

Abstract
Fibroblast growth factors (FGFs) and FGF receptors (FGFRs) are known for their potent effects on cell proliferation/differentiation and cortical patterning in the developing brain. However, little is known regarding the roles of FGFs/FGFRs in cortical circuit formation. Here we show that Fgfr1/2/3 and Fgf7/9/10/22 mRNAs are expressed in the developing primary somatosensory (S1) barrel cortex. Barrel cortex layer IV spiny stellate cells (bSCs) are the primary recipients of ascending sensory information via thalamocortical axons (TCAs). Detail quantification revealed distinctive phases for bSC dendritogenesis: orienting dendrites toward TCAs, adding de novo dendritic segments, and elongating dendritic length, while maintaining dendritic patterns. Deleting Fgfr1/2/3 in bSCs had minimal impact on dendritic polarity but transiently increased the number of dendritic segments. However, 6 d later, FGFR1/2/3 loss of function reduced dendritic branch numbers. These data suggest that FGFs/FGFRs have a role in stabilizing dendritic patterning. Depolarization of cultured mouse cortical neurons upregulated the levels of several Fgf/Fgfr mRNAs within 2 h. In vivo, within 6 h of systemic kainic acid administration at postnatal day 6, mRNA levels of Fgf9, Fgf10, Fgfr2c, and Fgfr3b in S1 cortices were enhanced, and this was accompanied by exuberant dendritogenesis of bSCs by 24 h. Deleting Fgfr1/2/3 abolished kainic acid-induced bSC dendritic overgrowth. Finally, FGF9/10 gain of function also resulted in extensive dendritogenesis. Together, our data suggest that FGFs/FGFRs can be regulated by glutamate transmission to modulate/stabilize bSC dendritic complexity. Both male and female mice were used for our study.
SIGNIFICANCE STATEMENT Glutamatergic transmission plays critical roles in cortical circuit formation. Its dysregulation has been proposed as a core factor in the etiology of many neurological diseases. We found that excessive glutamate transmission upregulated mRNA expression of Fgfrs and their ligands Fgfs. Deleting Fgfr1/2/3 not only impaired bSC dendritogenesis but also abolished glutamate transmission-induced dendritic overgrowth. Overexpressing FGF9 or FGF10 in cortical glutamatergic neurons results in excessive dendritic outgrowth within 24 h, resembling the changes induced by excessive glutamate transmission. Our findings provide strong evidence for the physiological role of fibroblast growth factors (FGFs) and FGF receptors (FGFRs) in establishing and maintaining cortical circuits. Perturbing the expression levels of FGFs/FGFRs by excessive glutamatergic neurotransmission could lead to abnormal neuronal circuits, which may contribute to neurological and psychiatric disease.
Keywords: dendritogenesis, FGF, FGFR, glutamatergic, neural activity, whisker barrel map
Introduction
One remarkable feature of cortical glutamatergic neurons is their distinctive dendritic arbor, where most of their presynaptic connections reside. The spatial arrangement of dendritic segments affects how synaptic inputs are propagated and integrated at the soma and subsequently affect synaptic plasticity and behavior (Häusser et al., 2000; Sjöström et al., 2008). During early brain development, both intrinsic and extrinsic signals can modulate dendritogenesis (McAllister, 2000; Gao, 2007; Jan and Jan, 2010; Joo et al., 2014). In particular, neuronal activity plays a crucial role in modulating dendritic morphogenesis (McAllister, 2000; Cline, 2001; Wong and Ghosh, 2002; Chen and Ghosh, 2005).
The rodent whisker-barrel map in the primary somatosensory (S1) cortex is a popular model system to explore the role of neural activity in sculpting cortical sensory circuits (Simons and Woolsey, 1979; Feldman and Brecht, 2005; Petersen, 2007; Wu et al., 2011). Barrel cortex layer IV spiny stellate cells (bSCs) project their dendrites toward the clusters of thalamocortical axons (TCAs) relaying sensory information from their corresponding whiskers. Studies using several “barrelless” mutant mice lacking barrel cytoarchitecture, indicate a key role for glutamate transmission in bSC dendritogenesis (Datwani et al., 2002; Espinosa et al., 2009; Ballester-Rosado et al., 2010; Narboux-Nême et al., 2012; Matsui et al., 2013; Mizuno et al., 2014). For example, deleting presynaptic active zone protein RIM1/2 in the thalamus (Narboux-Nême et al., 2012) or deleting postsynaptic glutamate receptors NR1, NR2B, or mGluR5 impair the dendritic morphogenesis of bSCs (Espinosa et al., 2009; She et al., 2009; Ballester-Rosado et al., 2010, 2016; Mizuno et al., 2014). Despite these striking results, the cellular mechanisms by which glutamate receptors regulate dendritic morphogenesis are poorly understood.
FGF signaling plays diverse roles in neurogenesis, brain patterning, and establishing of axonal projections during embryonic development (Thisse and Thisse, 2005; Iwata and Hevner, 2009; Dorey and Amaya, 2010; Guillemot and Zimmer, 2011). The majority of fibroblast growth factors (FGFs; 22 known members) act through FGF receptors (FGFRs) 1–3, all of which contain an intracellular tyrosine kinase domain that can trigger several different signaling pathways, thus having diverse impacts on the cell (Itoh and Ornitz, 2004; Guillemot and Zimmer, 2011). The functions of FGFs/FGFRs in neural circuit formation during postnatal development are only just now being revealed (Umemori et al., 2004; Terauchi et al., 2010; Singh et al., 2012). For example, Umemori et al. (2004) found that different FGF ligands exerted differential impacts on dendritic branching, axon elongation, and vesicle aggregation/synapse formation in chicken embryonic motor neurons. In addition, FGF1 application promotes neurite branching of cultured spinal ganglion cells (Aletsee et al., 2003) and intracerebroventricular FGF2 infusion promotes dendritic growth of adult-born hippocampal granule cells (Rai et al., 2007). Furthermore, cultured FGF2 knock-out neurons exhibit shorter dendritic length (Baum et al., 2016). Together, these findings suggest that FGF–FGFR signaling can regulate dendritic growth; however, whether endogenous FGF/FGFRs are regulators in dendritogenesis in vivo is unknown.
In this study, we explored whether FGF–FGFR signaling is involved in bSC dendritogenesis and whether neural activity regulates FGF/FGFR levels. Using both in vitro and in vivo approaches, we found that Fgf9, Fgf10, Fgfr2c, and Fgfr3b mRNAs were present in the developing S1 cortex and were upregulated by neural activity. Mosaic animals were generated to conduct both gain-of-function and loss-of-function (LOF) experiments. We found that deleting Fgfr1/2/3 in bSCs not only perturbs their dendritic outgrowth but also impairs the maintenance of a stable dendritic pattern. In addition, FGFR1/2/3 LOF abolishes the dendritogenesis induced by excessive glutamate receptor activation. Furthermore, expressing FGF9 or FGF10 in bSCs results in exuberant dendritic branching, similar to the morphology of bSCs following excessive synaptic activity. Together, our results suggest that FGFs/FGFRs play a key role in activity-dependent dendritic patterning of glutamatergic neurons in the developing cortex.
Materials and Methods
Chemicals and antibodies.
All reagents and chemicals were purchased from Sigma-Aldrich, unless otherwise stated. Alexa Fluor-conjugated secondary antibodies were purchased from Invitrogen. Rabbit anti-red fluorescent protein antibody was purchased from Rockland Antibodies & Assays. 2-Methyl-6-(phenylethynyl) pyridine hydrochloride (MPEP), and 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX) were purchased from Tocris Bioscience. d-(−)-2-Amino-5-phosphonopentanoic acid (d-APV) and nifedipine were purchased from Abcam.
Experimental design and animals.
The conditional alleles for Fgfr1 (Fgfr1flox/flox), Fgfr2 (Fgfr2flox/flox), and Fgfr3 (Fgfr3flox/flox) were generated as described previously (maintained in an FVB background; Trokovic et al., 2003; Yu et al., 2003; Su et al., 2010). Male and female triple transgenic mice homozygous for Fgfr1flox/flox;Fgfr2flox/flox;Fgfr3flox/flox (abbreviated as Fgfr1/2/3 triple conditional mice) were originally generated in Dr. Jean M. Hébert's laboratory (Kang and Hébert, 2015) and transferred to the Lu laboratory to establish breeding colonies, initially at Baylor College of Medicine and then at Indiana University. Wild-type ICR mice from Envigo were used for cortical neuron culture and FGF overexpression studies. Both male and female mice were used for data analysis. Animals were treated in compliance with the U.S. Department of Health and Human Services, Baylor College of Medicine, and Indiana University, Bloomington, guidelines and procedures approved by the relevant institutional animal care and use committee.
Genotyping.
Tail lysates were prepared by immersing tail pieces in tail digestion buffer (50 mm KCl, 10 mm Tris-HCl, Triton X-100, 0.1 mg/ml proteinase K, pH 9.0) and vortexing gently for 3 h at 60°C to lyse the tail tissue. Tail lysates were heated to 94°C for 10 min to denature the proteinase K (Thermo Fisher Scientific) then centrifuged at 16,100 × g for 15 min. The supernatants were used as DNA templates for PCRs (EconoTaq Plus Green 2X Master Mix, Lucigen). The genotyping primers are as previously described (Trokovic et al., 2003; Yu et al., 2003; Su et al., 2010).
Administration of kainic acid.
Kainic acid (KA) was dissolved in sterilized saline (0.9% NaCl) and administered intraperitoneally or subcutaneously to neonatal ICR pups [postnatal day 6 (P6)] at a dose of 1.5 mg/kg, according to previous studies that have demonstrated that this dose reliably induces electrographic and behavioral seizures (Stafstrom et al., 1992; Lynch et al., 2000; Howland et al., 2004). Pups were separated from their dam immediately before saline or KA administration. Littermates injected with saline served as controls. All pups injected with KA displayed swimming seizure-like behaviors within 15–30 min of injection as previously reported for rat pups (Stafstrom et al., 1992; Lynch et al., 2000; Howland et al., 2004). The seizing behavior lasted for 3–4 h. During this time, saline-injected pups were placed together with their littermates treated with KA. Once seizing behaviors stopped in all pups, they were returned simultaneously to their dam.
