

Abstract
In response to varying stress signals, the p53 tumor suppressor is able to promote repair, survival, or elimination of damaged cells – processes that have great relevance to organismal aging. Although the link between p53 and cancer is well established, the contribution of p53 to the aging process is less clear. Delineating how p53 regulates distinct aging hallmarks such as cellular senescence, genomic instability, mitochondrial dysfunction, and altered metabolic pathways will be critical. Mouse models have further revealed the centrality and complexity of the p53 network in aging processes. While naturally aged mice have linked longevity with declining p53 function, some accelerated aging mice present with chronic p53 activation, whose phenotypes can be rescued upon p53 deficiency. Further, direct modulation of the p53-MDM2 axis has correlated elevated p53 activity with either early aging or with delayed-onset aging. We speculate that p53-mediated aging phenotypes in these mice must have (1) stably active p53 due to MDM2 dysregulation or chronic stress or (2) shifted p53 outcomes. Pinpointing which p53 stressors, modifications, and outcomes drive aging processes will provide further insights into our understanding of the human aging process and could have implications for both cancer and aging therapeutics.
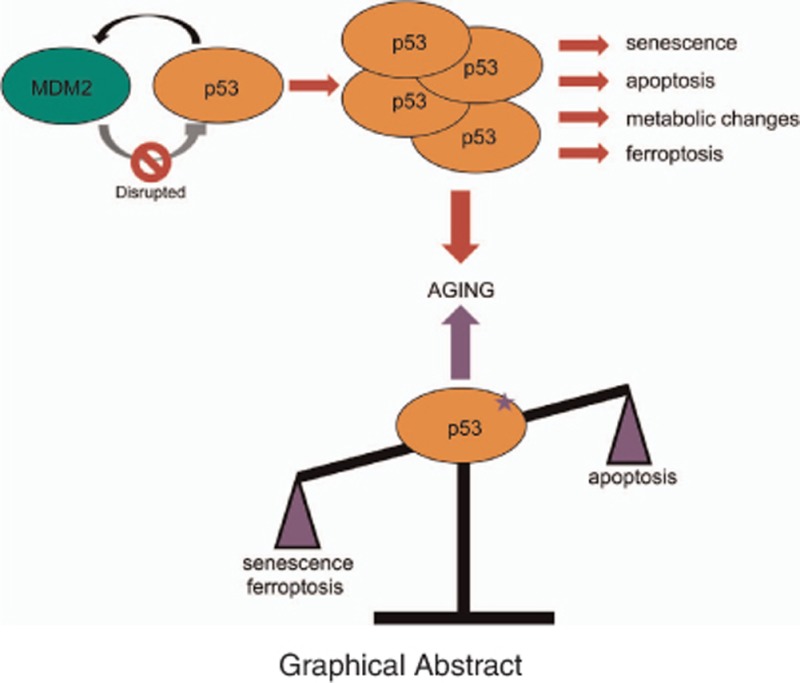
Facts
Proper regulation of p53 is essential for normal development and aging processes.
Cancer and aging are distinct manifestations of the same underlying cause of cumulative genomic changes.
Genomic damage and stress modulate p53 activity differentially.
Senescence has a critical role in the aging process.
Open Questions
Is p53 signaling a driver or downstream effector of aging?
Which stressors and p53 modifications are responsible for aging phenotypes?
Can aging effects be reversed and fitness restored at the cellular level?
Do we need to reconsider the side effects of p53-related therapies?
Aging is the gradual process of cellular deterioration due to cumulative molecular changes over time. At the organismal level, the aging phenomenon is often characterized by outward phenotypes such as shortened stature, decreased lifespan, frailty, skin abnormalities, and graying of the hair.1, 2 The underlying molecular mechanisms of aging feature the depletion of healthy, functional cells leading to tissue and organismal degeneration.3 This cellular turnover is often attributed to stem cell exhaustion and cellular senescence. Cells undergo senescence upon telomere shortening (replicative senescence) or exposure to genomic damage.4, 5 Instead of continually dividing, senescent cells adopt more flattened morphologies, exhibit declined stress response signaling, and lose homeostatic balance.6 Over time, the number of senescent cells in an organism can accumulate and develop a senescence-associated secretory phenotype (SASP), producing a chronic inflammatory microenvironment that can accelerate the aging process.7, 8
Cumulative genomic damage generates chronic stress that can activate stress response pathways. The p53 pathway is one such pathway that strives to maintain genomic integrity and cellular homeostasis by initiating tumor-suppressing cell survival processes such as cell cycle arrest, senescence, and DNA damage repair (Figure 1).5 p53 can also promote apoptosis and ferroptosis to eliminate damaged cells. p53 functions as a sequence-specific transcriptional regulator of myriad target genes, some of which are regulated by basal levels of p53, while others require increased levels of p53 most often elicited by numerous endogenous and exogenous stressors.5 The most well-validated negative regulator of p53 is MDM2.9, 10 MDM2, an E3 ubiquitin ligase, is able to downregulate p53 activity through three mechanisms: (1) poly-ubiquitylation to target p53 for proteasome-mediated degradation; (2) mono-ubiquitylation to export p53 out of the nucleus; and (3) direct binding to p53 to block transactivation of key targets.11 Though a broad link between the p53–MDM2 axis and aging has been established, the mechanism has not yet been elucidated. In this review, we aim to address the connections between p53–MDM2 axis and human aging disorders, aging-related pathways, aging mouse models, and therapeutic implications.
Figure 1.

Stress levels regulate alternate p53 responses. The tumor suppressor p53 mediates both survival and killer cellular outcomes in response to stressors. Under low stress conditions, p53 will initiate repair and cell cycle arrest mechanisms that will promote cell survival. Under acute stress conditions, p53 will eliminate the damaged cells from the proliferative pool through apoptosis, senescence, and more
Aging and Longevity
The study of age-related diseases and accelerated aging disorders (progeroid syndromes) are not only valuable in furthering our understanding of specific pathologies but also for understanding the broader aging phenomenon. In contrast to cancer in which cells gain fitness to proliferate rapidly and uncontrollably, aging features cells that lose fitness.3, 12 How cells respond to DNA damage is usually the determining factor between these two choices. The master regulator p53 determines cellular response to stress and has been linked to human diseases from development through old age.13 Wild-type p53 function is required for tumor suppression in humans, as evidenced by Li-Fraumeni patients who have germline p53 mutations leading to increased cancer incidence.14 Cells derived from aged individuals, such as astrocytes, fibroblasts, and retinal pigment epithelial cells, express increased levels of p53 protein,15, 16, 17 despite the intuitive expectation that aged populations may have reduced p53 activity leading to increased tumor incidence. In line with this, individuals with the p53 polymorphism Arg72Pro have increased longevity, though this p53 has reduced apoptotic potential. Dysregulated p53 has also been implicated in neurodegenerative disorders – p53 is upregulated in Alzheimer’s disease and is able to interact with mutant huntingtin protein to mediate aberrant transcriptional regulation in Huntington’s disease.18, 19, 20 Altogether, the question of whether p53 aids or negates the aging process in humans remains complex, as p53 activity has been associated with both premature aging and longevity.
