

Abstract
Background
Haloplex targeted resequencing is a popular method to analyze both germline and somatic variants in gene panels. However, involved wet-lab procedures may introduce false positives that need to be considered in subsequent data-analysis. No variant filtering rationale addressing amplicon enrichment related systematic errors, in the form of an all-in-one package, exists to our knowledge.
Results
We present pyAmpli, a platform independent parallelized Python package that implements an amplicon-based germline and somatic variant filtering strategy for Haloplex data. pyAmpli can filter variants for systematic errors by user pre-defined criteria. We show that pyAmpli significantly increases specificity, without reducing sensitivity, essential for reporting true positive clinical relevant mutations in gene panel data.
Conclusions
pyAmpli is an easy-to-use software tool which increases the true positive variant call rate in targeted resequencing data. It specifically reduces errors related to PCR-based enrichment of targeted regions.
Electronic supplementary material
The online version of this article (10.1186/s12859-017-1985-1) contains supplementary material, which is available to authorized users.
Keywords: Targeted resequencing, Variant filtering, Somatic, Germline, Next-generation sequencing
Background
Low-cost targeted resequencing using specific gene panels in large sample cohorts is widely used in diagnostic settings and forms the current gold standard for multiple reasons. For instance, in hearing loss, screening of specific genes can be more efficient than whole exome, or whole genome sequencing due to reduced sequencing and analysis costs [1]. Second, data interpretation outside known disease genes is difficult and has limited added value in clinical settings. Finally, it is a cost-effective technique for ultra-deep sequencing which enables detection of low-allelic variants, for instance needed to pinpoint subclonal IgHV rearrangements in chronic lymphocytic leukemia [2].
Target enrichment methods can be divided into amplicon or multiplex PCR-based approaches, showing vertical enrichment blocks of identical fragments, and hybridization capture-based techniques, showing more bell-shaped enrichment of random fragments (Fig. 1) [3]. Here, we focused specifically on the analysis of the Haloplex Target Enrichment System, which can enrich up to thousands of exons. The Haloplex technology was originally developed by Olink Bioscience (prof. Olle Ericsson, Uppsala, Sweden) from where it has been commercialized by the spin-off company Halo Genomics. To date, the technology is further developed and supplied by Agilent Technologies (Santa Clara, USA). Although the technique is hybridization based, it results in amplicon-like data due to non-random restriction enzyme fragmentation and subsequent PCR amplification. The ligation-dependent selection for circular fragments increases target specificity towards fragments where the start and end positions correspond to restriction sites. However, a significant fraction of aspecific amplicons, not corresponding to predicted restriction fragments, is often present in the library, and can induce spurious variants. These variants can be visually recognised by not being present in genuine amplicons (Fig. 2a). Second, coverage is not uniform across the captured fragments, possibly resulting in false-negative heterozygous variants when both alleles are not sufficiently captured. Finally, PCR duplicates cannot be removed without the usage of molecular barcode tags, as these are inherent to the technology. Here, one could hypothesize that true variants should be present in multiple overlapping amplicons, as these correspond to independent captures by definition [4]. Introduction of incorrect nucleotides during PCR is therefore indistinguishable from true subclonal variants with low-allelic frequency.
Fig. 1.
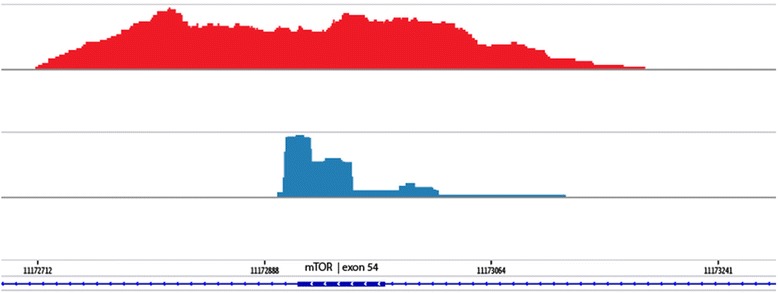
Sashimi target enrichment plots. mTOR Exon 54 coverage for two different target enrichment methods is represented by a Sashimi plot: 1) hybridization capture-based technique, showing more bell-shaped enrichment of random fragments (red histogram) and 2) amplicon- or multiplex PCR-based approach, showing vertical enrichment blocks of identical fragments (blue histogram)
Fig. 2.
