

Abstract
Strain CCMM B554, also known as FSM-MA, is a soil dwelling and nodule forming, nitrogen-fixing bacterium isolated from the nodules of the legume Medicago arborea L. in the Maamora Forest, Morocco. The strain forms effective nitrogen fixing nodules on species of the Medicago, Melilotus and Trigonella genera and is exceptional because it is a highly effective symbiotic partner of the two most widely used accessions, A17 and R108, of the model legume Medicago truncatula Gaertn. Based on 16S rRNA gene sequence, multilocus sequence and average nucleotide identity analyses, FSM-MA is identified as a new Ensifer meliloti strain. The genome is 6,70 Mbp and is comprised of the chromosome (3,64 Mbp) harboring 3574 predicted genes and two megaplasmids, pSymA (1,42 Mbp) and pSymB (1,64 Mbp) with respectively 1481 and 1595 predicted genes. The average GC content of the genome is 61.93%. The FSM-MA genome structure is highly similar and co-linear to other E. meliloti strains in the chromosome and the pSymB megaplasmid while, in contrast, it shows high variability in the pSymA plasmid. The large number of strain-specific sequences in pSymA as well as strain-specific genes on pSymB involved in the biosynthesis of the lipopolysaccharide and capsular polysaccharide surface polysaccharides may encode novel symbiotic functions explaining the high symbiotic performance of FSM-MA.
Electronic supplementary material
The online version of this article (doi: 10.1186/s40793-017-0298-3) contains supplementary material, which is available to authorized users.
Keywords: Ensifer meliloti, Root nodule bacteria, Nitrogen-fixation, Symbiosis
Introduction
To secure their nitrogen supply, legumes such as alfalfa, pea, (soy−/faba-)bean establish an endosymbiotic interaction with soil bacteria collectively called rhizobia that can reduce atmospheric nitrogen gas and produce reduced nitrogen molecules metabolizable by the plants. This symbiosis between legumes and rhizobia is of ecological and economic importance because of its contribution to the global nitrogen cycle, its impact on sustainable agriculture and its biotechnological potential to ensure nitrogen supply in agriculture [1].
The reduction of atmospheric nitrogen by rhizobia takes place in a specific niche, within the cells of de novo formed organs called nodules found usually on the roots and in some cases on the stem of the plants. Nodule development is initiated when flavonoids released by the plants induce the expression of the bacterial nodulation (nod) genes resulting in the production of the lipo-chitooligosaccharide signal molecules, the Nod factors. Nod factors cause a change in the direction of polar growth in developing root hairs and simultaneously induce cell division in the root cortex cells. As a result, a nodule primordium is formed that turns into meristematic tissue to produce the cells of the nodule and bacteria become entrapped in the curled root hair where they form an infection pocket. From the site of the infection pocket, a tubular structure, called infection thread, is formed in the root hair that grows toward the cells of the developing nodule. In the infection thread, bacteria multiply and finally they are released into the cytoplasm of the nodule cells via a mechanism resembling endocytosis resulting in organelle-like structures called symbiosomes. Symbiosomes have a membrane of plant origin which surrounds one or more bacteria. After bacterial release, the cells of both partners differentiate into mature symbiotic cells. The nodule cells become enlarged polyploid cells which host several tens of thousands of bacteria that are themselves differentiated into a nitrogen-fixing form called bacteroid [2–4]. Interestingly, in Medicago and closely related species like Pisum and Vicia, the host imposes a terminal differentiation on the bacterial partner that is accompanied by the increase in the DNA content and size of the bacteroids and results in the loss of their cell division capacity [5]. This terminal differentiation is orchestrated by nodule-specific cysteine-rich peptides that are expressed exclusively in the infected cells of the nodule [6, 7].
To effectively investigate these interactions, two genetic model legume species, Lotus japonicus (Regel) K. Larsen (bird’s-foot trefoil) and Medicago truncatula Gaertn. (barrel clover/barrel medic) have been chosen for which structural and functional genomics tools and databases have been developed [8, 9]. M. truncatula is a diploid, self-pollinating annual plant belonging to the Medicago genus, which contains species that are among the most extensively cultivated forage and pasture plants. Medicago plants establish symbiosis only with a limited number of bacterial species, mainly with 10.1601/nm.1328 (synonym 10.1601/nm.1339) meliloti and 10.1601/nm.1334, and with certain 10.1601/nm.1331 strains and 10.1601/nm.1298 [10–12]. However, some combinations of wild-type plants (species, sub-species and ecotypes) and bacterial strains of the most-studied bacterial species, 10.1601/nm.1335 and 10.1601/nm.1334, often lead to incompatible interactions [13–17], i.e. nodule formation is initiated but bacteria cannot invade nodules or cannot persist and fix nitrogen in the symbiotic organ. The incompatibility can be caused by functions/proteins encoded by genes in the accessory genome of the bacteria [14] such as the strain-specific HrrP peptidase [18], strain specific exopolysaccharide production [19] and/or allelic variants of the host genes like the NFS1 and NFS2 genes encoding NCR peptides in M. truncatula [20, 21]. Strikingly, the model bacterium 10.1601/nm.1335 strain 1021 (with the reference genome and most of the available mutants) is poorly matched for nitrogen fixation with the most widely used M. truncatula accessions Jemalong A17 and M. truncatula ssp. tricycla R108 [22, 23].