Progeroid syndromes are often characterized by early onset and rapid progression, with patients displaying accelerated aging phenotypes and shorter lifespans.21 Although progeroid syndromes only partially phenocopy normal aging, their study provides insights into the mechanisms underlying aging. In many cases, progeroid syndromes are monogenic and driven by defects in DNA repair mechanisms. For example, aberrant nucleotide excision repair has been associated with cerebro-oculo-facio-skeletal syndrome, trichothiodystrophy, and Cockayne syndrome.22 In addition, deficiencies in non-homologous end joining (NHEJ) have been associated with Werner’s syndrome and ataxia telangiectasia.23 Still other progeroid syndromes such as laminopathies feature disruption of the nuclear architecture via lamin processing, leading to chronic stress and activation of the p53 pathway.21, 24, 25 Recently, we reported studies on a patient with accelerated aging symptoms who harbors a homozygous anti-termination mutation in MDM2 that leads to aberrant regulation of p53.26 Our data indicate that a functional disturbance of the p53–MDM2 axis can contribute to human aging phenotypes.
Findings in model organisms have further connected p53 with aging, although some appear to contradict progeroid studies. For example, naturally aged mice experience a decline in p53 function at the transcriptional and protein levels, suggesting that their tumor suppressive functions may be compromised.27 In addition, model systems such as C. elegans and Drosophila associate loss of wild-type p53 with longevity and extended lifespan, and have implicated p53 activity in autophagy and crosstalk with pathways like the insulin-like growth factor/mTOR pathway.28, 29, 30, 31
p53 and Aging at the Cellular Level
In their landmark review, Lopez-Otin et al.12 enumerated nine key hallmarks of aging. Notably, the p53 pathway is a master regulator of at least three of these hallmarks: genomic instability; mitochondrial dysfunction; and cellular senescence. p53 is also a central player in regulating pathways involved in metabolic signaling and the immune system, which could contribute to regulating aging mechanisms.
Genome instability
As genomic damage accumulates over time, genomic instability becomes more prevalent. One of the main functions of p53 is to maintain genomic integrity through repair and cell cycle arrest mechanisms to limit proliferation of damaged cells. This potentially explains why many progeroid syndromes with defective repair DNA mechanisms exhibit genomic instability and subsequent elevated p53 activity.32 Activation of certain p53 target genes is stress- and tissue-dependent and will lead to manifestation of differential phenotypes in mice.33, 34
Mitochondrial dysfunction
Mitochondria are cellular energy generators that require regular maintenance through mitochondrial DNA (mtDNA) repair and mitophagy, an organelle-specific clearance of damaged mitochondria.35 As organisms age, mitochondrial dysfunction increases and respiration efficiency decreases.36 Thus, defects in proofreading mtDNA enzymes have led to accelerated aging phenotypes in mice.37 Mitochondrial deterioration can also be partially attributed to increased production of reactive oxygen species (ROS). However, simple elevation of ROS is insufficient to accelerate aging, and has in fact been associated with prolonged lifespan in yeast and C. elegans.38, 39, 40 p53 is a key regulator of ROS and is also linked to mitochondrial regulation. For example, the histone deacetylase SIRT1 is involved in mitochondrial biogenesis through cooperation with the transcriptional co-activators PGC-1α and PBC-1β, but can also negatively modulate p53 activity.41, 42 In response to telomere attrition, p53 is able to mediate repression of PGC-1α and PGC1-β, thereby inhibiting mitochondrial biogenesis and function, leading to excess ROS, which can further activate p53 in a feedback loop.43 Therefore, p53 can be both an upstream mediator and downstream effector of ROS.
The different modes by which the p53 transcriptional program can regulate metabolism and mitochondrial health can be paradoxical such that p53 can induce opposing cellular outcomes. In some cases, this relates to the type or extent of the cellular stress. Normal basal activity of p53 has been implicated in optimizing mitochondrial respiration and reducing ROS through SCO2, TIGAR, and sestrins.44, 45 Upon conditions of mild stress, p53 is able to induce expression of antioxidant genes such as sestrins, TIGAR, GLS2, SCO2, and others.45 However, upon acute stress p53 can activate pro-oxidant targets to mediate apoptosis and senescence.46 It is likely that collective mitochondrial dysfunction due to increased damage and reduced turnover could affect cell signaling, autophagy, and aging.
Crosstalk with the insulin/IGF-1 and mTOR pathways further complicates the p53-metabolism link.47 Diminished signaling of the insulin/IGF-1 and mTOR pathways has been linked with longevity.48, 49 p53 is able to activate expression of IGF-BP3 and PTEN to downregulate insulin/IGF-1 signaling, as well as TSC-2, AMPK-β1, sestrins, and REDD1 to downregulate mTOR signaling.46 Moreover, insulin/IGF-1 signaling through IRS-1 can activate PI3K/AKT, which in turn can activate MDM2. MDM2 is able to directly bind to IRS-1 and IGF-1R to target these substrates for degradation.50, 51 Thus, the interplay of p53 and MDM2 with these metabolic pathways are frequently context-dependent and highlight the complexity of p53 in metabolic regulation.
Cellular senescence
Perhaps the most commonly associated phenotype of aging is senescence, a process by which cells undergo a stable, irreversible cell cycle arrest.52 Although primarily triggered by telomere attrition, exposure to physiological stresses can also lead to senescence. Two key pathways have been linked to senescence: p16INK4a/RB and p53/p21. p16INK4a is able to block cell cycle progression by inhibiting cyclin D–CDK4/6 complexes, thereby activating Rb to inhibit E2F transcriptional effects.53 p53 is able to directly activate p2154, 55, 56 and PAI-157 to mediate cell cycle arrest and senescence. The accumulation of senescent cells, combined with reduced healthy cell turnover and slowed division, is largely thought to drive aging, while clearance of senescent cells has been associated with delayed aging.58
Accumulation of senescent cells often leads to chromatin reorganization and the SASP, which includes secretion of pro-inflammatory chemokines, including cytokines (e.g., interleukin-6 and tumor necrosis factor), growth factors, matrix metalloproteases, and others.3 SASP can also lead to paracrine signaling to induce further cellular senescence, extracellular matrix deterioration, and disruption of the local microenvironment.59, 60 Although SASP-derived inflammation can attract an immune response and subsequent clearance of senescent scells,61, 62 chronic SASP can also lead to immune evasion through microRNAs and other mechanisms, which may lead to limited clearance and further propagation of senescence and aging phenotypes.63, 64
The role of p53 in immune regulation, particularly in inflammation and immunosurveillance, may also reinforce SASP and aging processes.65, 66 In fact, p53 loss has been shown to accelerate the aging of the immune system through accumulation of memory T cells, enhanced cytokine production, and limited T-cell proliferation capacity.67 Wild-type p53 can upregulate targets such as Toll-like receptors, cytokines, and natural killer cell ligands (ULBP1 and ULBP2) that mediate innate immune responses, inflammation, and apoptosis.68, 69 p53 can also induce expression of CCL2 and DD1α to attract natural killer cells and macrophages for senescent and apoptotic cell clearance, respectively.66, 70, 71, 72 In addition, p53 can downregulate PDL1 expression via activation of miR-34, thereby effectively inhibiting the PD1 checkpoint.73 Nevertheless, though p53 activity can engage the innate immune system to facilitate cellular clearance, p53 could potentially promote an inflammatory microenvironment that could aide chronic SASP to drive further tissue deterioration. Therefore, the role of p53 in senescence and immune regulation could be contributing to aging processes.
Aging Mouse Models
Premature aging models
Mouse models are useful in understanding human physiological systems, particularly as genome maintenance genes are conserved across species. Several mouse models have been developed to model progeroid syndromes, including Hutchinson–Gilford progeria syndrome (HGPS) and TTD (Table 1). HGPS is driven primarily by lamin A-processing defects that lead to chronic nuclear architectural stress. Zmpste24−/− mice that lack a protease that is required for lamin A processing exhibit HGPS symptoms, upregulated p53 target gene expression, and premature senescence, though basal levels of p53 remain unchanged.25 The Zmpste24−/− aging phenotype is partially rescued by crossing mice into a p53-null background, indicating that p53 is a key driver of the phenotype. Similarly, elimination of p53 or p16INK4A to reduce senescence can rescue the aging phenotype in mice with defects in double strand break repair and spindle assembly checkpoints caused by mutations in Brca1 and Bub1b, respectively.58, 74 Ku86−/− mice with defective double-strand break repair also present with aging-related characteristics, including dermal atrophy, hair follicle, and early senescence.75 However, when generated in a p53-deficient background, Ku80−/− mice no longer display early replicative senescence that is p53-dependent, but the mice do experience shortened lifespans due to enhanced tumor incidence.76 Similarly, SnoNm/m mice that can no longer antagonize TGFβ signaling display accelerated aging phenotypes that can be partially rescued by p53 deficiency.77 Conversely, unlike other accelerated aging models that are rescued upon p53 knockout, ATR-Seckel mice display exacerbated phenotypes when p53 is lost.78 This could potentially be due to p53 loss overwhelming the mouse with additional genomic instability, as AtrS/S mice already exhibit high levels of embryonic replicative stress and shortened lifespans.
Table 1. Premature aging mouse model.
| Alteration | Disease model | Phenotype | Reference |
|---|---|---|---|
| Lmna−/− | HGPS Defect in lamin processing | Accelerated aging Increased p53 activity, elevated p21 levels | Sullivan et al.132 |
| Zmpste24−/− | HGPS Defect in lamin processing | Accelerated aging Phenotype can be partially rescued by p53-null background p53 target genes upregulated despite normal p53 protein levels Premature senescence | Varela et al.25 |
| AtrS/S | ATR Seckel Defect in single-stranded break repair | Accelerated aging Phenotype aggravated by p53 loss High levels of embryonic replicative stress, genomic instability | Murga et al.78 |
| XpdR722W | Trichothiodystrophy (TTD) Defect in NER | Accelerated aging Osteoporosis, early graying, infertility Defect in additional Xpa gene accelerates phenotype | de Boer et al.129 |
| Csb−/− | Cockayne’s syndrome Defect in NER | Normal aging UV-sensitive | Lu et al.133 |
| Ercc1*292 | Defect in NER | Accelerated aging Reduced lifespan, reduced growth, increased senescence, nuclear abnormalities, accumulation of p53 and p21 | Weeda et al.134 |
| Wrn−/− | Werner’s syndrome Defect in double-stranded break repair | Normal aging Moderately accelerated senescence Mortality accelerated in p53−/− background | Lebel et al.135 Lombard et al.136 |
| Ku86−/− | Defect in NHEJ | Accelerated aging Early onset senescence, earlier tumorigenesis, hair graying, skin atrophy | Vogel et al.75 |
| Xrcc4−/− | Defect NHEJ | Embryonic lethal Lethality rescued in p53-null background | Gao et al.103 |
| BrcaΔ11/Δ11; p53+/− | Defect in double-strand break repair and MMR | Accelerated aging Skeletal abnormalities, reduced dermal thickness MEFs exhibit senescence, mediated by p53–p21 axis | Cao et al.74 |
| Bub1bH/H | Defect in spindle assembly checkpoint protein | Accelerated aging Loss of fat, reduced wound healing, cataracts Early onset senescence; accumulation of p53/p21/p16/p19 | Baker et al.137 |
| SnoNm/m | Block of antagonist of TGF-β signaling | Accelerated aging Shortened lifespan, osteoperosis, premature senescence, p53 cannot be regulated by Mdm2 | Pan et al.77 |
| mTR−/− | Defect in telomerase | Accelerated aging Decreased lifespan, infertility at G6, increased genomic instability Increased senescence | Blasco et al.79 Rudolph et al.80 Chin et al.81 |
| Sp53/Sp16/SArf/TgTert | Overexpress telomerase Super-p53/p16/ARF | Delayed-onset aging Improved intestinal and skin fitness, increased lifespan | Tomas-Loba et al.83 |
List of accelerated aging mouse models associated with defects in lamin processing, ATR signaling, genome maintenance, and telomerase.
Mouse models focusing on the role of telomere shortening have also directly connected senescence to aging. When the RNA component of telomerase (mTR) is knocked out, the resulting mTR−/− mice have decreased lifespans, shortened telomere lengths, increased senescence, and infertility by the sixth generation.79, 80 In addition, mTR−/− mice have more skin lesions, delayed wound healing, and increased genomic instability. Moreover, late-generation mTR−/− mice have greater induction of p53 and p21, and experience more extensive apoptosis in certain tissues. This phenotype can be rescued by p53 loss, despite the telomere dysfunction.81 However, another mouse model featuring telomere dysfunction demonstrates that conditional p53 deletion is unable to rescue the aging phenotype, but rather inhibits the depletion of chromosomally unstable stem cells.82 On the opposite end of the spectrum, constitutive expression of telomerase reverse transcriptase (TERT) in mice with overexpressed p53/p16INK4A6/p19ARF improves fitness and delays aging.83 Altogether, these mouse models indicate that p53 has somewhat conflicting roles in aging and is context-dependent.
Mdm2–p53 mouse models
Mouse models manipulating the p53 axis directly have also provided insight into how p53 is linked to aging (Table 2). Mounting evidence suggest that simple elevation of p53 levels is insufficient to drive the accelerated aging phenotype in mice. Mice expressing ‘super-p53’ via introduction of multiple transgenes of wild-type p53 are highly cancer-resistant, but do not display signs of early aging.84 Furthermore, ‘super-ARF/super-p53’ mice exhibit delayed-onset aging.85 Mdm2puro/Δ7–12 mice modulate p53 levels through Cre-mediated Mdm2 ablation.86 Despite exhibiting 70% ablation of wild-type MDM2 protein, cells from Mdm2puro/Δ7–12 mice express normal basal p53 protein levels, though they have heightened p53 activity that is correlated with increased tumor resistance. Although these Mdm2puro/Δ7–12 mice are about 15% smaller in size, radiosensitive, and display hematopoietic deficiencies, the mice do not age prematurely. Combined, these studies indicate that total p53 levels are not directly responsible for aging phenotypes, rather, specific functions of p53 may be contributing factors. In addition, mice that have genetic alterations shifting the balance of p53-dependent cellular outcomes (senescence, ferroptosis, apoptosis, etc.) also appear to display accelerated aging phenotypes. Therefore, we propose that two factors are crucial to driving p53-dependent aging phenotypes: (1) aberrant p53 regulation leading to increased p53 stability; or (2) shifted p53-dependent cellular outcomes (Figure 2).
Table 2. Mdm2/p53 mouse models.
| Mouse model | p53 activity | Phenotype | Reference |
|---|---|---|---|
| p53+/m (Δexons 1–6) | Normal basal p53 levels Hyper-stable p53, robust p21 activation | Accelerated aging More tumor resistant | Tyner et al.87 |
| p44 TG | Normal basal p53 levels, elevated p44 levels Elevated p21, Mdm2, IGFBP-3 | Accelerated aging More tumor resistant Hyperactive IGF-1 signaling | Maier et al.88 |
| p53TSD/− (T21D/S23D) | Normal basal p53 levels Hyper-stable p53, constitutive p53 activation | Accelerated aging Phenotype rescued by Puma knockout | Liu et al.93 |
| p53S18A | Normal basal p53 levels Defective Puma induction and apoptosis | Accelerated aging in non-tumor-bearing mice Premature senescence | Armata et al.101 |
| Mdm2Δ/Δ (conditional mouse) | Increased p53 levels Increased expression of p53 senescence targets | Accelerated aging Epidermal senescence | Gannon et al.97 |
| Super p53 (+1 p53 transgene) | Normal basal p53 levels Increased DNA damage response Increased apoptotic response | Normal aging More tumor resistant | Garcia-Cao et al.84 |
| Super-Arf/p53 (+1 ARF, +1 p53 transgenes) | Normal basal p53 levels Increased expression of Sesn1/2 | Delayed onset aging More tumor resistant Protects against insulin resistance/glucose intolerance | Matheu et al.85 |
| Xrcc4−/−; p53S18/S23A | Normal basal p53 levels Resistant to apoptosis | Accelerated aging Not as tumor prone as Xrcc4-/- p53-/- mice | Chao et al.102 |
| Xrcc4−/−; p533KR3KR | Normal basal p53 levels p53 target genes: high Ptgs2, low SLC7A11 | Accelerated aging More tumor resistant High genomic instability, ferroptosis | Li et al.104 |
| Mdm2−/− | Unrestricted p53 activity by Mdm2 | Embryonic lethal Lethality rescued in p53 null background | Jones et al.94 Montes de Oca Luna et al.95 |
| Mdm2C462A (disrupted RING domain of MDM2) | p53 cannot be degraded | Embryonic lethal Lethality rescued in p53 null background MDM2 has no E3 ligase activity | Itahana et al.96 |
| Mdm2Y487A | Increased basal p53 levels Low p53 transcriptional activity unless stressed | Normal aging MDM2 has decreased E3 ubiquitin ligase activity, can still bind to MDMX | Tollini et al.138 |
| Mdm2puro/Δ7-12 (Mdm2 hypomorph) | Normal basal p53 levels More radiosensitive Increased p53-mediated apoptotic response | Normal aging Smaller mice | Mendrysa et al.86 |
| Mdm2p2/p2 (mutated p2 promoter) | Normal basal p53 levels More radiosensitive | Normal aging | Pant et al.100 |
| Mdm2PND/PND (mutant P2 promoter, neomycin gene, 184 bp deletion in intron 3) | Increased basal p53 levels | Accelerated aging Phenotype rescued in p53 null background Partial rescue with Puma deletion | Pant et al.33 |
List of aging-related Mdm2–p53 axis mouse models and their associated p53 activities and phenotypes.
Figure 2.

p53-mediated accelerated aging requires stable p53 or differential p53 outcomes. Speculation as to how p53 regulation is linked to aging in mouse models. When p53 is properly regulated, it leads to normal aging phenotypes in mice. However, constitutive p53 activity due to aberrant regulation through deletion or modifications of the N terminus of p53 (the primary interaction site with Mdm2) leads to accelerated aging phenotypes. Mice with defects in double-strand break repair and HGPS mouse models that display chronic nuclear structural stress lead to chronic p53 activation and accelerated aging phenotypes. Engineered mice that lack the entire spectrum of p53 outcomes but can promote differential outcomes (apoptosis, ferroptosis, and senescence) also display accelerated aging phenotypes. This figure is adapted from Serrano and Blasco130
Mouse models with aberrant p53 regulation
Two studies featuring mice with one functional copy of wild-type p53 and one copy of an N-terminal truncation of p53 display accelerated aging phenotypes.87, 88 The site of the p53 alteration is revealing, as the N terminus of p53 harbors the primary interaction site with MDM2, suggesting that MDM2 is unable to properly regulate p53 in these mice. The Donehower group, generated mice (p53+/m) with a heterozygous p53 mutation that deleted the first six exons of p53, leading to hyperactive p53.87 Although basal p53 protein levels were similar at least in p53+/m and p53+/+ kidney cells, the p53 response in these p53+/m mice were augmented. The p53+/m mice were also highly resistant to cancer, with a sporadic tumor formation of 6% compared to 45% for p53+/+ mice. Most notably, the p53+/m mice displayed an unexpected phenotype of accelerated aging, with skeletal defects and organ atrophy. A follow-up study indicated that the m allele produced a p53 protein that was able to interact with, stabilize, and facilitate the nuclear localization of wild-type p53 under basal conditions.89 A second aging mouse model (p44 TG) from the Scrable group demonstrated that overexpression of a naturally occurring p53 isoform, p44, in the presence of full-length p53 also leads to an accelerated aging phenotype.88 The p44 protein lacks the N-terminal transactivation domain of p53, as translation is initiated from codon 41 in p53. Interestingly, MEFs from both p53+/m and p44 TG mice express normal p53 protein levels but have constitutively active p53, likely stemming from aberrant regulation of p53 by MDM2 due to the N-terminal truncation. An important clue as to the aging phenotype of the p44 mouse was derived from the finding that these mice have deregulated insulin/insulin-like growth factor signaling. Specifically, cells from these mice displayed increased expression of some p53 targets (p21, MDM2, and IGFBP-3) while others (GADD45 and IGF-1-R) were relatively decreased. These observations imply that mice with one N-terminally truncated p53 allele along with one full-length p53 allele display altered, but not across-the-board elevated p53 activity in their cells.
Not only gross deletions, but missense mutations of modification sites within p53 can also predispose mice to premature aging in some cases. Phosphorylation of the N terminus of p53 at key residues (S15, T18, and S20) disrupts the MDM2–p53 interaction and can also lead to aberrant p53 regulation.90, 91, 92 p53TSD/– mice with phosphorylation-mimicking mutations of human T18 and S20 (in mice: T21 and S23, respectively) indicate that this region is critical for mouse development.93 While homozygous p53 T21D/S23D mice (p53TSD/TSD) mice are embryonic lethal, p53TSD/– knock-in mice are born and die by 6 weeks of age, exhibiting accelerated aging phenotypes.93 Constitutive p53 activation, hematopoietic degeneration, testicular atrophy, and adult stem cell depletion were observed in these heterozygous mice. Elimination of the p53 target Puma is able to rescue the segmental progeroid phenotype in p53TSD/− mice, indicating that the aging phenotype in these mice is primarily mediated through p53-dependent apoptosis of the adult stem cell pool.
Direct manipulation of MDM2 adds an additional layer of complexity to understanding the p53 pathway connection to aging (Figure 3). Complete liberation of p53 from the repressive hold of MDM2 leads to massive apoptosis and developmental deficiencies. MDM2 knockout (Mdm2−/−) mice are embryonic lethal unless generated in a p53-null background, indicating that p53 regulation by MDM2 is essential for development.94, 95 Concordantly, engineered mice that express a key MDM2 RING domain mutation that abrogates E3 ligase activity (Mdm2C462A) display an early embryonic lethal phenotype unless present in a p53-null background.94, 95, 96 However, in a conditional mouse model, deletion of Mdm2 in the epidermis induced p53-mediated senescence and accelerated aging phenotypes in the skin, including decreased wound-healing ability and skin thinning.97 Notably, p53 activation in the epidermis did not lead to upregulation of pro-apoptotic genes, just senescent targets. As conditional loss of Mdm2 in several other tissues leads to cell death,98, 99 it is likely that tissue-specific differences account for the senescence outcome observed.
Figure 3.

Mdm2–p53 balance is required for normal development. Proper regulation (balance) of p53 by MDM2 is needed for normal aging. Aberrant regulation can lead to tumorigenic, accelerated aging, or lethal phenotypes. Figure is taken and adapted from Poyurovsky and Prives131
Mouse models with differential p53 outcomes
Activation of specific programs of p53 downstream targets to shift the balance of p53-dependent outcomes could manifest in different phenotypes. Indeed, the p53TSD/– accelerated aging phenotype observed is dependent on apoptosis. Nevertheless, altering the transcriptional program of p53 can have unanticipated effects. Engineered homozygous mice that lack the Mdm2 p2 promoter (Mdm2P2/P2), which abolishes Mdm2 as a p53 transcriptional target, age normally, indicating that full activation of Mdm2 by p53 is not necessary for aging.100 However, homozygous Mdm2PND/PND mice that were serendipitously generated from a mutant P2 promoter harboring a neomycin cassette and partial intron 3 deletion display certain aging-related characteristics, including shorter lifespans, skin hyperpigmentation, and hematopoietic defects.33, 100 Interestingly, Puma deficiency rescued some of the reproductive defects, but p21 loss did not rescue any aging-related abnormalities, indicating that the apoptotic pathway was likely responsible for this particular phenotype as well.
Shifting the p53-dependent response away from apoptosis can also lead to accelerated aging. Knock-in mice p53 mice with an S18A mutation (p53S18A) that removes the ATM phosphorylation site that is necessary for Puma-mediated apoptosis have a high cancer incidence. Surprisingly, the non-tumor-bearing mice from this study presented with accelerated aging signs. Cells from the non-tumor-bearing p53S18A mice have diminished apoptotic responses and undergo premature senescence.101 Speculatively, epigenetic variations in the mice may contribute to the differences seen. Similar to the p53S18A mice, p53S18A/S23A mice are also apoptosis-resistant, and thus highly tumorigenic.102 However, in another model, the p53S18A/S23A background can rescue the lethality of XRCC4−/− mice, and the crossed mice (Xrcc4−/−;p53S18A/S23A) display accelerated aging phenotypes.102 XRCC4 is a protein involved in the stabilization of a key ligase in the NHEJ pathway that repairs double-strand breaks. XRCC4 knockout mice are embryonic lethal, unless rescued via co-committal knockout of p53.103 These models suggest that the apoptotic functions of p53 are not required for genomic instability-driven aging phenotypes. This idea is further supported by recent evidence demonstrating that Xrcc4−/− lethality can also be rescued in a p533KR/3KR background in mice (Xrcc4−/−;p533KR/3KR).104 p53 3KR is an acetylation-defective mutant that is unable to induce cell cycle arrest, senescence, and apoptosis, but can still undergo ferroptosis, an iron-dependent form of cell death that results from lipid peroxidation.105 The authors speculate that the high levels of ferroptosis could be a contributing factor to the aging process.
Altogether, these p53 mouse models implicate a significant role for p53 in accelerated aging processes. However, as mentioned before, simple excess of p53 is insufficient, perhaps because the MDM2–p53 circuitry is intact. Instead, p53 stability may be the critical factor in the lifespan of these mice (Figure 4). We speculate that for p53-mediated aging, p53 must (1) be stably active due to MDM2 dysregulation or chronic stress or (2) shift the balance of its transcriptional program to facilitate specific outcomes. These stress- and modification-dependent responses must vary across tissue types and could account for variable accelerated aging abnormalities. There are also conflicting lines of evidence relating apoptosis to aging phenotypes. Though depletion of stem progenitor cells could be a driver, experimental contexts must be carefully considered to gain further understanding of the roles of programmed cell death in aging. Finally, we propose that the lack of exposure to stressors is another factor that needs to be considered when analyzing these mouse models. Specifically, mice living under laboratory conditions are not exposed to everyday physiological stresses such as UV radiation. Super-p53 and super-ARF/p53 mice have augmented DNA damage responses, which if chronically stimulated, could speculatively lead to altered aging-related phenotypes.
Figure 4.

Lifespan and cancer resistance are related to p53 stability. Model for proposed relationship between lifespan and cancer resistance and their links to p53 protein stability. Mice with low p53 or no p53 due to deletion of one or two TP53 alleles are tumor prone and have shortened lifespans due to cancer. Mice with normal p53 levels or stability have normal lifespans, while super-p53/super-ARF mice have delayed-onset aging. Mice with increased p53 stability can lead to hyper-activation and accelerated aging phenotypes (dip in lifespan), but greater cancer resistance. Finally, mice with likely super-stable p53 due to Mdm2 or MdmX deletion leads to embryonic lethal phenotypes. Dotted line refers to inability to assess cancer resistance due to embryonic lethality. Lifespan and cancer resistance curves are diagrammatic rather than quantitative in terms of actual lifespan and resistance of mice.
Therapeutic Implications
Side effects of cancer therapeutics?
The MDM2–p53 axis presents an attractive target for cancer therapeutics, particularly those that wish to restore wild-type p53 function.9 Stapled peptides, small-molecule inhibitors, and p53 vaccines have all undergone clinical trials to test the efficacy of targeting this pathway. Compounds such as Nutlin-3 and RITA activate p53 by disrupting the MDM2–p53 interaction and can induce p53-mediated cell cycle arrest and senescence in some cases.106, 107, 108, 109 However, while activation of p53 through pharmacological release from MDM2 has been actively sought for cancer therapy, there may be unanticipated aging consequences due to aberrant p53 activation and altered pathway signaling. Both Nutlin-3 and RITA may induce MDM2-mediated downregulation of IGF-1R signaling through ubiquitylation, which could have potential effects on regulation of the IGF-1 pathway and aging.110, 111 Furthermore, patients with MDM2 amplifications hyper-progressed upon immunotherapy treatment, suggesting that the p53–MDM2 axis could have a role in immune evasion and limit cellular clearance.112 As several mouse models have showed that non-aberrant regulation of p53 by MDM2 can restrain p53 activity and promote tumor resistance without the damaging lifespan effects113 we can remain cautiously optimistic about aging-related deleterious effects of short-term cancer therapies.
Aging therapeutics
Two general questions frame the search for aging therapeutics: can we curtail the aging process and can we reverse the effects of aging? Studies using caloric restriction and mTOR inhibitors have investigated how remodeling metabolic pathways can be used to extend longevity.114, 115 In addition, proof-of-principle studies have demonstrated that aging effects could be reversed in HGPS by a modified oligonucleotide that corrects the aberrant splice site in LMNA116 and by a rho-associated protein kinase inhibitor (Y-27362) that dampens ROS-associated mitochondrial dysfunction.117
Other studies have explored direct rejuvenation in mice. Specifically, researchers have attempted to determine whether ‘young environments’ can produce factors or stimuli that can restore youthfulness by using heterochronic parabioses to connect the circulatory systems of old and young mice.118 In particular, growth differentiation factor 11 (GDF11) expression was discovered to reverse skeletal and cardiac aging-related dysfunction in mice,119, 120, 121 though other studies imply that GDF11 inhibits muscular regeneration instead.122, 123 The discrepancies seem to stem from antibody reactivity, technical differences, and model organisms.124 Recently, an HGPS mouse model (Zmpste24−/−) was used to test in vivo GDF11 therapy, revealing that while circulating GDF11 declined in aged mice, daily GDF11 intraperitoneal injections were unable to prolong lifespans.125 However, combination therapy with GDF11 injection and p53 depletion could potentially be an avenue of exploration to attempt to fully rescue aging symptoms.
Clearance of senescent cells is another therapeutic approach to reverse aging.58 Multiple compounds have been identified to preferentially target senescence. For example, the compound KU-60019 was discovered as an effective inhibitor of ATM-driven senescence that yielded decreased levels of p53,126 while the small molecule ABT233 selectively eliminates senescent cells and leads to rejuvenated hematopoietic stem cells.127 More recently, a FOXO4–p53 interfering peptide was used to selectively clear senescent cells from an aged mouse.128 By perturbing the interaction between FOXO4 and p53, the peptide was able to induce p53 nuclear exclusion and p53-dependent apoptosis in senescent cells. Intriguingly, the peptide did not raise total p53 protein levels, but did induce accumulation of serine-15-phosphorylated p53. Along with diminished senescence, a reduction in p21 levels was observed. The resulting clearance of senescent cells led to restoration of fitness in both naturally aged and XPDTTD/TTD-accelerated aged mice, effectively demonstrating that p53 could be a promising target for aging therapeutics.128, 129
Taken together, aging mouse models have revealed the complexity of the p53–Mdm2 axis and have solidly placed the p53 network as being key to many aspects of both pathological aging conditions and normal aging. Further research is necessary to determine which p53 stressors, modifications, and outcomes drive aging processes. The p53–MDM2 axis poses new aging-related considerations for p53-activating cancer therapeutics, but has exciting implications for next-generation aging therapeutics.
Acknowledgments
This review was prepared under the aegis of a grant from the National Cancer Institute, CA58316.
Footnotes
The authors declare no conflict of interest.
References
- Kaeberlein M, Rabinovitch PS, Martin GM. Healthy aging: the ultimate preventative medicine. Science 2015; 350: 1191–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passarino G, Montesanto A, De Rango F, Garasto S, Berardelli M, Domma F et al. A cluster analysis to define human aging phenotypes. Biogerontology 2007; 8: 283–290. [DOI] [PubMed] [Google Scholar]
- Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol 2013; 75: 685–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell 2005; 120: 483–495. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell 2009; 137: 413–431. [DOI] [PubMed] [Google Scholar]
- Linskens MH, Harley CB, West MD, Campisi J, Hayflick L. Replicative senescence and cell death. Science 1995; 267: 17. [DOI] [PubMed] [Google Scholar]
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 2008; 6: 2853–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 2009; 11: 973–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer 2013; 13: 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karni-Schmidt O, Lokshin M, Prives C. The roles of MDM2 and MDMX in cancer. Annu Rev Pathol 2016; 11: 617–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade M, Wang YV, Wahl GM. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol 2010; 20: 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013; 153: 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyurovsky MV, Prives C. P53 and aging: a fresh look at an old paradigm. Aging (Albany NY) 2010; 2: 380–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkin D, Li FP, Strong LC, Fraumeni JF Jr., Nelson CE, Kim DH et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990; 250: 1233–1238. [DOI] [PubMed] [Google Scholar]
- Bitto A, Sell C, Crowe E, Lorenzini A, Malaguti M, Hrelia S et al. Stress-induced senescence in human and rodent astrocytes. Exp Cell Res 2010; 316: 2961–2968. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Chaum E, Johnson DA, Johnson LR. Age-related susceptibility to apoptosis in human retinal pigment epithelial cells is triggered by disruption of p53-Mdm2 association. Invest Ophthalmol Vis Sci 2012; 53: 8350–8366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulju KS, Lehman JM. Increased p53 protein associated with aging in human diploid fibroblasts. Exp Cell Res 1995; 217: 336–345. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H et al. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci USA 2000; 97: 6763–6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper C, Meimaridou E, Tavassoli M, Melino G, Lovestone S, Killick R. p53 is upregulated in Alzheimer's disease and induces tau phosphorylation in HEK293a cells. Neurosci Lett 2007; 418: 34–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Checler F, Alves da Costa C. p53 in neurodegenerative diseases and brain cancers. Pharmacol Ther 2014; 142: 99–113. [DOI] [PubMed] [Google Scholar]
- Navarro CL, Cau P, Levy N. Molecular bases of progeroid syndromes. Hum Mol Genet 2006; 2: R151–R161. [DOI] [PubMed] [Google Scholar]
- Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol 2014; 15: 465–481. [DOI] [PubMed] [Google Scholar]
- Hasty P, Campisi J, Hoeijmakers J, van Steeg H, Vijg J. Aging and genome maintenance: lessons from the mouse? Science 2003; 299: 1355–1359. [DOI] [PubMed] [Google Scholar]
- Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X et al. Genomic instability in laminopathy-based premature aging. Nat Med 2005; 11: 780–785. [DOI] [PubMed] [Google Scholar]
- Varela I, Cadinanos J, Pendas AM, Gutierrez-Fernandez A, Folgueras AR, Sanchez LM et al. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature 2005; 437: 564–568. [DOI] [PubMed] [Google Scholar]
- Lessel D, Wu D, Trujillo C, Ramezani T, Lessel I, Alwasiyah MK et al. Dysfunction of the MDM2/p53 axis is linked to premature aging. J Clin Invest 2017; 127: 3598–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Hu W, Teresky AK, Hernando E, Cordon-Cardo C, Levine AJ. Declining p53 function in the aging process: a possible mechanism for the increased tumor incidence in older populations. Proc Natl Acad Sci USA 2007; 104: 16633–16638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavernarakis N, Pasparaki A, Tasdemir E, Maiuri MC, Kroemer G. The effects of p53 on whole organism longevity are mediated by autophagy. Autophagy 2008; 4: 870–873. [DOI] [PubMed] [Google Scholar]
- Bauer JH, Chang C, Morris SN, Hozier S, Andersen S, Waitzman JS et al. Expression of dominant-negative Dmp53 in the adult fly brain inhibits insulin signaling. Proc Natl Acad Sci USA 2007; 104: 13355–13360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer JH, Poon PC, Glatt-Deeley H, Abrams JM, Helfand SL. Neuronal expression of p53 dominant-negative proteins in adult Drosophila melanogaster extends life span. Curr Biol 2005; 15: 2063–2068. [DOI] [PubMed] [Google Scholar]
- Arum O, Johnson TE. Reduced expression of the Caenorhabditis elegans p53 ortholog cep-1 results in increased longevity. J Gerontol A Biol Sci Med Sci 2007; 62: 951–959. [DOI] [PubMed] [Google Scholar]
- Reinhardt HC, Schumacher B. The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet 2012; 28: 128–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant V, Xiong S, Chau G, Tsai K, Shetty G, Lozano G. Distinct downstream targets manifest p53-dependent pathologies in mice. Oncogene 2016; 35: 5713–5721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn HF, Vousden KH. Coping with stress: multiple ways to activate p53. Oncogene 2007; 26: 1306–1316. [DOI] [PubMed] [Google Scholar]
- Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 2013; 20: 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005; 309: 481–484. [DOI] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004; 429: 417–423. [DOI] [PubMed] [Google Scholar]
- Doonan R, McElwee JJ, Matthijssens F, Walker GA, Houthoofd K, Back P et al. Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev 2008; 22: 3236–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee C, Yang W, Hekimi S. The intrinsic apoptosis pathway mediates the pro-longevity response to mitochondrial ROS in C. elegans. Cell 2014; 157: 897–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesquita A, Weinberger M, Silva A, Sampaio-Marques B, Almeida B, Leao C et al. Caloric restriction or catalase inactivation extends yeast chronological lifespan by inducing H2O2 and superoxide dismutase activity. Proc Natl Acad Sci USA 2010; 107: 15123–15128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001; 107: 137–148. [DOI] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001; 107: 149–159. [DOI] [PubMed] [Google Scholar]
- Sahin E, DePinho RA. Axis of ageing: telomeres, p53 and mitochondria. Nat Rev Mol Cell Biol 2012; 13: 397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budanov AV. The role of tumor suppressor p53 in the antioxidant defense and metabolism. Subcell Biochem 2014; 85: 337–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol 2015; 16: 393–405. [DOI] [PubMed] [Google Scholar]
- Feng Z, Lin M, Wu R. The regulation of aging and longevity: a new and complex role of p53. Genes Cancer 2011; 2: 443–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA 2005; 102: 8204–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluher M, Kahn BB, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 2003; 299: 572–574. [DOI] [PubMed] [Google Scholar]
- Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even PC et al. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003; 421: 182–187. [DOI] [PubMed] [Google Scholar]
- Girnita L, Girnita A, Larsson O. Mdm2-dependent ubiquitination and degradation of the insulin-like growth factor 1 receptor. Proc Natl Acad Sci USA 2003; 100: 8247–8252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui I, Imamura T, Huang J, Satoh H, Shenoy SK, Lefkowitz RJ et al. beta-arrestin-1 competitively inhibits insulin-induced ubiquitination and degradation of insulin receptor substrate 1. Mol Cell Biol 2004; 24: 8929–8937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufini A, Tucci P, Celardo I, Melino G. Senescence and aging: the critical roles of p53. Oncogene 2013; 32: 5129–5143. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell 2002; 2: 103–112. [DOI] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM et al. WAF1, a potential mediator of p53 tumor suppression. Cell 1993; 75: 817–825. [DOI] [PubMed] [Google Scholar]
- Dulic V, Kaufmann WK, Wilson SJ, Tlsty TD, Lees E, Harper JW et al. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell 1994; 76: 1013–1023. [DOI] [PubMed] [Google Scholar]
- Di Leonardo A, Linke SP, Clarkin K, Wahl GM. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev 1994; 8: 2540–2551. [DOI] [PubMed] [Google Scholar]
- Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol 2006; 8: 877–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011; 479: 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deursen JM. The role of senescent cells in ageing. Nature 2014; 509: 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 2013; 15: 978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011; 479: 547–551. [DOI] [PubMed] [Google Scholar]
- Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE et al. Non-cell-autonomous tumor suppression by p53. Cell 2013; 153: 449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaumik D, Scott GK, Schokrpur S, Patil CK, Orjalo AV, Rodier F et al. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging (Albany NY) 2009; 1: 402–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 2010; 5: 99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooks T, Harris CC, Oren M. Caught in the cross fire: p53 in inflammation. Carcinogenesis 2014; 35: 1680–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente R, Mausset-Bonnefont AL, Jorgensen C, Louis-Plence P, Brondello JM. Cellular senescence impact on immune cell fate and function. Aging Cell 2016; 15: 400–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkusu-Tsukada K, Tsukada T, Isobe K. Accelerated development and aging of the immune system in p53-deficient mice. J Immunol 1999; 163: 1966–1972. [PubMed] [Google Scholar]
- Munoz-Fontela C, Mandinova A, Aaronson SA, Lee SW. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat Rev Immunol 2016; 16: 741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez D, Shatz M, Resnick MA. Interactions between the tumor suppressor p53 and immune responses. Curr Opin Oncol 2013; 25: 85–92. [DOI] [PubMed] [Google Scholar]
- Iannello A, Thompson TW, Ardolino M, Lowe SW, Raulet DH. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J Exp Med 2013; 210: 2057–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L, Kroemer G. CANCER. A p53-regulated immune checkpoint relevant to cancer. Science 2015; 349: 476–477. [DOI] [PubMed] [Google Scholar]
- Yoon KW, Byun S, Kwon E, Hwang SY, Chu K, Hiraki M et al. Control of signaling-mediated clearance of apoptotic cells by the tumor suppressor p53. Science 2015; 349: 1261669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez MA, Ivan C, Valdecanas D, Wang X, Peltier HJ, Ye Y et al. PDL1 regulation by p53 via miR-34. J Natl Cancer Inst. 2016; 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Li W, Kim S, Brodie SG, Deng CX. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev 2003; 17: 201–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel H, Lim DS, Karsenty G, Finegold M, Hasty P. Deletion of Ku86 causes early onset of senescence in mice. Proc Natl Acad Sci USA 1999; 96: 10770–10775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim DS, Vogel H, Willerford DM, Sands AT, Platt KA, Hasty P. Analysis of ku80-mutant mice and cells with deficient levels of p53. Mol Cell Biol 2000; 20: 3772–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Zhu Q, Conboy MJ, Conboy IM, Luo K. SnoN activates p53 directly to regulate aging and tumorigenesis. Aging Cell 2012; 11: 902–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murga M, Bunting S, Montana MF, Soria R, Mulero F, Canamero M et al. A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat Genet 2009; 41: 891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA et al. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 1997; 91: 25–34. [DOI] [PubMed] [Google Scholar]
- Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C et al. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell 1999; 96: 701–712. [DOI] [PubMed] [Google Scholar]
- Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ et al. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 1999; 97: 527–538. [DOI] [PubMed] [Google Scholar]
- Begus-Nahrmann Y, Lechel A, Obenauf AC, Nalapareddy K, Peit E, Hoffmann E et al. p53 deletion impairs clearance of chromosomal-instable stem cells in aging telomere-dysfunctional mice. Nat Genet 2009; 41: 1138–1143. [DOI] [PubMed] [Google Scholar]
- Tomas-Loba A, Flores I, Fernandez-Marcos PJ, Cayuela ML, Maraver A, Tejera A et al. Telomerase reverse transcriptase delays aging in cancer-resistant mice. Cell 2008; 135: 609–622. [DOI] [PubMed] [Google Scholar]
- Garcia-Cao I, Garcia-Cao M, Martin-Caballero J, Criado LM, Klatt P, Flores JM et al. "Super p53" mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J 2002; 21: 6225–6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheu A, Maraver A, Klatt P, Flores I, Garcia-Cao I, Borras C et al. Delayed ageing through damage protection by the Arf/p53 pathway. Nature 2007; 448: 375–379. [DOI] [PubMed] [Google Scholar]
- Mendrysa SM, O'Leary KA, McElwee MK, Michalowski J, Eisenman RN, Powell DA et al. Tumor suppression and normal aging in mice with constitutively high p53 activity. Genes Dev 2006; 20: 16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H et al. p53 mutant mice that display early ageing-associated phenotypes. Nature 2002; 415: 45–53. [DOI] [PubMed] [Google Scholar]
- Maier B, Gluba W, Bernier B, Turner T, Mohammad K, Guise T et al. Modulation of mammalian life span by the short isoform of p53. Genes Dev 2004; 18: 306–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore L, Lu X, Ghebranious N, Tyner S, Donehower LA. Aging-associated truncated form of p53 interacts with wild-type p53 and alters p53 stability, localization, and activity. Mech Ageing Dev 2007; 128: 717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig AL, Burch L, Vojtesek B, Mikutowska J, Thompson A, Hupp TR. Novel phosphorylation sites of human tumour suppressor protein p53 at Ser20 and Thr18 that disrupt the binding of mdm2 (mouse double minute 2) protein are modified in human cancers. Biochem J 1999; 342(Pt 1): 133–141. [PMC free article] [PubMed] [Google Scholar]
- Meek DW. Regulation of the p53 response and its relationship to cancer. Biochem J 2015; 469: 325–346. [DOI] [PubMed] [Google Scholar]
- Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997; 91: 325–334. [DOI] [PubMed] [Google Scholar]
- Liu D, Ou L, Clemenson GD Jr., Chao C, Lutske ME, Zambetti GP et al. Puma is required for p53-induced depletion of adult stem cells. Nat Cell Biol 2010; 12: 993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995; 378: 206–208. [DOI] [PubMed] [Google Scholar]
- Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 1995; 378: 203–206. [DOI] [PubMed] [Google Scholar]
- Itahana K, Mao H, Jin A, Itahana Y, Clegg HV, Lindstrom MS et al. Targeted inactivation of Mdm2 RING finger E3 ubiquitin ligase activity in the mouse reveals mechanistic insights into p53 regulation. Cancer Cell 2007; 12: 355–366. [DOI] [PubMed] [Google Scholar]
- Gannon HS, Donehower LA, Lyle S, Jones SN. Mdm2-p53 signaling regulates epidermal stem cell senescence and premature aging phenotypes in mouse skin. Dev Biol 2011; 353: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boesten LS, Zadelaar SM, De Clercq S, Francoz S, van Nieuwkoop A, Biessen EA et al. Mdm2, but not Mdm4, protects terminally differentiated smooth muscle cells from p53-mediated caspase-3-independent cell death. Cell Death Differ 2006; 13: 2089–2098. [DOI] [PubMed] [Google Scholar]
- Ringshausen I, O'Shea CC, Finch AJ, Swigart LB, Evan GI. Mdm2 is critically and continuously required to suppress lethal p53 activity in vivo. Cancer Cell 2006; 10: 501–514. [DOI] [PubMed] [Google Scholar]
- Pant V, Xiong S, Jackson JG, Post SM, Abbas HA, Quintas-Cardama A et al. The p53-Mdm2 feedback loop protects against DNA damage by inhibiting p53 activity but is dispensable for p53 stability, development, and longevity. Genes Dev 2013; 27: 1857–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armata HL, