cruysberg JR, Willemsen MA, et al. Subclinical changes in the juvenile crystalline macular dystrophy in Sjögren-Larsson syndrome detected by optical coherence tomography. Ophthalmology. 2008;115:870–5. doi: 10.1016/j.ophtha.2007.05.063. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.