Plasmid construction.
The full-length cDNA coding regions for mouse Fgf9 (mFGF9) and Fgf10 (mFGF10) were PCR amplified from pMD18-mFGF9 and pMD18-mFGF10 (Sino Biological) using the following primers for mFGF9: forward-5′-CTAGCTAGCATGGCTCCCTTAGGTGAAGTTGGG-3′, reverse-5′-TTGGCGCGCCTCAGCTTTGGCTTAGAATATCCTTA-3′; and for mouse FGF10: forward-5′-CTAGCTAGCATGTGGAAATGGATACTGACACATT-3′, reverse-5′-TTGGCGCGCCCTATGTTTGGTATCGTCATGGGGAG-3′. (Italics indicate the restriction sites for NheI and AscI.) The PCR fragment was cloned between NheI and AscI sites of the pAAV-EF1-DIO-hCHR2(H134R)-EYFP-WPRE plasmid (a gift from Mingshan Xue, Baylor College of Medicine and Jan and Dan Duncan Neurological Research Institute at Texas children's hospital, Houston, TX) and designated as pAAV-EF1α-DIO-mFGF9 and pAAV-EF1α-DIO-mFGF10.
In utero electroporation.
In utero electroporation (IUE) was performed as previously described (Shimogori, 2006; Rice et al., 2010). Briefly, pregnant female dams 14.5 d postgestation were anesthetized by isoflurane inhalation, and a small incision was made in the abdominal wall to expose the uterine horns. Approximately 0.5–1 μl of the DNA solution was injected into the lateral ventricle of the embryo using a pulled glass micropipette. For deleting FGFR1/2/3 from cortical neurons, pAAV-EF1α-icre-P2A-tdTomato (0.5 μg/μl) was electroporated into the right hemisphere of 50% of the Fgfr1/2/3 triple conditional embryos. pAAV-EF1α-tdTomato (0.25 ∼ 0.5 μg/μl) was electroporated into the left hemisphere of the other 50% Fgfr1/2/3 triple conditional mice that served as littermate controls. For the FGF9/FGF10 gain-of-function experiments, a DNA mixture of pCAG-ERT2CreERT2 (Matsuda and Cepko, 2007), pAAV-EF1α-DIO-tdTomato (a gift from Dr. Mingshan Xue), and pAAV-EF1α-DIO-mFgf9/10 (mixed in a molar ratio of 10:1:10, with a final concentration of 1 μg/μl) was infused into the right hemisphere of 50% wild-type embryos (ICR mice). The remaining embryos received a DNA mixture of pCAG-ERT2CreERT2 and pAAV-EF1α-DIO-tdTomato to serve as littermate controls (mixed in a molar ratio of 10:1). Each embryo within the uterus was placed between platinum tweezers-type electrodes (5 mm diameter; Harvard Apparatus). Square electric pulses (30–35 V, 50 ms) were passed five times at 1 s intervals using an electroporator (catalog #ECM830, Harvard Apparatus). After electroporation, the wall of the abdominal cavity and skin were then sutured, and embryos were allowed to develop to term, depending on the experiment.
Primary neuronal culture.
Cerebral cortices were dissected from embryonic day 15.5 (E15.5) ICR mouse embryos and dissociated, and the dissociated cells were seeded at 8 × 105/cm2 and maintained in Neurobasal medium (Invitrogen) supplemented with 2% B27 supplement (Invitrogen), 50 U/ml penicillin, and 50 μg/ml streptomycin, as described previously (Huang et al., 2009). Neurons were cultured for 7 d in vitro (DIV7) for pharmacological experiments.
Total RNA extraction and RT-PCR.
Total RNA was extracted from neuronal cultures or brain tissue by RNeasy Mini Kit (Qiagen) and followed by on column DNase digestion according to the manufacturer instructions. One microgram total RNA was converted to cDNA by using an iScript TM cDNA Synthesis Kit (Bio-Rad). For real-time PCR, cDNA was mixed with PCR master mix solution (Bio-Rad) and 0.4 μm of each forward and reverse primer in a final volume of 20 μl. Real-time PCR primers were as previously described (Fon Tacer et al., 2010). The PCR was reacted in the CFX96 Touch Real-time PCR Detection System (Bio-Rad). The Δ Δ Ct method was used to calculate relative expression levels.
Immunostaining.
Mice were anesthetized and perfused with PBS followed by 4% paraformaldehyde (PFA)/PBS. Brains were harvested and postfixed with 4% PFA/PBS overnight at 4°C. Fixed brains were sectioned into 100-μm-thick sections in the coronal plane by using a vibrating microtome (VT-1000 Vibrating Microtome, Leica Microsystems). Free-floating sections were used in all subsequent steps. Sections were permeabilized with 0.2% Triton X-100 in PBS for 20 min at room temperature, incubated with blocking solution (3% normal goat serum prepared in PBS with 0.01% Triton X-100), and then incubated overnight with rabbit anti-red fluorescent protein antibody (1:2000 dilution in blocking solution) to identify tdTomato protein. Goat anti-rabbit IgG-conjugated with Alexa Fluor 555 antibody (1:2000 dilution) and 4′,6′-diamidino-2-phenylindole dihydrochloride (5 μg/ml) were used to detect primary antibody and visualize nuclei, respectively.
Reconstruction and quantification of neuronal morphology.
In brain sections, tdTomato-expressing layer IV bSCs were identified as those neurons with nuclei located near barrel borders and lacking apical dendrites projecting toward the pial surface. The z-stack images of these layer IV cortical neurons were acquired with a Leica SP8 confocal microscope with a 25× numerical aperture 0.95 objective. The z-stacks were taken at 0.5 μm intervals to image all the dendritic segments connected to the cell bodies. Neuronal morphology was traced and reconstructed using Imaris software (Bitplane) with the Neurofilament module. Dendritic order was defined using the centrifugal method, as described previously (Uylings et al., 1975). Polarized and nonpolarized neurons were defined as previously described (Espinosa et al., 2009; Ballester-Rosado et al., 2010). The greatest sum of dendritic length in a continuous 180° sector was defined as the dendritic length inside the barrel, while the remainder was defined as the dendritic length outside the barrel. All imaging and analysis was performed in a blinded manner.
Statistical analysis.
Data were acquired from both male and female animals and were combined for data analysis. Data analysis and determination of statistical significance were performed using GraphPad Prism 6.0 software (GraphPad Software). In the figures, data are expressed as the mean ± SEM. We used the unpaired t test, Pearson correlation coefficients, one-way ANOVA, or two-way ANOVA to examine data, as presented in the figure legends. Original data values and detail statistical comparisons for all figures are contained in extended data.
Results
Simultaneously eliminating Fgfr1/2/3 perturbed activity-guided dendritogenesis in vivo
Fgfr1/2/3 are expressed in the developing S1 cortex based on in situ hybridization data shown in the Allen Brain Atlas (http://developingmouse.brain-map.org/), while Fgfr4 expression has not been detected in neocortical precursors during development (Kang et al., 2009). Fgfr5 lacks an intracellular kinase domain and is thought to act as a negative regulator of FGF–FGFR signaling (Sleeman et al., 2001). Despite the differential binding affinities of an individual FGF for FGFRs, it is proposed that different FGFRs can compensate for each other's functions (Itoh and Ornitz, 2004; Beenken and Mohammadi, 2009; Guillemot and Zimmer, 2011; Hébert, 2011). Considering the redundant function of FGFR1/2/3, in vivo loss-of function experiments that simultaneously remove Fgfr1/2/3 were conducted to explore the potential contribution of FGFR signaling in bSC dendritogenesis. Specifically, Fgfr1/2/3 was deleted from a small number of bSCs by using IUE to introduce icre (nuclear cre) and tdTomato-expressing constructs into cortical layer IV precursor cells of Fgfr1/2/3 triple conditional embryos (Fgfr1flox/flox;Fgfr2flox/flox;Fgfr3flox/flox; Fig. 1A). Control tdTomato-positive bSCs were generated by electroporating a tdTomato-expressing construct with the IUE procedure.
Figure 1.

Eliminating Fgfr1/2/3 perturbed activity-guided dendritogenesis processes in vivo. A, Schematic representation of experimental procedures. B–G, Examples of original images and computer-aided reconstructions. B1–G1 show the locations of barrels (dashed lines) and reconstructed neurons (white arrows). II–V, Cortical layers. The projected images from each confocal image stack are shown in B2–G2. B3–G3 show the traced images of neurons in B2–G2. B4–G4 show color-coded segments according to their branch orders. H–J, Quantitation of total length (H), branch points (I), and mean length (J) of dendrites. K–N, Summaries of segment length (K, M) and number (L, N) per branch order. Cell numbers and animal numbers for each group are as listed in the figure, and there are 28 cells/4 animals in the P6-LOF group. One-way ANOVA with post hoc Tukey's multiple-comparisons test was conducted for H to J. Two-way ANOVA with post hoc Tukey's multiple-comparisons test was conducted for K to N. The statistical analysis (#) compared the indicated groups with control (Ctrl) group with younger age: #p < 0.05; ###p < 0.001. The statistical analysis (*) compared the indicated groups: *p < 0.05; **p < 0.01; ***p < 0.001. Original data values and detailed statistical comparisons for this part of the figure are contained in extended data file Figure 1-1.