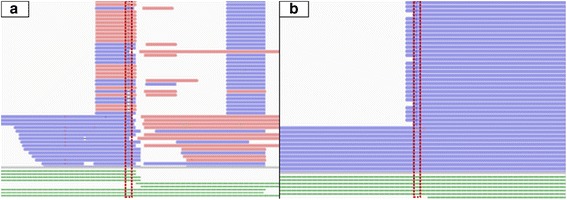
Vertical read blocks and variant calling bias visualization. Typical vertically enriched read blocks are illustrated in (2a and 2b). Aspecific fragments, not corresponding to predicted amplicons are shown in (2a) and variants restricted to read ends are shown in (2b). Called variants are indicated by a red dashed rectangular. Reads are given in blue and pink colored horizontal bars, indicating read orientation. Theoretical manufacturer designed Haloplex probes are presented in green colored horizontal bars below their corresponding enriched reads
To the best of our knowledge, no variant filtering rationale, in the format of an all-in-one package, exists that takes these targeted resequencing specific biases into account to differentiate false-positive from true-positive variants. Here, we present pyAmpli, a platform independent parallelized Python package that leverages amplicon specific information during variant filtering. Although user applied variant calling algorithms (e.g. VarScan2 and GATK Unified Genotyper) return various variant quality and reliability scores, these parameters are limited in amplicon- or PCR-based enrichment methods as they do not include amplicon information. Further, they are only suitable for hard filtering of variants. As such, variant hard filtering is exclusively based on the information available in the variant calling file generated by the chosen variant caller algorithm. pyAmpli uses solely the variant calling file for extraction of the variant’s position and further uses the sample’s alignment file to extract amplicon information.
pyAmpli can be applied in an oncological setting, after somatic tumor-normal variant calling as well in germline disease-gene screening projects. Our variant filtering algorithm ensures an enrichment of true-positive variants via a seven-step multi-staged categorization pipeline (Additional file 1).
Implementation
The pyAmpli package is developed using Python 2.7 [5]. The package is freely available for downstream variant analysis across various computing platforms. pyAmpli requires the following dependencies: pysam [6] is required for reading alignment files, PyVCF [7] for reading, formatting and generating variant files and pyYAML [8] for reading the user configuration file.
Input
The package provides both somatic and germline variant filtering. The default somatic mode of pyAmpli requires an amplicon-design file provided by the manufacturer, a paired tumor-normal variant calling file (VCF) and a normal and tumor alignment file (BAM) as input. The amplicon-design file is a BED file containing genomic location and probe identifier names for each included restriction fragment. Germline variants filtering requires an amplicon-design file, a single sample VCF and alignment file. Default and optimized settings for both somatic and germline parameters are included in the software as YAML configuration files. Command-line usage of pyAmpli in somatic mode is illustrated by Listing 1.
pyAmpli . py somatic
bn normal_sample_chr1.bam.
bt tumor_sample_chr1.bam.
v somatic_variants_chr1.vcf.
d amplicon_design_chr1.bed.
od output_directory
Listing 1
Variant processing
The variant processing workflow of pyAmpli can be summarized as follows. If supplied, a configuration file with user-defined thresholds is read-in, otherwise default settings are used. Next, the amplicon-design file provided by the manufacturer is processed into an easy accessible dictionary. Subsequently, every input variant present in the VCF is subjected to variant filtering analysis. The main variant analysis starts by assigning aligned read pairs overlapping the variant position to amplicons specified in the design file, discarding aspecific amplicons. Second, the ratio of variant-containing amplicons over all predicted amplicons covering the target position is calculated. Based on this ratio, variants are then categorized in 7 categories, as discussed below: DepthFail, OneAmpPass, LowAmpFail, MatchAmpPass, PositionFail, NormalFail and AmpPass. pyAmpli adds the final variant category to the FILTER field of the output VCF file (v4.1-formatted). Additional metrics, including the amplicon ratio and several amplicon counts are added to the INFO field to allow users to easily perform further downstream selection of variant categories (Table 1). A detailed decision diagram of pyAmpli’s filter logic is given in Additional file 1.