10.1601/nm.1335 strain FSM-MA (first catalogued as 10.1601/nm.1330 strain 10.1601/strainfinder?urlappend=%3Fid%3DCCMM+B554, also known as LMG-R33403 and MR372) was isolated from the nodules of Medicago arborea L. (moontrefoil/tree medic) in Maamora Forest between Rabat and Meknes, Morocco, and is stored in The Moroccan Coordinated Collections of Microorganisms as 10.1601/strainfinder?urlappend=%3Fid%3DCCMM+B554. Recently, Kazmierczak et al. [22] identified 10.1601/nm.1335 strain FSM-MA as a highly effective symbiotic partner of the two most widely used M. truncatula ecotypes, A17 and R108, as well as all tested Medicago sativa L. (alfalfa) cultivars. To gain the potential to identify novel bacterial symbiotic genes and genes associated with FSM-MA’s exceptional symbiotic performance, we sequenced the genome of the strain FSM-MA. Here we present a summary classification and a set of general features for 10.1601/nm.1335 strain FSM-MA, together with a description of its genome sequence and annotation.
Organism information
Classification and features
10.1601/nm.1335 FSM-MA is a motile, non-sporulating, Gram-negative strain (Fig. 1) in the order 10.1601/nm.1277 of the class 10.1601/nm.809. This fast growing strain forms colonies within 3 days on YEB agar plates [22] at 30 °C. The colonies (Fig. 1a, b) are light beige colored on YEB plates, slightly doomed, mucoid and have a smooth margin. The rod shaped free-living form (Fig. 1c, d) has dimensions of 1.0–2.0 μm in length and approximately 0.5 μm in width, while bacteroids in M. truncatula Jemalong A17 nodules (Fig. 1e, f) have the same width and are elongated to 5–8 μm. A summary of the classification is provided in Table 1.
Fig. 1.

Colony morphology of E. meliloti strain FSM-MA on solid medium (a,b) at 5× (A) and 50× (b) magnifications as well as scanning electron microscopic images at 4000× (c,e) and 20,000× magnifications (d,f) of free-living cells (c,d) and bacteroids isolated from M. truncatula Jemalong A17 nodules (e,f)
Table 1.
Classification and general features of E. meliloti strain FSM-MA
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Current classification | Domain Bacteria | TAS [32] | |
| Phylum Proteobacteria | TAS [33] | ||
| Class Alphaproteobacteria | TAS [34, 35] | ||
| Order Rhizobiales | TAS [35–37] | ||
| Family Rhizobiaceae | TAS [37, 38] | ||
| Genus Ensifer | TAS [39–43] | ||
| Species Ensifer meliloti | TAS [40, 42] | ||
| Strain FSM-MA (B554) | |||
| Gram stain | Negative | IDA | |
| Cell shape | Rod | IDA | |
| Motility | Motile | IDA | |
| Sporulation | Non-sporulating | NAS | |
| Temperature range | Mesophile | NAS | |
| Optimum temperature | 28–37 °C | IDA | |
| pH range | 5.5–9.5 | IDA | |
| Carbon source | Various | TAS [44] | |
| GS-6 | Habitat | Soil, root nodule on hosts | IDA |
| MIGS-6.3 | Salinity | Unknown | NAS |
| MIGS-22 | Oxygen requirement | Aerobic | NAS |
| MIGS-15 | Biotic relationship | Free living, Symbiotic | IDA |
| MIGS-14 | Pathogenicity | Non-pathogen | TAS [45] |
| Energy source | Chemoorganotroph | NAS | |
| MIGS-14 | Pathogenicity | Non-pathogenic | NAS |
| MIGS-4 | Geographic location | Maamora Forest, Morocco | NAS |
| MIGS-5 | Sample collection | 2004 | NAS |
| MIGS-4.1 | Latitude | Not reported | NAS |
| MIGS-4.2 | Longitude | Not reported | NAS |
| MIGS-4.4 | Altitude | Not reported | NAS |
aEvidence codes - IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [46] (http://geneontology.org/page/guide-go-evidence-codes)
Extended feature descriptions
Phylogenetic analysis of 10.1601/nm.1335 strain FSM-MA was performed by aligning the 16S rRNA sequence to the 16S rRNA sequences (consensus sequence length of 1346 basepairs (bp)) of other 10.1601/nm.1328 strains (Fig. 2). The FSM-MA 16S rRNA sequence has 100% sequence identity with those of the widely used 10.1601/nm.1335 strains such as strain 1021 or Rm41, while four mismatches can be observed with the 10.1601/nm.1334 strain 10.1601/strainfinder?urlappend=%3Fid%3DWSM+419 sequence. Moreover, there are five mismatches between the 16S rRNA sequence fragments of strain FSM-MA and 10.1601/nm.1330 strain 10.1601/strainfinder?urlappend=%3Fid%3DLMG+14919 T. A Multilocus Sequence Analysis (Additional file 1: Figure S1) using 14 chromosomal genes further confirmed FSM-MA as an 10.1601/nm.1335 strain and clearly separated it from 10.1601/nm.1330 strain 10.1601/strainfinder?urlappend=%3Fid%3DLMG+14919 T, 10.1601/nm.1334 strain 10.1601/strainfinder?urlappend=%3Fid%3DWSM+419 and the 10.1601/nm.1331 strains 10.1601/strainfinder?urlappend=%3Fid%3DNGR+234, 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+257 and HH103. Among the 10.1601/nm.1335 strains, strain FSM-MA is most closely related to strains BO21CC and BL225C which were isolated from M. sativa nodules in Lodi, Italy [24]. Finally, the two-way average nucleotide identity [25] was calculated between genomes using the default settings of the ANI calculator. The genome of strain FSM-MA showed 99,42% identity with the genome of 10.1601/nm.1335 strain 1021 and only 90,09% identity with the genome of 10.1601/nm.1330 strain 10.1601/strainfinder?urlappend=%3Fid%3DLMG+14919 T , 87,09% identity with the genome of 10.1601/nm.1334 strain 10.1601/strainfinder?urlappend=%3Fid%3DWSM+419 and 83,16% and 83,31% identity with the genomes of 10.1601/nm.1331 strains 10.1601/strainfinder?urlappend=%3Fid%3DNGR+234 and HH103, respectively. Once more this analysis showed that FSM-MA is an 10.1601/nm.1335 strain and not an 10.1601/nm.1330 strain, considering a cut-off for species delineation at 95% identity [25].