Original data values and detailed statistical comparison for Figure 1. Download Figure 1-1, XLSX file (37.3KB, xlsx)
bSCs project the majority of their dendrites toward the barrel hollow, where they form synaptic connections with their corresponding TCAs. The polarized dendritic pattern of bSCs is established before P6 and is followed by substantial addition and outgrowth of dendritic segments (Espinosa et al., 2009; Matsui et al., 2013; Mizuno et al., 2014). The dendritic morphologies of individual tdTomato-positive bSCs were reconstructed in three dimensions (Fig. 1B–G) and analyzed for the following characteristics: total dendritic length, total branch point number, average dendritic length, segment number/length per branch order, and polarity (see Materials and Methods). We found that control bSCs acquired a polarized pattern by P6 (Huang and Lu, 2017) and progressively increased their total dendritic length (Fig. 1H), similar to previous studies (Espinosa et al., 2009; Ashby and Isaac, 2011). This increase in total dendritic length is the result of both developmental additions of branch point numbers (Fig. 1I) and the augmentation of mean dendritic length (Fig. 1J). The progressive increase of mean dendritic length during development for most branch orders suggests that the overall dendritic coverage of bSCs expands from P6 to P15 (Fig. 1K). Interestingly, bSC dendritic segment complexity increases substantially from P6 to P9, with no further increase after P9 (Fig. 1I,L). For dendritic segments, de novo branches were added at multiple branch orders between P6 and P9 (p < 0.0001; Fig. 1L). Together, our detailed morphological characterization revealed at least three distinctive phases of dendritic morphogenesis, as follows: (1) bSCs orient their dendrites toward TCAs by the end of the first postnatal week, (2) bSCs add de novo dendritic segments and reach their final dendritic branch pattern by P9, and (3) individual segments get progressively longer throughout the first 2 postnatal weeks.
FGFR1/2/3 LOF neurons deviate from this pattern of dendritic morphogenesis as they develop (Fig. 1H–J,M–N). FGFR1/2/3 LOF bSCs display normal total dendritic length, branch points, and mean segment length at P6 (Fig. 1H–J). By P9, FGFR1/2/3 LOF bSCs have significantly more branch points (p = 0.0037), but total dendritic length is similar to that in P9 controls (p = 0.4928). Surprisingly, between P9 and P15 in FGFR1/2/3 LOF bSCs, the branch point number (p < 0.0001; Fig. 1I) is significantly reduced. At P15, FGFR1/2/3 LOF bSCs are less complex than P15 control bSCs with regard to total dendritic length (p = 0.0001; Fig. 1H) and dendritic branch numbers (p = 0.0274; Fig. 1I). The mean dendritic length of FGFR1/2/3 LOF bSCs is comparable to that of control bSCs at P6 (p = 0.9795), P9 (p = 0.1298), and P15 (p > 0.9999; Fig. 1J). There is also no significant difference in dendritic polarities between P15 control bSCs (0.7935 ± 0.0221) and FGFR1/2/3 LOF bSCs (0.7691 ± 0.0192; p = 0.9586). Our finding supports the endogenous role of FGFR1/2/3 in regulating dendritic segment addition from P6 to P9 and also in maintaining the dendritic complexity after P9.
To examine the temporal profiles of Fgfr1/2/3 mRNA expression during barrel map formation, real-time PCR experiments were conducted with P3–P9 S1 cortex (Fig. 2A,B) using the experimental procedures described in Huang et al. (2017). The threshold cycle value of real-time PCR amplification curve (Ct value; a lower Ct value corresponds to higher expression) was used to represent relative expression. Ct values of the Fgfr1/2/3-b isoforms (Ct value range, 28–31; Fig. 2A) are much higher than those of the Fgfr1/2/3-c isoforms (Ct value range, 22–25; Fig. 2B). These data suggest that Fgfr1/2/3-c isoforms are much more abundant compared with the b isoforms in the developing S1 cortex. Moreover, Fgfr1b (r = −0.7913), Fgfr2b (r = −0.7930), Fgfr2c (r = −0.8397), and Fgfr3c (r = −0.9135) expression levels increase, while Fgfr3b (r = 0.9953) and Fgfr1c (r = 0.953) decrease from P3 to P9 (Fig. 2A,B). Together, our results suggest that Fgfrs are dynamically expressed in developing somatosensory cortex. The dendritic deficits in Fgfr1/2/3 LOF bSCs support a physiological role of FGF–FGFR signaling in instructing bSC dendritogenesis.
Figure 2.
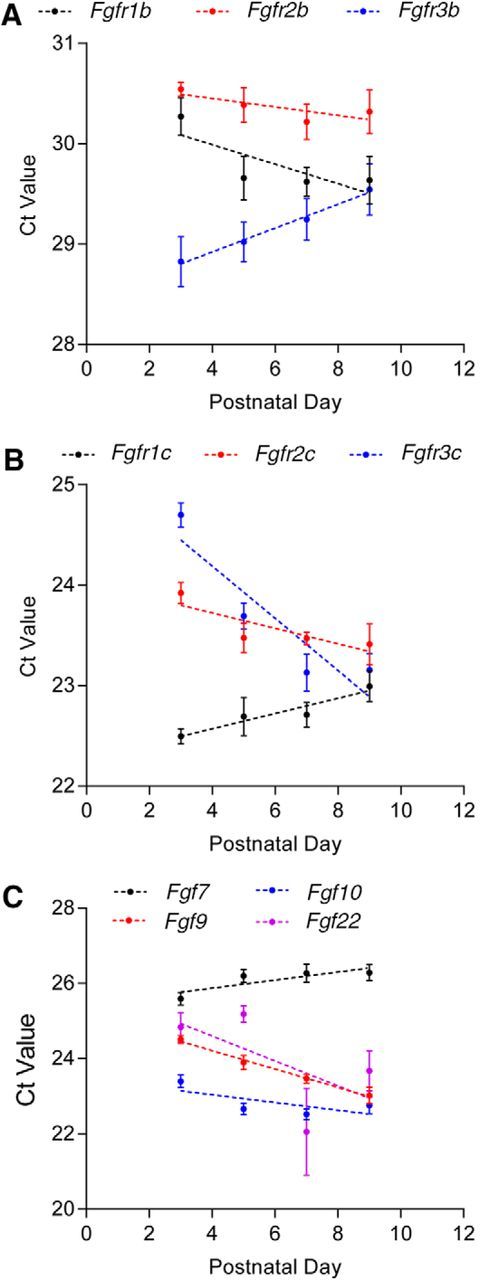
Developmental expression of FGFs and FGFRs. A–C, The mRNA expression of the b splicing variant of Fgfr1/2/3 (A); the c splicing variant of Fgfr1/2/3 (B); as well as Fgf7, Fgf9, Fgf10, and Fgf22 (C) were measured at the indicated developmental stage. The threshold cycle values of the real-time PCR amplification curve [Ct value (the lower the Ct value corresponds to a higher expression level)] are plotted to represent the relative expression of each gene. Pearson correlation coefficients were calculated to examine the correlation between gene expression level and developmental stage. Original data values and detailed statistical comparisons for this part of the figure are contained in extended data file Figure 2-1.
Original data values and detailed statistical comparison for Figure 2. Download Figure 2-1, XLSX file (14.6KB, xlsx)
Several FGFs and FGFRs are upregulated by synchronized glutamate transmission
Our 3D reconstruction studies point to the involvement of FGFR1-3 in dendritogenesis after P6. This timing coincides with the stage when there is a substantial increase in spontaneous activity due to synaptogenesis and augmentation of synaptic strength (AMPAfication; Mierau et al., 2004; Allène et al., 2008; Yang et al., 2009; Unichenko et al., 2015). Our previous studies found that deleting mGluR5 in cortical glutamatergic neurons results in an increase of calcium-permeable AMPAR in the thalamocortical synapses (Huang and Lu, 2017). Interestingly, mRNA levels of Fgf 7/9/10/22 and Fgfr1, Fgfr3 are increased in the S1 cortex of mGluR5 knock-out mice (Huang and Lu, 2017). Here we also found dynamic regulation of Fgf7/9/10/22 expression from P3 to P9. The abundance of Fgf9 (r = −0.9963), Fgf10 (r = −0.6873), and Fgf22 (r = −0.6058) mRNAs gradually increased, while the Fgf7 (r = 0.8361) mRNA level gradually decreased (Fig. 2C). Hence, we hypothesized that neural activity regulates FGF/FGFR expression in developing cortical neurons and modulates dendritic morphogenesis.
To test this hypothesis, primary cortical cultured neurons at DIV7 were treated with 30 mm KCl to trigger depolarization and glutamate release. Fgf7/9/10/22 and Fgfr1/2/3b/c mRNA levels were examined after 1–24 h treatment. We found that 4 h of KCl treatment significantly upregulated Fgf7 (p = 0.0184), Fgf9 (p = 0.0003), and Fgf10 (p = 0.0003; Fig. 3A). Fgf7/9/10 mRNA upregulation lasted at least until 24 h (the last time point examined). KCl treatment also upregulated Fgfr1–3b and c splicing variants with different temporal dynamics: Fgfr2b/c were significantly upregulated after 2 h, while Fgfr1b/c were significantly upregulated only after 8 h (Fig. 3A,B).
Figure 3.
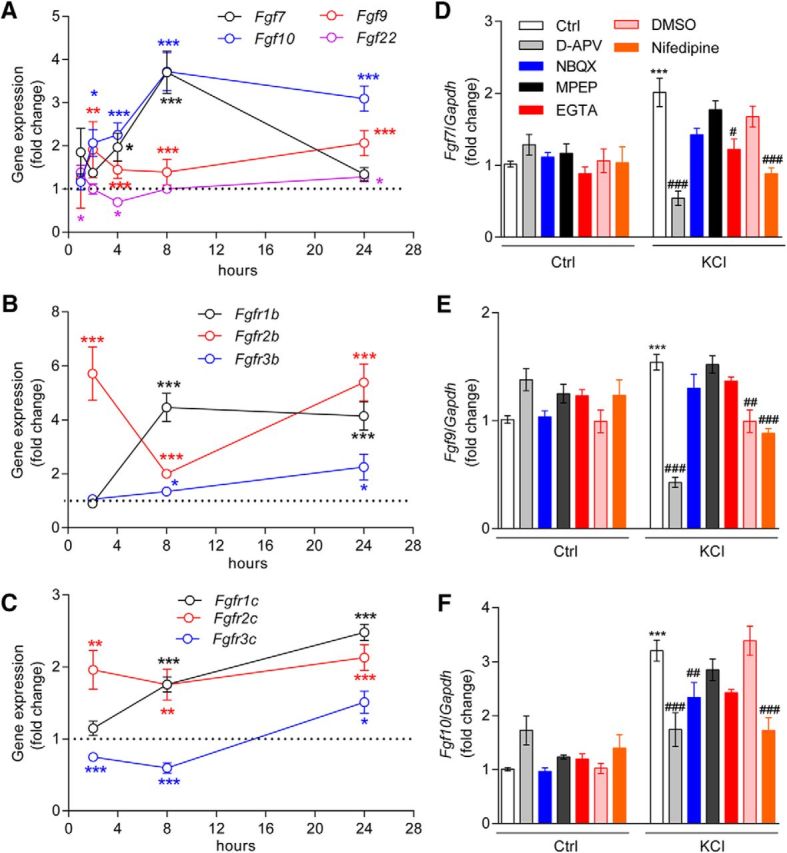
Glutamate transmission is required for KCl-induced upregulation of Fgf7, Fgf9, and Fgf10 mRNA expression. A–C, Time course of Fgf7, Fgf9, Fgf10, and Fgf22 mRNA expression (A), b splicing variant of Fgfr1/2/3 (B), and c splicing variant of Fgfr1–3 (C) were measured at the indicated time points after KCl (30 mm) treatment in DIV7 primary cortical neurons. An unpaired t test was used to assess statistical significance. In A, the statistical analysis indicated by black (Fgf7), blue (Fgf10), red (Fgf9), and purple (Fgf22) symbols refers to the KCl-treated group compared with the control (Ctrl) group at that time point. In B, the statistical analysis indicated by black (Fgfr1b), red (Fgfr2b), and blue (Fgfr3b) symbols referred to the KCl-treated group compared with the Ctrl group at that time point. In C, the statistical comparisons indicated by black (Fgfr1c), red (Fgfr2c), and blue (Fgfr3c) symbols compared the KCl-treated group and the Ctrl group at the indicated time point. *p < 0.05; **p < 0.01; ***p < 0.001. Primary cortical neurons were pretreated with an NMDAR antagonist (d-APV, 50 μm), an AMPAR antagonist (NBQX, 10 μm), an mGluR5 antagonist (MPEP, 2 μm), an extracellular calcium chelator (EGTA, 1 mm), nifedipine (l-type calcium channel inhibitor, 10 μm), or DMSO (vehicle of nifedipine, 0.01%) for 30 min, which was followed with KCl (30 mm) treatment for 8 h. D–F, Real-time PCR was conducted to measure the mRNA expression of Fgf7 (D), Fgf9 (E), and Fgf10 (F). One-way ANOVA with post hoc Tukey's multiple-comparisons test was used to assess statistical significance. * refers to the indicated group compared with control group: *p < 0.05; **p < 0.01; ***p < 0.001. # refers to the indicated group compared with KCl-treated neurons: #p < 0.05; ##p < 0.01; ###p < 0.0001. Original data values and detailed statistical comparisons for this part of the figure are contained in extended data file Figure 3-1.
Original data values and detailed statistical comparison for Figure 3. Download Figure 3-1, XLSX file (71.1KB, xlsx)
KCl treatment increases extracellular calcium influx through l-type calcium channels and enhances glutamate neurotransmission (Wang and Zhuo, 2012). To determine whether calcium influx or glutamate receptors are required for KCl-induced Fgf-Fgfr mRNA changes, we pretreated cortical neurons with l-type calcium channel blocker nifedipine (10 μm), AMPAR antagonist NBQX (10 μm), NMDAR antagonist d-APV (50 μm), or mGluR5 antagonist MPEP (2 μm) before KCl treatment. We found that blocking l-type calcium channels or NMDARs significantly attenuated Fgf7/9/10 upregulation after 8 h of KCl treatment, while inhibiting AMPAR or mGluR5 had no impact (Fig. 3D–F). Interestingly, Fgf7 upregulation, but not Fgf9/10 upregulation, was also affected by an extracellular calcium blocker, EGTA (1 mm; p = 0.0206).
To explore whether Fgf-Fgfr mRNA levels can be regulated by neural activity in vivo, we further examined whether increasing glutamate transmission can upregulate Fgf/Fgfr expression in the developing S1 cortex. P6 pups were injected once intraperitoneally or subcutaneously with KA (a glutamate analog; 1.5 mg/10 ml/kg) or saline. We found that mRNA expression of Fgf9 (p = 0.0208), Fgf10 (p = 0.0133), Fgfr2c (p = 0.004), and Fgfr3b (p = 0.0162) in the S1 cortex was significantly upregulated in KA-treated neonatal mice 6 h after KA administration (Table 1, Fig. 4). We did not observe significant changes in Fgf7 (p = 0.6407), Fgf22 (p = 0.5531), Fgfr1b (p = 0.8646), Fgfr1c (p = 0.295), Fgfr2b (p = 0.5099), and Fgfr3c (p = 0.7034) expression levels (Table 1). Bdnf (p = 0.0031) was also upregulated (Table 1), as previously reported (Zafra et al., 1990; Rocamora et al., 1996). Together, these data suggest that mRNA expression of Fgfr2c, Fgfr3b, Fgf9, and Fgf10 is upregulated in response to enhanced glutamatergic transmission in vivo.
Table 1.
Summary of normalized mRNA levels of Fgfs and Fgfrs in S1 cortex
| Gene | Ctrl group (n = 5) | KA group (n = 5) | p value |
|---|---|---|---|
| Fgf7 | 1 ± 0.13 | 1.11 ± 0.12 | 0.6407 |
| Fgf10 | 1 ± 0.08 | 1.32 ± 0.05 | 0.0133* |
| Fgf22 | 1 ± 0.07 | 1.11 ± 0.15 | 0.5531 |
| Fgf9 | 1 ± 0.02 | 1.34 ± 0.12 | 0.0208* |
| Fgfr1 b | 1 ± 0.05 | 1.04 ± 0.17 | 0.8646 |
| Fgfr1 c | 1 ± 0.08 | 1.29 ± 0.15 | 0.295 |
| Fgfr2 b | 1 ± 0.12 | 0.91 ± 0.11 | 0.5099 |
| Fgfr2 c | 1 ± 0.08 | 2.18 ± 0.28 | 0.004** |
| Fgfr3 b | 1 ± 0.06 | 1.32 ± 0.09 | 0.0162* |
| Fgfr3 c | 1 ± 0.05 | 1.04 ± 0.05 | 0.7034 |
| Bdnf | 1 ± 0.10 | 2.82 ± 0.41 | 0.0031** |
Somatosensory cortex samples were collected after KA injection for 6 h and subjected to quantitative PCR experiments. The statistical analysis refers to the control (Ctrl) group compared with the KA group. Bold highlights the p values that are statistically significant. Original data values and detailed statistical comparisons for this part of the figure are contained in extended data file Figure 4-1 available at https://doi.org/10.1523/JNEUROSCI.1174-17.2017.f4-1.
*p < 0.05,
**p < 0.01 by unpaired t test.
Figure 4.

KA treatment upregulated Fgf9, Fgf10, Fgfr2c, and Fgfr3b mRNA expression in S1 cortex. Wild-type pups were administered KA at postnatal day 6. S1 cortices were collected after 6 h treatment, and real-time PCR was conducted to measure mRNA expression (n = 5 for each group). An unpaired t test was used to assess statistical significance. The statistical analysis compared the KA-treated group to the control (Ctrl) group: *p < 0.05; **p < 0.01. Original data values and detailed statistical comparisons for this part of the figure are contained in extended data file Figure 4-1.
Original data values and detailed statistical comparison for Figure 4 and Table 1. Download Figure 4-1, XLSX file (15.8KB, xlsx)
KA promotes dendritogenesis in bSCs through FGFR in vivo
The fact that glutamate transmission modulated the dendritogenesis of bSCs (Datwani et al., 2002; Espinosa et al., 2009; Mizuno et al., 2014; Ballester-Rosado et al., 2016) motivated us to investigate whether the massive glutamate transmission elicited by KA administration promoted dendritogenesis in an FGFR-dependent manner. Simultaneous removal of Fgfr1/2/3 was achieved as described in Figure 1 followed by KA injection at P6 (Fig. 5A). Afterward, detailed dendritic morphology was examined at P7 (Fig. 5B). We first examined the impacts of KA-induced seizures on bSC dendritic morphology. Scattered bSCs in the S1 cortex of FGFR1/2/3 conditional mice were labeled with tdTomato using IUE as described above, and their dendritic morphology was examined 1 d after KA administration at P6. We found that KA injection significantly increased total dendritic length (p < 0.0001; Fig. 5C) and branch point number (p < 0.0001; Fig. 5D) but did not alter average segment length (p = 0.3399; Fig. 5E). Branch order analysis revealed that KA significantly increased segment number (p < 0.0001; Fig. 5F) but had no impact on the segment length of any branch orders (p = 0.5492; Fig. 5G). KA administration did not affect the polar ratio (p = 0.3124; Fig. 5J), it promoted dendritic growth both inside the barrel (p < 0.0001; Fig. 5H) and outside the barrel (p < 0.0001; Fig. 5I). Notably, KA failed to induce dendritic outgrowth in Fgfr1/2/3 LOF bSCs. These results suggest that FGFR1/2/3 signaling can be upregulated to promote dendritic outgrowth by excessive neural activity.