Table 1.
Additional VCF information fields. After running pyAmpli a new VCF is generated with additional INFO fields. These fields provide the user information on amplicon fractions, counts and offsets of reference and alternative alleles
| INFO ID | Description |
|---|---|
| AmpFR | Amplicon fraction for reference allele |
| AmpFA | Amplicon fraction for alternative allele |
| AmpCR | Amplicon count for reference allele |
| AmpCA | Amplicon count for alternative allele |
| AmpC | Amplicon total count |
| AmpF_OA | Amplicon count offset compared to allelic depth, for alternative allele |
| AmpF_OR | Amplicon count offset compared to allelic depth, for reference allele |
Variant categories
Variants are evaluated for each of the following criteria, in the order given here, and assigned to the first matching category (Additional file 1). When all criteria are passed, a variant is classified as high quality, corresponding to the label AmpPass.
DepthFail: variants with low read evidence
In a first step, variants with insufficient coverage by genuine read-pairs are flagged as low read evidence variants, and not subjected to further variant filtering. FILTER field flags DepthFail and DepthFailTumor/Normal are set, respectively in germline and somatic modes. Users have the flexibility to define their own DepthFail cut-off by adjusting the min_depth_normal and/or min_depth_tumor values in the configuration file.
OneAmpPass: variants with panel design limitations
Variants covered by and present in a single theoretical amplicon, as a design limitation, might be more prone for systemic enrichment artefacts. As we have insufficient information to evaluate the reliability of these variants, they are flagged as OneAmpPass, and not subjected to further filtering.
LowAmpFail: variants with low amount of covered amplicons
When variants are covered by multiple theoretical amplicons, we can infer variant reliability based on the number of amplicons containing the variant. Variants covered by more than two overlapping theoretical amplicons are flagged as LowAmpFail if the alternative allele is present in reads corresponding to less than three of these amplicons.
Variants covered by just two theoretical amplicons are handled separately. These variants are flagged as LowAmpFail if the alternative allele is present in reads corresponding to only one of both amplicons.
MatchAmpPass: variants with low amount of covered amplicons
Variants covered by just two theoretical amplicons are handled separately as MatchAmpPass if the alternative allele is present in reads from both amplicons, to indicate the limited discriminative power.
PositionFail: positional biases
Variants only present in the first two positions of either 3′ or 5′ read ends are flagged as PositionFail. This enrichment artefact is typically seen in Haloplex gene panels, because fragments are reproducibly generated by restriction enzymes, which cut only recognized sequences and generate non-random fragments [9]. Users can adjust the min_read_pos (default 2) and min_read_pos_fraction (default 10) in the configuration file, i.e. variants will be flagged as PositionFail if more than 10% of the total reads contain the alternative allele in the first two positions of either 3′ or 5′ read ends.
NormalFail: low-fraction variants in normal samples
This filter is only applied in somatic mode and is more subjective to user settings. When considering paired tumor-normal samples, somatic variants are not expected to be present in the patient’s paired normal tissue sample. First, this can be indicative for a false-positive somatic variant in the tumor tissue sample, that is in fact a true-positive low-fraction germline variant in the normal sample. Secondly, it might be a systemic enrichment artefact that is more pronounced in the tumor sample and therefore called as somatic. Lastly, it could be a reliable somatic variant. This may be explained by field cancerization, which is the occurrence of genetic, epigenetic and biochemical aberrations in structurally intact cells in histologically normal tissue adjacent to cancerous lesions [10]. By default, somatic variants present in more than 1% of reads from the normal sample are flagged as NormalFail. To allow the effect of field cancerization, the user can adjust the threshold (min_frac) for flagging these variants in the configuration file.