Fig. 2.
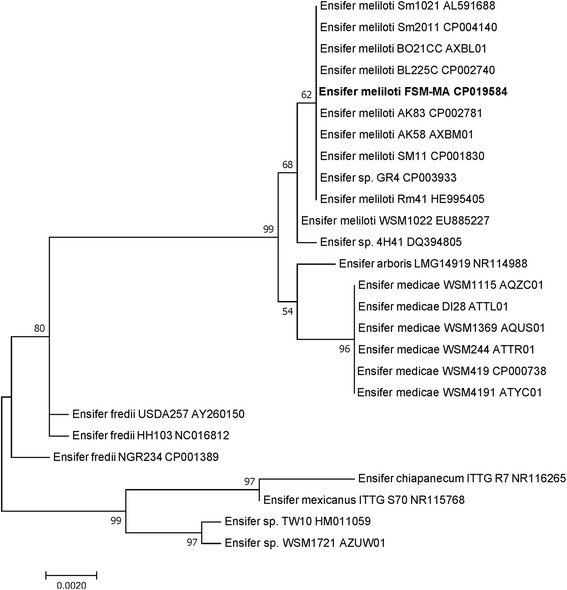
Phylogenetic tree showing the relationship of E. meliloti strain FSM-MA. The evolutionary history was inferred by using the Maximum Likelihood method based on the General Time Reversible model [30]. The tree with the highest log likelihood (−2208.71) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. Evolutionary analyses were conducted in MEGA7 [31]
Symbiotaxonomy
Strain FSM-MA forms effective nitrogen fixing nodules on Medicago species M. sativa L., M. truncatula, M. arborea L., M. sativa subsp. x varia, M. ruthenica (L.) Trautv. as well as on Trigonella calliceras Fisch., Melilotus albus (L.) Lam. (white sweetclover) and Melilotus officinalis (L.) Lam. (yellow sweetclover). Moreover, in agreement with its classification as 10.1601/nm.1335, it nodulates Medicago polymorpha L. (burclover) –that forms nitrogen-fixing symbiosis with 10.1601/nm.1334 strains – but there is no nitrogen fixation in the formed nodules.
Genome sequencing information
Genome project history
This organism was selected for sequencing on the basis of its superior symbiotic performance [22] with the most widely used accessions (A17 and R108) of the model legume M. truncatula. The genome project and the sequence of the three replicons are deposited in the National Center for Biotechnology Information (NCBI; accession numbers: CP019584, CP019585, CP019586). Genome sequencing and sequence assembling were performed at the University of Malaya (Kuala Lumpur, Malaysia) and at the Seqomics Biotechnology Ltd. (Mórahalom, Hungary). Annotation was carried out at Seqomics Biotechnology Ltd. A summary of the project information can be found in Table 2.
Table 2.
Genome sequencing project information for E. meliloti strain FSM-MA
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Finished |
| MIGS-28 | Libraries used | Illumina mate-paired library |
| PacBio SMRTbell library | ||
| MIGS-29 | Sequencing platforms | Illumina MiSeq |
| PacBio RS II | ||
| MIGS-31.2 | Fold coverage | 249.2× |
| MIGS-30 | Assemblers | CLC Genomic Worknbench v. 9.5; HGAP v. 3 |
| MIGS-32 | Gene calling methods | Genemark S+, used as part of the NCBI Prokaryotic Genome Annotation Pipeline PGAP |
| Locus Tag | SMB554 | |
| Genbank ID | CP019584-CP019586 | |
| Genbank Date of Release | 2017.07.01 | |
| GOLD ID | Gp0258805 | |
| BIOPROJECT | PRJNA369312 | |
| MIGS-13 | Source Material Identifier | FSM-MA |
| Project relevance | Symbiotic Nitrogen-fixation, agriculture |
Growth condition and genomic DNA preparation
10.1601/nm.1335 strain FSM-MA was grown on solid YEB medium (0.5% beef extract; 0.1% yeast extract; 0.5%peptone; 0.5% sucrose; 0.04% MgSO4.7H2O; pH 7.5) for 3 days and a single colony was used to inoculate 3 ml YEB broth medium. The culture was grown for 24 h on a gyratory shaker at 225 rpm at 30 °C, then 0.5 ml of the starter culture was used to inoculate 50 ml YEB broth medium. The culture was grown at 30 °C at 225 rpm until OD600 = 0.6 was reached. DNA was isolated from the cells with the MasterPure Complete DNA and RNA Purification Kit (Epicentre). The integrity of the extracted genomic DNA was analyzed by 0.7% agarose gel electrophoresis. The final concentration of the DNA, estimated with the help of a Qubit Fluorometer (ThermoFisher Scientific), was 0.45 mg ml−1.
Genome sequencing and assembly
The genome sequence of 10.1601/nm.1335 strain FSM-MA was generated using Pacific BioScience (PacBio) and Illumina technologies. An Illumina Mate Paired library (average insert length 7 kbp) was constructed and sequenced using the Illumina MiSeq platform, which generated 3,387,162 reads. Similarly, a PacBio SMRTbell library was constructed and sequenced on the PacBio RS II platform to generate 254,443 filtered reads (N50 value at 8643 bp and total bases at 1,726,776,880 bp). Assembly was then carried out using HGAP version 3 [26] yielding three contigs with an average coverage of 186.71×. Subsequently, Illumina reads were aligned to the PacBio assembly with the help of the CLC Genomics Workbench version 9.5 and the observed 17 InDels were corrected. The final assembly contains three circular contigs corresponding to the three replicons (the chromosome and the pSymA and pSymB megaplasmids) totaling 6,703,999 bp and total input read coverage was at 249.2×.