Figure 5.

KA treatment promoted bSC dendritogenesis within 24 h in an FGFR1/2/3-dependent manner. A, Schematic representation shows the experimental procedures and developmental stages for IUE and KA treatment. B, Examples of computer-aided reconstructions. C–E, The total dendritic length (C), branch point number (D), and mean dendritic length (E) are plotted. F, G, Summary of number (F) and length (G) per branch order. Two-way ANOVA with post hoc Tukey's multiple-comparisons test was conducted for F and G. The statistical analysis indicated by the red asterisk refers to the KA group compared with the control (Ctrl) group. The statistical analysis indicated by the purple asterisk refers to the KA+FGFR LOF group compared with the KA group: ***p < 0.001. H–J, Quantitative analysis of dendritic length inside (H) and outside (I) of the barrel that was used to calculate dendritic polarity (J). One-way ANOVA with post hoc Tukey's multiple-comparisons test was conducted for C–E and H–J. The statistical analysis compared the indicated groups: ***p < 0.001. Original data values and detailed statistical comparisons for this part of the figure are contained in extended data file Figure 5-1.
Original data values and detailed statistical comparison for Figure 5. Download Figure 5-1, XLSX file (48.8KB, xlsx)
Postnatal FGF9 and FGF10 overexpression promoted dendritic growth in vivo
If Fgf/Fgfr upregulation induced by KA leads to dendritic extension, FGF9 and FGF10 overexpression at P6 should result in a similar dendritic extension. To overexpress Fgf9 or Fgf10 in bSCs after P5/P6, we used the combination of IUE and an inducible Cre/loxP approach (Fig. 6A). Specifically, we used the Cre-On system to activate Fgf9 and Fgf10 expression by using inducible Cre (ERT2-Cre-ERT2) and Cre-dependent activation constructs (Fig. 6A; Matsuda and Cepko, 2007; Atasoy et al., 2008; Cardin et al., 2010). A Cre-On expression cassette was engineered by placing Fgf9 or Fgf10 cDNA into the double-floxed inverted orientation (DIO) construct (see Materials and Methods). Upon Cre-mediated recombination, the orientation is reversed to allow Fgf9 or Fgf10 transcription driven by the EF1α promoter. We conducted IUE to introduce DIO-Fgf9/DIO-Fgf10 and DIO-tdTomato (reporter) constructs into wild-type embryos at E14.5 to target bSCs (Fig. 6A). ERT2-Cre-ERT2 was activated by tamoxifen injection (100 mg/10 ml/kg, i.p.; single injection) at P5 to initiate Fgf9 and Fgf10 transcription in cortical neurons (Fig. 6A). Two days after tamoxifen treatment, the impact of Fgf9 or Fgf10 overexpression on bSCs was examined through a detailed morphological analysis of tdTomato-positive spiny stellate cells at P7 (Fig. 6B–D).
Figure 6.

FGF9 and FGF10 expression promoted dendritogenesis of layer IV cortical neurons. A, Schematic representations of the electroporation and tamoxifen treatment protocols that were used to express FGF9 and FGF10. B–D, Examples of original images and computer-aided reconstructions. B1–D1 show the locations of barrels (dashed lines) and reconstructed neurons (white arrows). II–V, Cortical layers. The projection images from confocal image stacks are shown in B2–D2. B3–D3 show the traced images of neurons in B2–D2. B4–D4 show color-coded segments according to their branch orders. E–G, Quantitation of total length (E), branch point numbers (F), and mean length (G) of dendrites. H, I, Summaries of segment number (H) and length (I) per branch order. Two-way ANOVA with post hoc Bonferroni's multiple-comparisons test was conducted for H and I. The statistical analysis indicated by the red asterisk refers to the FGF9 group compared with the control (Ctrl) group. The statistical analysis indicated by the blue asterisk refers to the FGF10 group compared with the Ctrl group. J–L, Quantitative analysis of dendritic length inside (J) and outside (K) the barrel that was used to calculate dendritic polarity (L). A Mann–Whitney test was conducted for E, F, G, J, K, and L. The statistical analysis compared the indicated groups: *p < 0.05; **p < 0.01; ***p < 0.001. Original data values and detailed statistical comparisons for this part of the figure are contained in extended data file Figure 6-1.
Original data values and detailed statistical comparison for Figure 6. Download Figure 6-1, XLSX file (34.3KB, xlsx)
We found that Fgf9 and Fgf10-expressing bSCs displayed significantly more dendritic outgrowth compared with their control neurons. Total dendritic length (p = 0.0007 and p = 0.034; Fig. 6E) and dendritic branch point number (p = 0.0037 and p = 0.0081; Fig. 6F) were significantly increased, while average dendritic length (p = 0.4979; p = 0.9674; Fig. 6G) was unchanged. The segment numbers per branch order were significantly increased in Fgf9-overexpressing (p = 0.0006) and Fgf10-overexpressing (p = 0.01) bSCs (Fig. 6H). Neither FGF9 (p = 0.7536) nor FGF10 (p = 0.6535) had significant impacts on segment length per branch order (Fig. 6I). Regarding polarity, FGF9 or FGF10 overexpression significantly increased dendritic growth inside the barrel (p = 0.0025; p = 0.0095; Fig. 6J), but not outside the barrel (p = 0.0581; p = 0.4857; Fig. 6K); thus, bSCs retained their polarized morphology (p = 0.6115; p = 0.7375; Fig. 6L). Together, we found that FGF9 or FGF10 overexpression is sufficient to promote dendritic growth of bSCs.
Discussion
Here we first show that the dendritic pattern of bSCs is established in several distinctive phases during the first 2 postnatal weeks. Fgfr1/2/3 are dynamically expressed in S1 cortex, and deleting these receptors in bSCs results in a reduction in dendritic branch numbers and total dendritic length from P9 to P15, while control bSCs maintain their dendritic branch pattern and increase their total dendritic length by the elongation of individual segments. Interestingly, mRNA levels of several Fgfs and Fgfrs can be upregulated by glutamate transmission. Glutamate transmission-driven dendritic growth depends on FGFR1/2/3, while FGF9 or FGF10 overexpression results in a similar dendritic growth. Together, our data suggest that FGF–FGFR signaling can be regulated by glutamate transmission to both establish and maintain the dendritic patterns of bSCs.
FGFRs in bSCs are required for activity-induced dendritogenesis during whisker-barrel map formation
Our dendritogenesis study revealed distinctive phases of dendritic patterning. First, as reported by Espinosa et al. (2009), bSCs acquire a TCA-oriented pattern by P6. Our previous studies show the importance of NGF–TrkA signaling in the establishment of the dendritic polarity of bSCs (Huang and Lu, 2017). Next, from P6 to P9, branch numbers increase more than twofold and then remain stable from P9 to P15. The segment addition at multiple branch orders from P6 to P9 indicates de novo branch addition (Fig. 1L). Despite the establishment of a basic dendritic pattern by P9, individual dendritic segments at almost all branch orders, except the first order, continue to elongate from P9 to P15 (Fig. 1K).
At P6, FGFR1/2/3 LOF bSCs (0.8264 ± 0.0161, p = 0.9798) acquire dendritic polarity similar to control bSCs (0.8479 ± 0.0267) and exhibit no detectable deficits. At P9, FGFR1/2/3 LOF bSCs display a small but significant increase in dendritic branch point numbers (Fig. 1I). However, this increase in dendritic complexity is transient. By P15, dendritic segment numbers in FGFR1/2/3 LOF bSCs are reduced compared with both P15 control bSCs and P9 FGFR1/2/3 LOF bSCs. The substantial reduction of FGFR1/2/3 LOF bSC dendritic branch point numbers from P9 to P15 argues (Fig. 1N) for the importance of FGFR1/2/3 bSCs in maintaining the established dendritic pattern. In other words, these data reveal the importance of neuronal/dendritic maintenance immediately after the establishment of dendritic morphogenesis.
FGFR signaling is commonly initiated by ligand-dependent dimerization of FGFRs, followed by the activation of several signaling cascades, including& extracellular signal-regulated kinases 1/2, phosphatidylinositol 3-kinase-AKT, protein kinase C, and small GTPases (Eswarakumar et al., 2005; Iwata and Hevner, 2009; Dorey and Amaya, 2010; Guillemot and Zimmer, 2011; Chuang et al., 2015). Our understanding of the function of each specific FGF ligand and FGFR remains limited. This is partially due to the highly redundant functions of the 22 FGF family members and complex interactions between ligands and b/c splicing variants of FGFR1/2/3. The high sequence similarity between FGFRs together with the promiscuity of FGF binding implies redundancy within the family members. However, targeted gene inactivation of different FGF and FGFR members yields a specific phenotype for each knockout. For example, FGF7 and FGF22 independently regulate inhibitory and excitatory synapse formation, respectively (Terauchi et al., 2010; Dabrowski et al., 2015). Our mosaic analysis of FGFR1/2/3 LOF bSCs within the wild-type environment supports the cell-autonomous requirement of FGFR1/2/3.
In addition to the studies revealing the endogenous role of FGF–FGFR signaling in dendritogenesis of bSCs during barrel map formation, we also find that FGFR1/2/3 are required for KA-induced dendritic outgrowth (Fig. 5). Taking a complementary approach of overexpressing FGFR ligands by using the Cre-ER/loxP system, we were able to reveal that FGF9 or FGF10 expression for 1–2 d had a dramatic impact on bSC dendritic outgrowth after P5/P6 (Fig. 6). Together, our data provide strong support for the involvement of FGF/FGFR in mediating neuronal activity-dependent dendritogenesis. Several important questions remain to be answered in future studies: which FGFRs are the key receptor; which FGFs or FGFs activate FGFRs on bSCs; what cells secrete FGFs; and what are the downstream mechanisms underlying FGFR-mediated dendritic formation and maintenance.
Neural activity regulated Fgf/Fgfr mRNA levels
During embryonic development, the expression of each individual FGF is tightly regulated regarding their expression levels, timing, and location (Ford-Perriss et al., 2001; Yaguchi et al., 2009). Little is known on what controls Fgf/Fgfr mRNA expression in postmitotic neurons. Our studies with primary cortical neurons show that neuronal depolarization (KCl treatment) upregulated Fgf/Fgfr mRNA levels very quickly in an NMDAR- and calcium-dependent manner. Interestingly, the activity regulates different Fgf/Fgfr mRNAs with distinct dynamics. It has been shown that epileptiform activity upregulates Fgf/Fgfr (Gómez-Pinilla et al., 1995; Kondratyev et al., 2002; Kim et al., 2010). Both Fgf3 and Fgfr1 transcription was increased 1 h after seizure onset in hippocampi of the KA-induced seizure model (Hermey et al., 2013). FGF2 protein expression was increased in whisker stimulation (Whitaker et al., 2007).
Several synaptic events are notable during the transition from the first to the second postnatal week for thalamocortical connections: synaptic surface AMPARs increase (AMPAfication); NMDAR subunits switch from NR2B dominant to NR2A dominant; and the critical period for whisker barrel map plasticity and synaptic plasticity closes (Wu et al., 2011). By combining whole-cell recording and single-cell resolution 2P glutamate uncaging, Ashby and Isaac (2011) mapped out local functional connectivity in layer IV barrel cortex during P3–P12 on a daily basis. They found that during the first postnatal week, the connectivity between bSCs was ∼4%. Interestingly, between P8 and P9, the connectivity between bSCs increases abruptly, approximately threefold, which persisted until P12, the oldest stage recorded. Before P8, bSC dendrites are almost entirely aspiny (Ashby and Isaac, 2011). However, from P8 to P13, dendritic spine counts for bSCs increase ∼250-fold. Sensory experience from whiskers is required for the increase in intraconnectivity but not for spinogenesis or total dendritic length. Thus, it is plausible that the rise in intracortical glutamatergic connectivity enhances FGF/FGFR signaling after P8/P9 to regulate and maintain dendritic morphology.
In this study, we provide the first in vivo demonstration for neural activity regulation on Fgf9, Fgf10, Fgfr2c, and Fgfr3b mRNA levels in the cortex. The precise mechanism underlying activity-regulated Fgf/Fgfr transcription remains to be determined. Using stromal cells, Chuang et al. (2006) showed that prostaglandin E2 upregulated Fgf9 mRNA expression via activating ELK1 (Chuang et al., 2006). Interestingly, ELK1 can be activated by increased glutamate transmission in either cortical neurons or brain slices (Vanhoutte et al., 1999; Madabhushi et al., 2015). Regarding Fgf10 transcription, studies using mouse embryos, trigeminal ganglion, and chick embryonic limb buds show that Tbx5 and POU transcriptional factors can upregulate Fgf10 expression by binding to its promoter region (Agarwal et al., 2003; Cox et al., 2006; Nishimoto et al., 2015).
Cortical circuit plasticity triggered by sensory/motor experience or learning has been documented in many species using different paradigms (Globus and Scheibel, 1967; Hensch, 2005). In the barrel cortex, the impacts of whisker trimming or whisker lesions on bSC dendritic morphology have been studied mostly using classic Golgi staining and 2D reconstruction of dendritic orientation and overall dendritic coverage (Steffen and Van der Loos, 1980; Lee et al., 2009). Here we triggered massive neurotransmission by applying KCl or kainic acid, which are very strong paradigms. It remains to be determined whether natural neural activity, for example through whisking or whisker stimulation, will similarly upregulate FGF/FGFR signaling and, if so, whether the dendritic changes induced by this level of neural activity will be long lasting.
Interestingly, saline-treated control bSCs exhibited more branch points and total length than the values extrapolated from naive P6 and P9 controls (compare Figs. 1H,I, 5C,D). Saline-treated pups encountered different sensory experiences than naive pups ∼20 h before being killed. As a part of the experimental procedures, saline-treated pups underwent maternal deprivation for 3–4 h, experienced handling, and were also housed together with their KA littermates undergoing seizures for 3–4 h (see Materials and Methods). It remains to be determined whether handling and extra sensory stimuli caused the differences noted between the two groups. Interesting, the values for saline-treated P7 FGFR1/2/3 LOF bSCs are in the range of expected values from naive P6 and P9 FGFR1/2/3 LOF bSCs. The differences between control and FGFR1/2/3 LOF bSCs are in the same direction we observed for KA-treated pups, providing additional support for our hypothesis that activity-driven dendritic morphological changes require FGFRs. Alternatively, the disparity between dendritic data from Figure 1 and Figure 5 could suggest a transient dendritic outgrowth between P6 and P7 that is refined between P7 and P9. Such transient overgrowth failed to occur in FGFR1/2/3 LOF neurons.
In summary, our data here suggest that FGF–FGFR signaling can be enhanced by glutamate transmission to promote dendritic arborization and physiological levels of glutamate transmission that are sufficient for FGF–FGFR signaling to maintain dendritic structures. Dysregulated FGF/FGFR expression (Gaughran et al., 2006; Turner et al., 2006, 2012, 2016; Terwisscha van Scheltinga et al., 2010; Williams and Umemori, 2014) has been observed in depression, anxiety, and schizophrenia. Thus, abnormal FGF/FGFR levels in these neurological disorders may account for the abnormal dendritic arborization patterns observed in these diseases (Kulkarni and Firestein, 2012).
Footnotes
This work was supported by National Institutes of Health Grants NS-048884 and NS-086794 to H.-C.L. Confocal images were taken in the Light Microscopy Imaging Center at Indiana University, Bloomington, and the Baylor Microscopy Core (supported by Baylor College of Medicine Intellectual and Developmental Disabilities Research Center Grant U54-HD-083092 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development). We thank Dr. Jean Hébert for FGFR1/2/3 triple conditional mice, and helpful comments from Drs. Chia-Chien Chen, Chia-Shan Wu, Chiaki Itami, Fumitaka Kimura, Ken Mackie, Mingshan Xue, and Wei-Hsiang Huang. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the Eunice Kennedy Shriver National Institute of Child Health and Human Development or the National Institutes of Health.
The authors declare no competing financial interests.
References
- Agarwal P, Wylie JN, Galceran J, Arkhitko O, Li C, Deng C, Grosschedl R, Bruneau BG (2003) Tbx5 is essential for forelimb bud initiation following patterning of the limb field in the mouse embryo. Development 130:623–633. 10.1242/dev.00191 [DOI] [PubMed] [Google Scholar]
- Aletsee C, Brors D, Mlynski R, Ryan AF, Dazert S (2003) Branching of spiral ganglion neurites is induced by focal application of fibroblast growth factor-1. Laryngoscope 113:791–796. 10.1097/00005537-200305000-00005 [DOI] [PubMed] [Google Scholar]
- Allène C, Cattani A, Ackman JB, Bonifazi P, Aniksztejn L, Ben-Ari Y, Cossart R (2008) Sequential generation of two distinct synapse-driven network patterns in developing neocortex. J Neurosci 28:12851–12863. 10.1523/JNEUROSCI.3733-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashby MC, Isaac JT (2011) Maturation of a recurrent excitatory neocortical circuit by experience-dependent unsilencing of newly formed dendritic spines. Neuron 70:510–521. 10.1016/j.neuron.2011.02.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasoy D, Aponte Y, Su HH, Sternson SM (2008) A FLEX switch targets Channelrhodopsin-2 to multiple cell types for imaging and long-range circuit mapping. J Neurosci 28:7025–7030. 10.1523/JNEUROSCI.1954-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballester-Rosado CJ, Albright MJ, Wu CS, Liao CC, Zhu J, Xu J, Lee LJ, Lu HC (2010) mGluR5 in cortical excitatory neurons exerts both cell-autonomous and -nonautonomous influences on cortical somatosensory circuit formation. J Neurosci 30:16896–16909. 10.1523/JNEUROSCI.2462-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballester-Rosado CJ, Sun H, Huang JY, Lu HC (2016) mGluR5 exerts cell-autonomous influences on the functional and anatomical development of layer IV cortical neurons in the mouse primary somatosensory cortex. J Neurosci 36:8802–8814. 10.1523/JNEUROSCI.1224-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum P, Vogt MA, Gass P, Unsicker K, von Bohlen und Halbach O (2016) FGF-2 deficiency causes dysregulation of Arhgef6 and downstream targets in the cerebral cortex accompanied by altered neurite outgrowth and dendritic spine morphology. Int J Dev Neurosci 50:55–64. 10.1016/j.ijdevneu.2016.03.002 [DOI] [PubMed] [Google Scholar]
- Beenken A, Mohammadi M (2009) The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov 8:235–253. 10.1038/nrd2792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardin JA, Carlén M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai LH, Moore CI (2010) Targeted optogenetic stimulation and recording of neurons in vivo using cell-type-specific expression of channelrhodopsin-2. Nat Protoc 5:247–254. 10.1038/nprot.2009.228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Ghosh A (2005) Regulation of dendritic development by neuronal activity. J Neurobiol 64:4–10. 10.1002/neu.20150 [DOI] [PubMed] [Google Scholar]
- Chuang JI, Huang JY, Tsai SJ, Sun HS, Yang SH, Chuang PC, Huang BM, Ching CH (2015) FGF9-induced changes in cellular redox status and HO-1 upregulation are FGFR-dependent and proceed through both ERK and AKT to induce CREB and Nrf2 activation. Free Radic Biol Med 89:274–286. 10.1016/j.freeradbiomed.2015.08.011 [DOI] [PubMed] [Google Scholar]
- Chuang PC, Sun HS, Chen TM, Tsai SJ (2006) Prostaglandin E2 induces fibroblast growth factor 9 via EP3-dependent protein kinase Cdelta and Elk-1 signaling. Mol Cell Biol 26:8281–8292. 10.1128/MCB.00941-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline HT. (2001) Dendritic arbor development and synaptogenesis. Curr Opin Neurobiol 11:118–126. 10.1016/S0959-4388(00)00182-3 [DOI] [PubMed] [Google Scholar]
- Cox E, Lanier J, Quina L, Eng SR, Turner EE (2006) Regulation of FGF10 by POU transcription factor Brn3a in the developing trigeminal ganglion. J Neurobiol 66:1075–1083. 10.1002/neu.20277 [DOI] [PubMed] [Google Scholar]
- Dabrowski A, Terauchi A, Strong C, Umemori H (2015) Distinct sets of FGF receptors sculpt excitatory and inhibitory synaptogenesis. Development 142:1818–1830. 10.1242/dev.115568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datwani A, Iwasato T, Itohara S, Erzurumlu RS (2002) NMDA receptor-dependent pattern transfer from afferents to postsynaptic cells and dendritic differentiation in the barrel cortex. Mol Cell Neurosci 21:477–492. 10.1006/mcne.2002.1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorey K, Amaya E (2010) FGF signalling: diverse roles during early vertebrate embryogenesis. Development 137:3731–3742. 10.1242/dev.037689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa JS, Wheeler DG, Tsien RW, Luo L (2009) Uncoupling dendrite growth and patterning: single-cell knockout analysis of NMDA receptor 2B. Neuron 62:205–217. 10.1016/j.neuron.2009.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswarakumar VP, Lax I, Schlessinger J (2005) Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev 16:139–149. 10.1016/j.cytogfr.2005.01.001 [DOI] [PubMed] [Google Scholar]
- Feldman DE, Brecht M (2005) Map plasticity in somatosensory cortex. Science 310:810–815. 10.1126/science.1115807 [DOI] [PubMed] [Google Scholar]
- Fon Tacer K, Bookout AL, Ding X, Kurosu H, John GB, Wang L, Goetz R, Mohammadi M, Kuro-o M, Mangelsdorf DJ, Kliewer SA (2010) Research resource: comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol Endocrinol 24:2050–2064. 10.1210/me.2010-0142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford-Perriss M, Abud H, Murphy M (2001) Fibroblast growth factors in the developing central nervous system. Clin Exp Pharmacol Physiol 28:493–503. 10.1046/j.1440-1681.2001.03477.x [DOI] [PubMed] [Google Scholar]
- Gao FB. (2007) Molecular and cellular mechanisms of dendritic morphogenesis. Curr Opin Neurobiol 17:525–532. 10.1016/j.conb.2007.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaughran F, Payne J, Sedgwick PM, Cotter D, Berry M (2006) Hippocampal FGF-2 and FGFR1 mRNA expression in major depression, schizophrenia and bipolar disorder. Brain Res Bull 70:221–227. 10.1016/j.brainresbull.2006.04.008 [DOI] [PubMed] [Google Scholar]
- Globus A, Scheibel AB (1967) The effect of visual deprivation on cortical neurons: a Golgi study. Exp Neurol 19:331–345. 10.1016/0014-4886(67)90029-5 [DOI] [PubMed] [Google Scholar]
- Gómez-Pinilla F, van der Wal EA, Cotman CW (1995) Possible coordinated gene expressions for FGF receptor, FGF-5, and FGF-2 following seizures. Exp Neurol 133:164–174. 10.1006/exnr.1995.1019 [DOI] [PubMed] [Google Scholar]
- Guillemot F, Zimmer C (2011) From cradle to grave: the multiple roles of fibroblast growth factors in neural development. Neuron 71:574–588. 10.1016/j.neuron.2011.08.002 [DOI] [PubMed] [Google Scholar]
- Häusser M, Spruston N, Stuart GJ (2000) Diversity and dynamics of dendritic signaling. Science 290:739–744. 10.1126/science.290.5492.739 [DOI] [PubMed] [Google Scholar]
- Hébert JM. (2011) FGFs: neurodevelopment's jack-of-all-trades—how do they do it? Front Neurosci 5:133. 10.3389/fnins.2011.00133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch TK. (2005) Critical period plasticity in local cortical circuits. Nat Rev Neurosci 6:877–888. 10.1038/nrn1787 [DOI] [PubMed] [Google Scholar]
- Hermey G, Mahlke C, Gutzmann JJ, Schreiber J, Blüthgen N, Kuhl D (2013) Genome-wide profiling of the activity-dependent hippocampal transcriptome. PLoS One 8:e76903. 10.1371/journal.pone.0076903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howland JG, Hannesson DK, Phillips AG (2004) Delayed onset of prepulse inhibition deficits following kainic acid treatment on postnatal day 7 in rats. Eur J Neurosci 20:2639–2648. 10.1111/j.1460-9568.2004.03731.x [DOI] [PubMed] [Google Scholar]
- Huang JY, Lu HC (2017) mGluR5 tunes NGF/TrkA signaling to regulate spiny stellate neuron dendrites towards thalamocortical axons during whisker-barrel map formation. Cereb Cortex. Advance online publication. Retrieved November 2, 2017. 10.1093/cercor/bhx105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JY, Hong YT, Chuang JI (2009) Fibroblast growth factor 9 prevents MPP+-induced death of dopaminergic neurons and is involved in melatonin neuroprotection in vivo and in vitro. J Neurochem 109:1400–1412. 10.1111/j.1471-4159.2009.06061.x [DOI] [PubMed] [Google Scholar]
- Itoh N, Ornitz DM (2004) Evolution of the Fgf and Fgfr gene families. Trends Genet 20:563–569. 10.1016/j.tig.2004.08.007 [DOI] [PubMed] [Google Scholar]
- Iwata T, Hevner RF (2009) Fibroblast growth factor signaling in development of the cerebral cortex. Dev Growth Differ 51:299–323. 10.1111/j.1440-169X.2009.01104.x [DOI] [PubMed] [Google Scholar]
- Jan YN, Jan LY (2010) Branching out: mechanisms of dendritic arborization. Nat Rev Neurosci 11:316–328. 10.1038/nrn2836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo W, Hippenmeyer S, Luo L (2014) Neurodevelopment. Dendrite morphogenesis depends on relative levels of NT-3/TrkC signaling. Science 346:626–629. 10.1126/science.1258996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang W, Hébert JM (2015) FGF signaling is necessary for neurogenesis in young mice and sufficient to reverse its decline in old mice. J Neurosci 35:10217–10223. 10.1523/JNEUROSCI.1469-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang W, Wong LC, Shi SH, Hébert JM (2009) The transition from radial glial to intermediate progenitor cell is inhibited by FGF signaling during corticogenesis. J Neurosci 29:14571–14580. 10.1523/JNEUROSCI.3844-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Gale K, Kondratyev A (2010) Effects of repeated minimal electroshock seizures on NGF, BDNF and FGF-2 protein in the rat brain during postnatal development. Int J Dev Neurosci 28:227–232. 10.1016/j.ijdevneu.2010.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondratyev A, Ved R, Gale K (2002) The effects of repeated minimal electroconvulsive shock exposure on levels of mRNA encoding fibroblast growth factor-2 and nerve growth factor in limbic regions. Neuroscience 114:411–416. 10.1016/S0306-4522(02)00266-X [DOI] [PubMed] [Google Scholar]
- Kulkarni VA, Firestein BL (2012) The dendritic tree and brain disorders. Mol Cell Neurosci 50:10–20. 10.1016/j.mcn.2012.03.005 [DOI] [PubMed] [Google Scholar]
- Lee LJ, Chen WJ, Chuang YW, Wang YC (2009) Neonatal whisker trimming causes long-lasting changes in structure and function of the somatosensory system. Exp Neurol 219:524–532. 10.1016/j.expneurol.2009.07.012 [DOI] [PubMed] [Google Scholar]
- Lynch M, Sayin U, Bownds J, Janumpalli S, Sutula T (2000) Long-term consequences of early postnatal seizures on hippocampal learning and plasticity. Eur J Neurosci 12:2252–2264. 10.1046/j.1460-9568.2000.00117.x [DOI] [PubMed] [Google Scholar]
- Madabhushi R, Gao F, Pfenning AR, Pan L, Yamakawa S, Seo J, Rueda R, Phan TX, Yamakawa H, Pao PC, Stott RT, Gjoneska E, Nott A, Cho S, Kellis M, Tsai LH (2015) Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell 161:1592–1605. 10.1016/j.cell.2015.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T, Cepko CL (2007) Controlled expression of transgenes introduced by in vivo electroporation. Proc Natl Acad Sci U S A 104:1027–1032. 10.1073/pnas.0610155104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui A, Tran M, Yoshida AC, Kikuchi SS, U M, Ogawa M, Shimogori T (2013) BTBD3 controls dendrite orientation toward active axons in mammalian neocortex. Science 342:1114–1118. 10.1126/science.1244505 [DOI] [PubMed] [Google Scholar]
- McAllister AK. (2000) Cellular and molecular mechanisms of dendrite growth. Cereb Cortex 10:963–973. 10.1093/cercor/10.10.963 [DOI] [PubMed] [Google Scholar]
- Mierau SB, Meredith RM, Upton AL, Paulsen O (2004) Dissociation of experience-dependent and -independent changes in excitatory synaptic transmission during development of barrel cortex. Proc Natl Acad Sci U S A 101:15518–15523. 10.1073/pnas.0402916101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno H, Luo W, Tarusawa E, Saito YM, Sato T, Yoshimura Y, Itohara S, Iwasato T (2014) NMDAR-regulated dynamics of layer 4 neuronal dendrites during thalamocortical reorganization in neonates. Neuron 82:365–379. 10.1016/j.neuron.2014.02.026 [DOI] [PubMed] [Google Scholar]
- Narboux-Nême N, Evrard A, Ferezou I, Erzurumlu RS, Kaeser PS, Lainé J, Rossier J, Ropert N, Südhof TC, Gaspar P (2012) Neurotransmitter release at the thalamocortical synapse instructs barrel formation but not axon patterning in the somatosensory cortex. J Neurosci 32:6183–6196. 10.1523/JNEUROSCI.0343-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimoto S, Wilde SM, Wood S, Logan MP (2015) RA Acts in a coherent feed-forward mechanism with Tbx5 to control limb bud induction and initiation. Cell Rep 12:879–891. 10.1016/j.celrep.2015.06.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen CC. (2007) The functional organization of the barrel cortex. Neuron 56:339–355. 10.1016/j.neuron.2007.09.017 [DOI] [PubMed] [Google Scholar]
- Rai KS, Hattiangady B, Shetty AK (2007) Enhanced production and dendritic growth of new dentate granule cells in the middle-aged hippocampus following intracerebroventricular FGF-2 infusions. Eur J Neurosci 26:1765–1779. 10.1111/j.1460-9568.2007.05820.x [DOI] [PubMed] [Google Scholar]
- Rice H, Suth S, Cavanaugh W, Bai J, Young-Pearse TL (2010) In utero electroporation followed by primary neuronal culture for studying gene function in subset of cortical neurons. J Vis Exp (44):e2103. 10.3791/2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocamora N, Welker E, Pascual M, Soriano E (1996) Upregulation of BDNF mRNA expression in the barrel cortex of adult mice after sensory stimulation. J Neurosci 16:4411–4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She WC, Quairiaux C, Albright MJ, Wang YC, Sanchez DE, Chang PS, Welker E, Lu HC (2009) Roles of mGluR5 in synaptic function and plasticity of the mouse thalamocortical pathway. Eur J Neurosci 29:1379–1396. 10.1111/j.1460-9568.2009.06696.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimogori T. (2006) Micro in utero electroporation for efficient gene targeting in mouse embryos. Cold Spring Harb Protoc 2006:prot4447. 10.1101/pdb.prot4447 [DOI] [PubMed] [Google Scholar]
- Simons DJ, Woolsey TA (1979) Functional organization in mouse barrel cortex. Brain Res 165:327–332. 10.1016/0006-8993(79)90564-X [DOI] [PubMed] [Google Scholar]
- Singh R, Su J, Brooks J, Terauchi A, Umemori H, Fox MA (2012) Fibroblast growth factor 22 contributes to the development of retinal nerve terminals in the dorsal lateral geniculate nucleus. Front Mol Neurosci 4:61. 10.3389/fnmol.2011.00061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöström PJ, Rancz EA, Roth A, Häusser M (2008) Dendritic excitability and synaptic plasticity. Physiol Rev 88:769–840. 10.1152/physrev.00016.2007 [DOI] [PubMed] [Google Scholar]
- Sleeman M, Fraser J, McDonald M, Yuan S, White D, Grandison P, Kumble K, Watson JD, Murison JG (2001) Identification of a new fibroblast growth factor receptor, FGFR5. Gene 271:171–182. 10.1016/S0378-1119(01)00518-2 [DOI] [PubMed] [Google Scholar]
- Stafstrom CE, Thompson JL, Holmes GL (1992) Kainic acid seizures in the developing brain: status epilepticus and spontaneous recurrent seizures. Brain Res Dev Brain Res 65:227–236. 10.1016/0165-3806(92)90184-X [DOI] [PubMed] [Google Scholar]
- Steffen H, Van der Loos H (1980) Early lesions of mouse vibrissal follicles:: their influence on dendrite orientation in the cortical barrelfield. Exp Brain Res 40:419–431. 10.1007/BF00236150 [DOI] [PubMed] [Google Scholar]
- Su N, Xu X, Li C, He Q, Zhao L, Li C, Chen S, Luo F, Yi L, Du X, Huang H, Deng C, Chen L (2010) Generation of Fgfr3 conditional knockout mice. Int J Biol Sci 6:327–332. 10.7150/ijbs.6.327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terauchi A, Johnson-Venkatesh EM, Toth AB, Javed D, Sutton MA, Umemori H (2010) Distinct FGFs promote differentiation of excitatory and inhibitory synapses. Nature 465:783–787. 10.1038/nature09041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwisscha van Scheltinga AF, Bakker SC, Kahn RS (2010) Fibroblast growth factors in schizophrenia. Schizophr Bull 36:1157–1166. 10.1093/schbul/sbp033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thisse B, Thisse C (2005) Functions and regulations of fibroblast growth factor signaling during embryonic development. Dev Biol 287:390–402. 10.1016/j.ydbio.2005.09.011 [DOI] [PubMed] [Google Scholar]
- Trokovic R, Trokovic N, Hernesniemi S, Pirvola U, Vogt Weisenhorn DM, Rossant J, McMahon AP, Wurst W, Partanen J (2003) FGFR1 is independently required in both developing mid- and hindbrain for sustained response to isthmic signals. EMBO J 22:1811–1823. 10.1093/emboj/cdg169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner CA, Akil H, Watson SJ, Evans SJ (2006) The fibroblast growth factor system and mood disorders. Biol Psychiatry 59:1128–1135. 10.1016/j.biopsych.2006.02.026 [DOI] [PubMed] [Google Scholar]
- Turner CA, Watson SJ, Akil H (2012) The fibroblast growth factor family: neuromodulation of affective behavior. Neuron 76:160–174. 10.1016/j.neuron.2012.08.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner CA, Eren-Koçak E, Inui EG, Watson SJ, Akil H (2016) Dysregulated fibroblast growth factor (FGF) signaling in neurological and psychiatric disorders. Semin Cell Dev Biol 53:136–143. 10.1016/j.semcdb.2015.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemori H, Linhoff MW, Ornitz DM, Sanes JR (2004) FGF22 and its close relatives are presynaptic organizing molecules in the mammalian brain. Cell 118:257–270. 10.1016/j.cell.2004.06.025 [DOI] [PubMed] [Google Scholar]
- Unichenko P, Yang JW, Luhmann HJ, Kirischuk S (2015) Glutamatergic system controls synchronization of spontaneous neuronal activity in the murine neonatal entorhinal cortex. Pflugers Arch 467:1565–1575. 10.1007/s00424-014-1600-5 [DOI] [PubMed] [Google Scholar]
- Uylings HB, Smit GJ, Veltman WA (1975) Ordering methods in quantitative analysis of branching structures of dendritic trees. Adv Neurol 12:347–354. [PubMed] [Google Scholar]
- Vanhoutte P, Barnier JV, Guibert B, Pagès C, Besson MJ, Hipskind RA, Caboche J (1999) Glutamate induces phosphorylation of Elk-1 and CREB, along with c-fos activation, via an extracellular signal-regulated kinase-dependent pathway in brain slices. Mol Cell Biol 19:136–146. 10.1128/MCB.19.1.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zhuo M (2012) Group I metabotropic glutamate receptor-mediated gene transcription and implications for synaptic plasticity and diseases. Front Pharmacol 3:189. 10.3389/fphar.2012.00189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker VR, Cui L, Miller S, Yu SP, Wei L (2007) Whisker stimulation enhances angiogenesis in the barrel cortex following focal ischemia in mice. J Cereb Blood Flow Metab 27:57–68. 10.1038/sj.jcbfm.9600318 [DOI] [PubMed] [Google Scholar]
- Williams AJ, Umemori H (2014) The best-laid plans go oft awry: synaptogenic growth factor signaling in neuropsychiatric disease. Front Synaptic Neurosci 6:4. 10.3389/fnsyn.2014.00004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RO, Ghosh A (2002) Activity-dependent regulation of dendritic growth and patterning. Nat Rev Neurosci 3:803–812. 10.1038/nrn941 [DOI] [PubMed] [Google Scholar]
- Wu CS, Ballester Rosado CJ, Lu HC (2011) What can we get from ‘barrels’: the rodent barrel cortex as a model for studying the establishment of neural circuits. Eur J Neurosci 34:1663–1676. 10.1111/j.1460-9568.2011.07892.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaguchi Y, Yu T, Ahmed MU, Berry M, Mason I, Basson MA (2009) Fibroblast growth factor (FGF) gene expression in the developing cerebellum suggests multiple roles for FGF signaling during cerebellar morphogenesis and development. Dev Dyn 238:2058–2072. 10.1002/dvdy.22013 [DOI] [PubMed] [Google Scholar]
- Yang JW, Hanganu-Opatz IL, Sun JJ, Luhmann HJ (2009) Three patterns of oscillatory activity differentially synchronize developing neocortical networks in vivo. J Neurosci 29:9011–9025. 10.1523/JNEUROSCI.5646-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM (2003) Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development 130:3063–3074. 10.1242/dev.00491 [DOI] [PubMed] [Google Scholar]
- Zafra F, Hengerer B, Leibrock J, Thoenen H, Lindholm D (1990) Activity-dependent regulation of BDNF and NGF mRNAs in the rat hippocampus is mediated by non-NMDA glutamate receptors. EMBO J 9:3545–3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Original data values and detailed statistical comparison for Figure 1. Download Figure 1-1, XLSX file (37.3KB, xlsx)
Original data values and detailed statistical comparison for Figure 2. Download Figure 2-1, XLSX file (14.6KB, xlsx)
Original data values and detailed statistical comparison for Figure 3. Download Figure 3-1, XLSX file (71.1KB, xlsx)
Original data values and detailed statistical comparison for Figure 4 and Table 1. Download Figure 4-1, XLSX file (15.8KB, xlsx)
Original data values and detailed statistical comparison for Figure 5. Download Figure 5-1, XLSX file (48.8KB, xlsx)
Original data values and detailed statistical comparison for Figure 6. Download Figure 6-1, XLSX file (34.3KB, xlsx)