AmpPass: threshold-passing variants
As mentioned above, variants passing all user-defined filters are flagged as high-quality variants, using the AmpPass label.
Performance evaluation
We benchmarked pyAmpli on VCF and BAM files generated on in-house data. We calculated and validated the true and false positive rates. Next, we estimated runtime for batch processing.
Pre-pyAmpli bioinformatic processing of benchmark samples
Haloplex libraries were generated following the manufacturers guidelines (Protocol F1, July 2015, Agilent, CA, USA) and sequenced on an Illumina HiSeq1500 platform. Reads were trimmed for adapter sequence with Trimmomatic v0.36, and aligned with BWA v0.7.4 to version hg19 of the human genome. Germline variants were called using GATK Unified Genotyper v3.3.0 on 21 normal colon tissue samples. Somatic/loss-of-heterozygosity (LOH) variants were called using VarScan2 v2.3.9 on 115 colon tumor-normal tissue pairs. Tumor sample is defined as either primary colon tumor or metastatic tissue.
Benchmarking
True and false positive rates were estimated as follows. Variants present in ExAc r1.0, COSMIC v81 or dbSNP v142 databases were assumed true positive, and false positive otherwise. Next, variants were categorized according to variant type (germline, somatic and LOH) and filtering status (i.e. passing pyAmpli filtering or failing). To validate pyAmpli variant classification, 37 somatic variants were selected and validated by Sanger sequencing on a 3130xl Genetic Analyzer platform (Applied Biosystems Inc.).
Results and discussion
Current variant calling algorithms return variant quality and reliability scores in their VCFs. The calculated scores do not provide any amplicon information for reliable variant filtering. Further, necessity for amplicon-based filtering was made clear in a Sanger sequencing validation experiment by Samorodnitsky and colleagues. They showed that alternative alleles covered by less amplicons than present in their design are prone to be false positive findings [9].
There are analysis pipelines optimized for amplicon sequencing data, such as SureCall (Agilent Technologies, USA) and SeqNext (JSI medical systems, Germany), available. Although, latter software packages are able to call variants, the downstream variant filtering relies on ‘hard’ filters and information regarding the amplicon itself is lacking. Further, researchers still need to visually inspect all the data and judge the validity, and eventually manually flag the variants, which is a time-consuming step. Another disadvantage of these tools is that they are incompatible for paired variant calling. Of course, we do not discourage using SureCall or SeqNext variant analysis pipeline. The software output can serve as input for pyAmpli. To the best of our knowledge, no downstream post-processing tools as pyAmpli exists. pyAmpli will add useful variant and amplicon parameters that will guide the end-user for a legit variant interpretation and a hopefully desired decrease in analysis time per patient.
We present a new convenient variant filtering tool pyAmpli targeted at the reduction of systematic biases present in resequencing data generated using amplicon-based enrichment protocols. These protocols give rise to recurrent artefacts, as illustrated for Haloplex enrichment in Fig. 2. First, aspecifically enriched amplicons can introduce false positive variant calls. In case of Haloplex, these can be identified by the absence of corresponding restriction sites in the design file. Consequently, variants present only in aspecific amplicons and absent from genuine amplicons, can be labelled as false positives (LowAmpFail category, Fig. 2a). Second, Fig. 2b shows variants restricted to read ends, likely corresponding to systemic enrichment artefacts (PositionFail category). Whereas these artefacts are relevant for both germline and somatic variant evaluation purposes, an additional filter is present to specifically evaluate somatic variants.
pyAmpli true and false positive rate
In general, applying the pyAmpli germline filter on GATK Unified Genotyper calls of 21 normal tissue samples, increases the true positive rate from 39% (15,673 variants) to 64% (11,368) (Table 2). VarScan2 is proven to be a sensitive caller, however the tumor-normal variant calls lack high specificity. Applying pyAmpli somatic filtering settings on somatic and LOH variants of 115 tumor-normal tissue pairs increases the true positive rate from 29% (4028) and 45% (934) to 37% (885) and 81% (208), respectively (Table 2). After validation by Sanger sequencing of 37 variants, 21, 12, 4 and 0 variants were categorized as true positive, true negative, false positive and false negative, respectively (Additional file 2).
Table 2.
pyAmpli true and false positive rates.False and true positive rates in percentages before (−) and after (+) pyAmpli germline, somatic and LOH variant filtering with corresponding total variant number for ratio calculation. Germline variants were called with the GATK Unified Genotyper. Somatic and LOH variants were called with VarScan2
| Variant | Filter | True positive rate (%) | False positive rate (%) | Number of variants |
|---|---|---|---|---|
| Germline | – | 39 | 61 | 15,673 |
| + | 64 | 36 | 11,368 | |
| Somatic | – | 29 | 71 | 4028 |
| + | 37 | 63 | 885 | |
| LOH | – | 45 | 55 | 934 |
| + | 81 | 19 | 208 |
pyAmpli allows the user to select for true positive variants in gene panel data. Further, user-defined settings, based on their in-house validation cohorts, can be implemented in the variant filtering by adjusting the YAML configuration file.
Performance
We obtained the time required for variant filtering using a set of 115 colon tumor-normal pairs with an average of 289 variants per sample. Using the available parallel variant filtering functionality with 16 processing threads, we obtained an average CPU runtime per variant of 16.03 ms (16-core AMD Opteron™ 6378, 64-bit Linux 4.4.0–22-generic) (Additional file 3). Further upscaling has marginal benefits due to I/O limitations.
Conclusions
pyAmpli is a fast and parallel python program tailored to improve moderate true positive rates and reduce high false positive rates observed in PCR-based targeted enrichment strategies, in comparison to hybridisation-based capturing approaches. Although it was validated on Haloplex data, its principles are applicable to all PCR-based methods, such as Molecular Inversion Probes (MIPs) or multiplex PCR. Usage requires minimal input and limited programming skills from the user and only commodity computational resources. Output is generated in VCF v4.1 format and can be easily post-processed by the user.
Availability and requirements
Project name: pyAmpli.
Project home page: https://mbeyens.github.io/pyAmpli. The repository provides the package, quick-start examples and command-line examples for easy testing and performing essential processing.
Operating system(s): any supporting Python 2.7 (tested on Ubuntu 14.04.4 LTS).
Programming language: Python 2.7.
Other requirements: pysam > =0.8.4, PyVCF > =0.6.8, pyYAML > =3.11, setuptools > =20.2.2, samtools > =0.1.18, pigz > =2.3.4.
License: The GPL-v3 license (https://opensource.org/licenses/GPL-3.0).
Any restrictions to use by non-academics: None
Additional files
pyAmpli variant filter decision diagram. (DOCX 209 kb)
Sanger sequencing variant validation. (DOCX 71 kb)
pyAmpli CPU runtime. (DOCX 132 kb)
Acknowledgements
Not applicable.
Funding
This work was supported by the Research Foundation Flanders (FWO, grant no. 12D1717N). The funding body did not influence in any way the study design and collection, analysis and interpretation of data. Nor did it participate in writing of the manuscript.
Availability of data and materials
The pyAmpli package is available under the GPL-v3 license from https://mbeyens.github.io/pyAmpli. The repository provides also quick-start examples and command-line scripts for easy testing and performing essential processing.
Abbreviations
- BAM
Binary alignment file
- LOH
Loss-of-heterozygosity
- MIPs
Molecular inversion probes
- PCR
Polymerase chain reaction
- VCF
Variant calling file
Authors’ contributions
The pyAmpli package was designed by MB and GV, implemented by MB, NB and GV and documented by MB and GV. The manuscript was written by MB, GV, KOdB and GVC. All authors revised and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12859-017-1985-1) contains supplementary material, which is available to authorized users.
Contributor Information
Matthias Beyens, Email: matthias.beyens@uantwerpen.be.
Nele Boeckx, Email: nele.boeckx@uantwerpen.be.
Guy Van Camp, Email: guy.vancamp@uantwerpen.be.
Ken Op de Beeck, Email: ken.opdebeeck@uantwerpen.be.
Geert Vandeweyer, Email: geert.vandeweyer@uantwerpen.be.
References
- 1.Sommen M, Schrauwen I, Vandeweyer G, Boeckx N, Corneveaux JJ, van den Ende J, Boudewyns A, De Leenheer E, Janssens S, Claes K, Verstreken M, Strenzke N, Predöhl F, Wuyts W, Mortier G, Bitner-Glindzicz M, Moser T, Coucke P, Huentelman MJ, Van Camp G. DNA diagnostics of hereditary hearing loss: a targeted resequencing approach combined with a mutation classification system. Hum Mutat. 2016;37:812–819. doi: 10.1002/humu.22999. [DOI] [PubMed] [Google Scholar]
- 2.Stamatopoulos B, Timbs A, Bruce D, Smith T, Clifford R, Robbe P, Burns A, Vavoulis DV, Lopez L, Antoniou P, Mason J, Dreau H, Schuh A. Targeted deep sequencing reveals relevant subclonal IgHV rearrangements in chronic lymphocytic leukemia. Leukemia. 2017;31:837–845. doi: 10.1038/leu.2016.307. [DOI] [PubMed] [Google Scholar]
- 3.Samorodnitsky E, Datta J, Jewell BM, Hagopian R, Miya J, Wing MR, Damodaran S, Lippus JM, Reeser JW, Bhatt D, Timmers CD, Roychowdhury S. Comparison of custom capture for targeted next-generation DNA sequencing. J Mol Diagn. 2015;17:64–75. doi: 10.1016/j.jmoldx.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leanne de Kock Y, Wang C, Revil T, Badescu D, Rivera B, Sabbaghian N, Wu M, Weber E, Sandoval C, Hopman SMJ, Merks JHM, van Hagen JM, Bouts AHM, Plager DA, Ramasubramanian A, Forsmark L, Doyle KL, Toler T, Callahan J, Engelenberg C, Soglio DB-D, Priest JR, Ragoussis J, Foulkes WD. High-sensitiviy sequencing reveals multi-organ somatic mosaicism causing DICER1 syndrome. J Med Genet. 2016;53:43–52. doi: 10.1136/jmedgenet-2015-103428. [DOI] [PubMed] [Google Scholar]
- 5.van Rossum G, de Boer J. Interactively testing remote servers using the python programming language. CWI Q. 1991;4:283–303. [Google Scholar]
- 6.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.James C. PyVCF. https://pyvcf.readthedocs.io (2012). Accessed 28 Jul 2017.
- 8.Kirill S. PyYAML. http://pyyaml.org (2006). Accessed 28 Jul 2017.
- 9.Samorodnitsky E, Jewell BM, Hagopian R, Miya J, Wing MR, Lyon E, Damodaran S, Bhatt D, Reeser JW, Datta J, Roychowdhury S. Evaluation of hybridization capture versus amplicon-based methods for whole-exome sequencing. Hum Mutat. 2015;36:903–914. doi: 10.1002/humu.22825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium: clinical implications of multicentric origin. Cancer. 1953;6:963–968. doi: 10.1002/1097-0142(195309)6:5<963::AID-CNCR2820060515>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
pyAmpli variant filter decision diagram. (DOCX 209 kb)
Sanger sequencing variant validation. (DOCX 71 kb)
pyAmpli CPU runtime. (DOCX 132 kb)
Data Availability Statement
The pyAmpli package is available under the GPL-v3 license from https://mbeyens.github.io/pyAmpli. The repository provides also quick-start examples and command-line scripts for easy testing and performing essential processing.