Genome annotation
Genes were identified and annotated using the NCBI Prokaryotic Genome Annotation Pipeline. The NCBI non-redundant database, UniProt, TIGR/Fam, Pfam, PRIAM, KEGG, COG, and InterPro databases were used to analyse the predicted coding sequences after translation. HMMER [27] and tRNAscan-SE [28] were used to identify the rRNA and tRNA genes, respectively.
Genome properties
The genome is 6,703,999 bp and comprised of three replicons (Table 3) with the size of 3,641,423 bp (chromosome), 1,422,736 bp (pSymA) and 1,639,840 bp (pSymB). The average GC content is 61.93%. Three rRNA operons, 67 RNA only genes were identified and 6583 protein coding genes were predicted in the genome. Five thousand thirty-two protein-coding genes were assigned a putative function and 1551 genes were predicted to code for hypothetical proteins (Table 4). The distribution of genes in COG functional categories is presented in Table 5.
Table 3.
Summary of genome: one chromosome and 2 plasmids
| Label | Size (Mb) | Topology | INSDC identifier | RefSeq ID |
|---|---|---|---|---|
| Chromosome | 3.641 | Circular | CP019584 | NZ_CP019584.1 |
| Plasmid 1 | 1.640 | Circular | CP019586 | NZ_CP019586.1 |
| Plasmid 2 | 1.423 | Circular | CP019585 | NZ_CP019585.1 |
Table 4.
Genome statistics for E. meliloti strain FSM-MA
| Attribute | Value | % of Total |
|---|---|---|
| Genome size (bp) | 6,703,999 | 100.00 |
| chromosome size (bp) | 3,641,423 | 54.32 |
| pSymA size (bp) | 1,422,736 | 21.22 |
| pSymB size (bp) | 1,639,840 | 24.46 |
| DNA coding region (bp) | 5,641,977 | 84.16 |
| DNA G + C content (bp) | 4,152,010 | 61.93 |
| DNA scaffolds | 3 | 100.00 |
| Total genes | 6650 | 100.00 |
| chromosomal genes | 3574 | 53.74 |
| pSymA genes | 1481 | 22.27 |
| pSymB genes | 1595 | 23.98 |
| Protein-coding genes | 6183 | 92.97 |
| RNA genes | 67 | 1.01 |
| Pseudo genes | 400 | 6.01 |
| Genes in internal clusters | 2341 | 35.20 |
| Genes with function prediction | 5032 | 75.67 |
| Genes assigned to COGs | 5801 | 87.23 |
| Genes with Pfam domains | 5167 | 77.70 |
| Genes with signal peptides | 534 | 8.03 |
| Genes with transmembrane helices | 1403 | 21.10 |
| CRISPR repeats | 0 | 0 |
Table 5.
Number of genes of Ensifer meliloti FSM-MA associated with general COG functional categories
| Code | chromosome | pSymA | pSymB | Genome | |||||
|---|---|---|---|---|---|---|---|---|---|
| Value | % age of total (3574) | value | % age of total (1481) | value | % age of total (1595) | value | % age of total (6650) | Description | |
| J | 164 | 4.59 | 7 | 0.47 | 16 | 1.00 | 187 | 2.81 | Translation, ribosomal structure and biogenesis |
| A | 0 | 0.00 | 0 | 0.00 | 0 | 0.00 | 0 | 0.00 | RNA processing and modification |
| K | 246 | 6.88 | 132 | 8.91 | 138 | 8.65 | 516 | 7.76 | Transcription |
| L | 140 | 3.92 | 40 | 2.70 | 26 | 1.63 | 206 | 3.10 | Replication, recombination and repair |
| B | 1 | 0.03 | 0 | 0.00 | 0 | 0.00 | 1 | 0.02 | Chromatin structure and dynamics |
| D | 30 | 0.84 | 5 | 0.34 | 10 | 0.63 | 45 | 0.68 | Cell cycle control, cell division, chromosome partitioning |
| V | 34 | 0.95 | 11 | 0.74 | 20 | 1.25 | 65 | 0.98 | Defense mechanisms |
| T | 135 | 3.78 | 77 | 5.20 | 71 | 4.45 | 283 | 4.26 | Signal transduction mechanisms |
| M | 148 | 4.14 | 32 | 2.16 | 104 | 6.52 | 284 | 4.27 | Cell wall/membrane/envelope biogenesis |
| N | 55 | 1.54 | 12 | 0.81 | 6 | 0.38 | 73 | 1.10 | Cell motility |
| U | 70 | 1.96 | 33 | 2.23 | 3 | 0.19 | 106 | 1.59 | Intracellular trafficking, secretion, and vesicular transport |
| O | 127 | 3.55 | 31 | 2.09 | 22 | 1.38 | 180 | 2.71 | Posttranslational modification, protein turnover, chaperones |
| C | 177 | 4.95 | 121 | 8.17 | 75 | 4.70 | 373 | 5.61 | Energy production and conversion |
| G | 236 | 6.60 | 94 | 6.35 | 245 | 15.36 | 575 | 8.65 | Carbohydrate transport and metabolism |
| E | 353 | 9.88 | 137 | 9.25 | 139 | 8.71 | 629 | 9.46 | Amino acid transport and metabolism |
| F | 82 | 2.29 | 7 | 0.47 | 21 | 1.32 | 110 | 1.65 | Nucleotide transport and metabolism |
| H | 133 | 3.72 | 31 | 2.09 | 35 | 2.19 | 199 | 2.99 | Coenzyme transport and metabolism |
| I | 117 | 3.27 | 37 | 2.50 | 53 | 3.32 | 207 | 3.11 | Lipid transport and metabolism |
| P | 140 | 3.92 | 81 | 5.47 | 78 | 4.89 | 299 | 4.50 | Inorganic ion transport and metabolism |
| Q | 76 | 2.13 | 35 | 2.36 | 42 | 2.63 | 153 | 2.30 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 399 | 11.16 | 172 | 11.61 | 169 | 10.60 | 740 | 11.13 | General function prediction only |
| S | 361 | 10.10 | 87 | 5.87 | 121 | 7.59 | 569 | 8.56 | Function unknown |
| W | 1 | 0.03 | 0 | 0.00 | 0 | 0.00 | 1 | 0.02 | Extracellular structures |
| – | 349 | 9.76 | 299 | 20.19 | 201 | 12.60 | 849 | 12.77 | Not in COGs |
Insights from the genome sequence
The genome size of FSM-MA falls within the expected size range of 6.65–8.94 Mbp observed in the 33 sequenced 10.1601/nm.1335 genomes that have been deposited in the Integrated Microbial Genomes (IMG) database. The genome of all 10.1601/nm.1335 strains is composed of a circular chromosome and two megaplasmids/chromids, however, certain strains harbour additional replicons too. In strain FSM-MA, however, no additional plasmid was detected. The strain contains three rRNA gene clusters as other 10.1601/nm.1335 strains do. Similarly to other 10.1601/nm.1328 strains, the highest number of genes is assigned to the COG functional categories amino acid transport and metabolism (9.46%), carbohydrate transport and metabolism (8.65%) and transcription (7.76%). An enrichment of the COG functional categories amino acid transport and metabolism, transcription and signal transduction mechanisms is observed in pSymA, while carbohydrate transport and metabolism and cell wall/membrane/envelope biogenesis are overrepresented on pSymB (Table 5).
Extended insights
Comparing the FSM-MA genome structure with that of other 10.1601/nm.1335 strains using the Mauve software [29] revealed high co-linearity of the chromosomes and the pSymB megaplasmids in contrast to the pSymA plasmids that are highly variable. For example, the average sequence identity between FSM-MA and strain 1021 is 99.4% and their chromosomes and pSymB plasmids are essentially co-linear. The major differences between the chromosomes originated from the insertion of three putative prophages/insertion elements into the FSM-MA genome at genes coding for tRNAs (SMB554__06910: tRNA-Thr, SMB554_09150: tRNA-Lys, SMB554_16265: tRNA-Met). These inserted elements are of approximately 48, 43 and 44 kbp and contains 70, 54 and 34 predicted ORFs, respectively. In the putative prophages at tRNA-Thr and tRNA-Lys, among hypothetical proteins, a number of phage related functions such as terminase, phage portal and capsid proteins (both prophages) as well as ORFs encoding endonucleases, transcriptional regulators, site-specific integrase, DNA ligase, peptidase or peptidoglycan-binding protein are encoded (prophage at tRNA-Lys). The inserted sequence at tRNA-Met seems to contain genes coding for type I restriction-modification system elements, an N6-DNA-methylase, chromosome segregation and AAA family ATPases as well as transcriptional regulators among hypothetical proteins. On the other hand, one putative prophage in the 1021 genome at a tRNA-Ser_CGA gene and the SMc01989-SMc02032 gene cluster coding for transcriptional regulators, membrane transporter and oxido-reductase elements are missing from the FSM-MA genome. The differences between the pSymB plasmids are mainly attributed to mobile genetic elements (IS elements, transposons) that are associated with strain-specific genes, essentially coding for proteins involved in the biosynthesis and transport of strain-specific LPS (lipopolysaccharide) and K-antigen (capsular polysaccharide) surface polysaccharides (discussed later). The pSymA plasmids – that are the carriers of major symbiotic functions such as genes encoding Nod factor biosynthesis and the nitrogenase enzyme and co-factor biosynthesis – have a number of co-linear blocks but have about 80 kbp size difference (FSM-MA > 1021), and more than 200 kbp (>1/7) of the sequences are absent in the other strain.
As the FSM-MA strain is interesting from the symbiotic point of view, we analysed those genes that are important for the development and functioning of the nitrogen-fixing symbioses. The initiation of the symbiotic interaction requires the production of Nod factors with proper chemical structure via the activity of the so-called Nod, Noe and Nol proteins. The FSM-MA genome contains all the known nod, noe and nol genes described in 10.1601/nm.1335. The nif and fix genes code for the structural elements of the nitrogenase complex (nitrogenase, nitrogenase reductase, electron transport proteins) performing the reduction of atmospheric nitrogen as well as for proteins required for the biosynthesis of co-factors and the assembly of the complex. All these genes – including the ones that are present in multiple copies such as the three fixNOQP operons – can be found in the FSM-MA genome. Notably, despite the high diversity of the 10.1601/nm.1335 pSymA plasmids harbouring these symbiotic genes, the arrangement and the genomic environment of the nodulation and nitrogen fixation genes in FSM-MA and strain 1021 are the same.
Surface polyasaccharides play an essential role during the infection process [4] when bacteria enter the cells of the developing nodules via the infection threads. In the Medicago - 10.1601/nm.1335 symbiosis, the production of the succinoglycan exopolysaccharide is required for the continuous growth of the infection threads and its lack can be suppressed by the production of galactoglycan or certain capsular polysaccharides. Lipopolysaccharides might also affect both the infection and bacteroid differentiation processes. The organization and genomic environment of genes for the production and transport of the species-specific polysaccahrides EPS I (exo and exs genes) and EPS II (exp genes) as well as of the conserved part (lipidA and O-antigene core) of LPS (chromosomal and pSymB-born genes) and the KPS transporters are the same in the two 10.1601/nm.1335 strains. In contrast, the genes responsible for the production of the strain-specific polysaccharide moieties of LPS and KPS (Additional file 2: Figure S2), located on pSymB, are unique for the given strains.
Conclusions
The genome sequence of FSM-MA is of particular interest because the strain is highly effective with the most widely used ecotypes, Jemalong and R108 of the model legume M. truncatula. Comparative genomics with less and similarly effective strains as well as the creation and use of genomic libraries from FSM-MA has the potential to identify novel symbiotic genes and genes/operons that contribute to the exceptional symbiotic performance of the strain.
Additional files
Multilocus Sequence Analysis of 14 genes, recA, gltA, glnA, ctrA, ftsA, ftsZ1, ftsZ2, gyrB, dnaK, pnp, rpoB, thrC, atpD and gap in E. meliloti strains FSM-MA, Sm1021, Su47, Rm41, AK58, AK83, SM11, GR4, BO21CC and BL225C, E. arboris strain LMG14919, E. medicae strain WSM419 and E. fredii strains USDA257, NGR234 and HH103. The concatenated gene sequences (total 23,220 bp) were aligned by ClustalW and a maximum likelihood tree was inferred from the aligned sequences using MEGA ver. 6.0.6 software (Tamura et al., 2007). The tree was estimated using the Tamura-Nei substitution model (Tamura and Nei, 1993). Bootstrap tests were performed with 1000 replications. The inset shows the topology of the maximum likelihood tree. Tamura K, Nei M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10: 512–526. Tamura K, Dudley J, Nei M, Kumar S. (2007). MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24: 1596–1599. (TIFF 14278 kb)
Comparison of the organization of genes responsible for the production of the strain-specifc KPS in E. meliloti strains FSM-MA, 1021 and Rm41. The gene clusters are located between conserved genes (red arrow) coding for a 3-methyl-2-oxobutanoate-hydroxymethyl transferase (MOBHMT) and a nucleotidyl transferase. Genes determining conserved functions in KPS production such as transport (RkpR, RkpS, RkpS) or chain-length determination (RkpZ) are drawn as solid blue boxes. Open arrows with blue line indicate strain-specific rkp genes. Mustard arrows indicate genes conserved between two strains in the region. Open arrows with black line show genes with unknown function or function that could not be related to KPS synthesis. The genes are not drawn to scale. HypProt: hypothetical protein; pAcetylT: putative acetyl transferase; pMethylT: putative methyl transferase; GlycosylT: glycosyl transferase; pLysozime: putative lysozyme; SecCaBProt: putative secreted calcium-binding protein; pMembProt: putative membrane protein. (TIFF 75 kb)
Acknowledgements
The authors thank Anne Willems (Laboratory of Microbiology, University Gent, Belgium) for initial characterization of the strain 10.1601/strainfinder?urlappend=%3Fid%3DCCMM+B554/FSM-MA.
Funding
This work was supported by a grant from the National Research, Development and Innovation Office of Hungary to AK (grant number: 120,122), by a grant from the Agence National de la Recherche to PM (grant number ANR-13-BSV7–0013) and by a CNRS MEDICRABAT PRAD grant (PRAD N° 06–10) to PR and MA.
Abbreviations
- Bp
Basepair
- EPS
Exopolysaccharide
- KPS
Capsular polysaccharide
- LPS
lipopolysaccharide
- NCR
Nodule-specific cysteine-rich
Authors’ contributions
JI isolated the strain; MA performed the taxonomic characterization of the strain; PR performed the first characterization of the strain’s symbiotic performance and with JI and MA assembled and provided the strain and the background information. Symbiotaxonomic evaluation of the strain was performed by QB (Melilotus and Trigonella species) with the supervision of VG and PM and by MN (Medicago species) with the supervision of EK and AK. AF performed all the imaging. MN also isolated the genomic DNA for sequencing. PacBio and Illumina sequencing were performed by TMC supervised by KGC and by PB, respectively. PacBio reads were analysed and assembled by KWH under the supervision of KGC. BMV created the final assembly supervised by BB while BH carried out the annotation of the genome supervised by BB and AK. PM and AK devised the experiments, analysed the data and wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi: 10.1186/s40793-017-0298-3) contains supplementary material, which is available to authorized users.
References
- 1.Oldroyd GE, Dixon R. Biotechnological solutions to the nitrogen problem. Curr Opin Biotechnol. 2014;26:19–24. doi: 10.1016/j.copbio.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 2.Oldroyd GE, Murray JD, Poole PS, Downie JA. The rules of engagement in the legume-rhizobial symbiosis. Annu Rev Genet. 2011;45:119–144. doi: 10.1146/annurev-genet-110410-132549. [DOI] [PubMed] [Google Scholar]
- 3.Kereszt A, Mergaert P, Kondorosi E. Bacteroid development in legume nodules: evolution of mutual benefit or of sacrificial victims? Mol Plant-Microbe Interact. 2011;24:1300–1309. doi: 10.1094/MPMI-06-11-0152. [DOI] [PubMed] [Google Scholar]
- 4.Gibson KE, Kobayashi H, Walker GC. Molecular determinants of a symbiotic chronic infection. Annu Rev Genet. 2008;42:413–441. doi: 10.1146/annurev.genet.42.110807.091427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mergaert P, Uchiumi T, Alunni B, Evanno G, Cheron A, Catrice O, et al. Eukaryotic control on bacterial cell cycle and differentiation in the Rhizobium-legume symbiosis. Proc Natl Acad Sci U S A. 2006;103:5230–5235. doi: 10.1073/pnas.0600912103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van de Velde W, Zehirov G, Szatmari A, Debreczeny M, Ishihara H, Kevei Z, et al. Plant peptides govern terminal differentiation of bacteria in symbiosis. Science. 2010;327:1122–1126. doi: 10.1126/science.1184057. [DOI] [PubMed] [Google Scholar]
- 7.Guefrachi I, Nagymihaly M, Pislariu CI, Van de Velde W, Ratet P, Mars M, et al. Extreme specificity of NCR gene expression in Medicago truncatula. BMC Genomics. 2014;15:712. doi: 10.1186/1471-2164-15-712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stacey G, Libault M, Brechenmacher L, Wan J, May GD. Genetics and functional genomics of legume nodulation. Curr Opin Plant Biol. 2006;9:110–121. doi: 10.1016/j.pbi.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 9.Suzaki T, Yoro E, Kawaguchi M. Leguminous plants: inventors of root nodules to accommodate symbiotic bacteria. Int Rev Cell Mol Biol. 2015;316:111–158. doi: 10.1016/bs.ircmb.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 10.Kuykendall LD, Hashem FM, Bauchan GR, Devine TE, Dadson RB. Symbiotic competence of Sinorhizobium fredii on twenty alfalfa cultivars of diverse dormancy. Symbiosis. 1999;27:1–16. [Google Scholar]
- 11.Rome S, Fernandez MP, Brunel MP, Normand P, Cleyet-Marel JC. Evidence that two genomic species of Rhizobium are associated with Medicago truncatula. Int J Syst Bacteriol. 1996;46:972–980. doi: 10.1099/00207713-46-4-972. [DOI] [PubMed] [Google Scholar]
- 12.Van Berkum P, Beyene D, Bao G, Campbell TA, Eardly BD. Rhizobium mongolense sp. nov. is one of three rhizobial genotypes identified which nodulate and form nitrogen-fixing symbioses with Medicago ruthenica (L.) Ledebour. Int J Syst Bacteriol. 1998;48:13–22. doi: 10.1099/00207713-48-1-13. [DOI] [PubMed] [Google Scholar]
- 13.Bena G, Lyet A, Huguet T, Olivieri I. Medicago-Sinorhizobium symbiotic specificity evolution and the geographic expansion of Medicago. J Evol Biol. 2005;18:1547–1558. doi: 10.1111/j.1420-9101.2005.00952.x. [DOI] [PubMed] [Google Scholar]
- 14.Crook MB, Lindsay DP, Biggs MB, Bentley JS, Price JC, Clement SC, et al. Rhizobial plasmids that cause impaired symbiotic nitrogen fixation and enhanced host invasion. Mol Plant-Microbe Interact. 2012;25:1026–1033. doi: 10.1094/MPMI-02-12-0052-R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu J, Yang S, Zheng Q, Zhu H. Identification of a dominant gene in Medicago truncatula that restricts nodulation by Sinorhizobium meliloti strain Rm41. BMC Plant Biol. 2014;14:167. doi: 10.1186/1471-2229-14-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tirichine L, de Billy F, Huguet T. Mtsym6, a gene conditioning Sinorhizobium strain-specific nitrogen fixation in Medicago truncatula. Plant Physiol. 2000;123:845–851. doi: 10.1104/pp.123.3.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sugawara M, Epstein B, Badgley BD, Unno T, Xu L, Reese J, et al. Comparative genomics of the core and accessory genomes of 48 Sinorhizobium strains comprising five genospecies. Genome Biol. 2013;14:R17. doi: 10.1186/gb-2013-14-2-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Price PA, Tanner HR, Dillon BA, Shabab M, Walker GC, Griffitts JS. Rhizobial peptidase HrrP cleaves host-encoded signaling peptides and mediates symbiotic compatibility. Proc Natl Acad Sci U S A. 2015;112:15244–15249. doi: 10.1073/pnas.1417797112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simsek S, Ojanen-Reuhs T, Stephens SB, Reuhs BL. Strain-ecotype specificity in Sinorhizobium meliloti-Medicago truncatula symbiosis is correlated to succinoglycan oligosaccharide structure. J Bacteriol. 2007;189:7733–7740. doi: 10.1128/JB.00739-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Q, Yang S, Liu J, Terecskei K, Abraham E, Gombar A, et al. Host-secreted antimicrobial peptide enforces symbiotic selectivity in Medicago truncatula. Proc Natl Acad Sci U S A. 2017;114:6854–6859. doi: 10.1073/pnas.1700715114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang S, Wang Q, Fedorova E, Liu J, Qin Q, Zheng Q, et al. Microsymbiont discrimination mediated by a host-secreted peptide in Medicago truncatula. Proc Natl Acad Sci U S A. 2017;114:6848–6853. doi: 10.1073/pnas.1700460114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kazmierczak T, Nagymihaly M, Lamouche F, Barriere Q, Guefrachi I, Alunni B, et al. Specific host-responsive associations between Medicago truncatula accessions and Sinorhizobium strains. Mol Plant-Microbe Interact. 2017;30:399–409. doi: 10.1094/MPMI-01-17-0009-R. [DOI] [PubMed] [Google Scholar]
- 23.Terpolilli JJ, O'Hara GW, Tiwari RP, Dilworth MJ, Howieson JG. The model legume Medicago truncatula A17 is poorly matched for N2 fixation with the sequenced microsymbiont Sinorhizobium meliloti 1021. New Phytol. 2008;179:62–66. doi: 10.1111/j.1469-8137.2008.02464.x. [DOI] [PubMed] [Google Scholar]
- 24.Carelli M, Gnocchi S, Fancelli S, Mengoni A, Paffetti D, Scotti C, et al. Genetic diversity and dynamics of Sinorhizobium meliloti populations nodulating different alfalfa cultivars in Italian soils. Appl Environ Microbiol. 2000;66:4785–4789. doi: 10.1128/AEM.66.11.4785-4789.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57:81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 26.Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10:563–569. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 27.Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nei M, Kumar S. Molecular evolution and phylogenetics. New York: Oxford University Press; 2000. [Google Scholar]
- 31.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl. Nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey's manual of systematic bacteriology. 2. New York: Springer; 2005. [Google Scholar]
- 34.Garrity GM, Bell JA, Lilburn T. Class I. Alphaproteobacteria class. Nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey's manual of systematic bacteriology. 2. New York: Springer; 2005. [Google Scholar]
- 35.Validation EJ, No L. 107. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol. 2006;56:1–6. doi: 10.1099/ijs.0.64188-0. [DOI] [PubMed] [Google Scholar]
- 36.Kuykendall LD. Order VI. Rhizobiales ord. Nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey's manual of systematic bacteriology. 2. New York: Springer; 2005. [Google Scholar]
- 37.Skerman VBD, McGowan V, Sneath PHA. Approved lists of bacterial names. Int J Syst Bacteriol. 1980;30:225–420. doi: 10.1099/00207713-30-1-225. [DOI] [PubMed] [Google Scholar]
- 38.Kuykendall LD. Family I. Rhizobiaceae. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey's manual of systematic bacteriology. 2. New York: Springer; 2005. [Google Scholar]
- 39.Casida LE. Ensifer adhaerens gen. Nov., sp. nov.: a bacterial predator of bacteria in soil. Int J Syst Bacteriol. 1982;32:339–345. doi: 10.1099/00207713-32-3-339. [DOI] [Google Scholar]
- 40.de Lajudie P, Willems A, Pot B, Dewettinck D, Maestrojuan G, Neyra M, et al. Polyphasic taxonomy of rhizobia: emendation of the genus Sinorhizobium and description of Sinorhizobium meliloti comb. nov., Sinorhizobium saheli sp. nov., and Sinorhizobium teranga sp. nov. Int J Syst Bacteriol. 1994;44:715–733. doi: 10.1099/00207713-44-4-715. [DOI] [Google Scholar]
- 41.Kuykendall LD, Hashem FM, Wang ET. Genus VII. Sinorhizobium. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey's manual of systematic bacteriology. New York: Springer; 2005. [Google Scholar]
- 42.Judicial Commission of the International Committee on Systematics of Prokaryotes The genus name Sinorhizobium Chen et al. 1988 is a later synonym of Ensifer Casida 1982 and is not conserved over the latter genus name, and the species name ‘Sinorhizobium adhaerens’ is not validly published. Opinion 84. Int J Syst Evol Microbiol. 2008;58:1973. doi: 10.1099/ijs.0.2008/005991-0. [DOI] [PubMed] [Google Scholar]
- 43.Chen WX, Yan GH, Li JL. Numerical taxonomic study of fast-growing soybean rhizobia and a proposal that Rhizobium fredii be assigned to Sinorhizobium gen. Nov. Int J Syst Bacteriol. 1988;28:392–397. doi: 10.1099/00207713-38-4-392. [DOI] [Google Scholar]
- 44.Geddes BA, Oresnik IJ. Physiology, genetics, and biochemistry of carbon metabolism in the alphaproteobacterium Sinorhizobium meliloti. Can J Microbiol. 2014;60:491–507. doi: 10.1139/cjm-2014-0306. [DOI] [PubMed] [Google Scholar]
- 45.Biological Agents: Technical rules for biological agents. [http://www.baua.de/en/Topics-from-A-to-Z/Biological-Agents/TRBA/TRBA.html].
- 46.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Multilocus Sequence Analysis of 14 genes, recA, gltA, glnA, ctrA, ftsA, ftsZ1, ftsZ2, gyrB, dnaK, pnp, rpoB, thrC, atpD and gap in E. meliloti strains FSM-MA, Sm1021, Su47, Rm41, AK58, AK83, SM11, GR4, BO21CC and BL225C, E. arboris strain LMG14919, E. medicae strain WSM419 and E. fredii strains USDA257, NGR234 and HH103. The concatenated gene sequences (total 23,220 bp) were aligned by ClustalW and a maximum likelihood tree was inferred from the aligned sequences using MEGA ver. 6.0.6 software (Tamura et al., 2007). The tree was estimated using the Tamura-Nei substitution model (Tamura and Nei, 1993). Bootstrap tests were performed with 1000 replications. The inset shows the topology of the maximum likelihood tree. Tamura K, Nei M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10: 512–526. Tamura K, Dudley J, Nei M, Kumar S. (2007). MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24: 1596–1599. (TIFF 14278 kb)
Comparison of the organization of genes responsible for the production of the strain-specifc KPS in E. meliloti strains FSM-MA, 1021 and Rm41. The gene clusters are located between conserved genes (red arrow) coding for a 3-methyl-2-oxobutanoate-hydroxymethyl transferase (MOBHMT) and a nucleotidyl transferase. Genes determining conserved functions in KPS production such as transport (RkpR, RkpS, RkpS) or chain-length determination (RkpZ) are drawn as solid blue boxes. Open arrows with blue line indicate strain-specific rkp genes. Mustard arrows indicate genes conserved between two strains in the region. Open arrows with black line show genes with unknown function or function that could not be related to KPS synthesis. The genes are not drawn to scale. HypProt: hypothetical protein; pAcetylT: putative acetyl transferase; pMethylT: putative methyl transferase; GlycosylT: glycosyl transferase; pLysozime: putative lysozyme; SecCaBProt: putative secreted calcium-binding protein; pMembProt: putative membrane protein. (TIFF 75 kb)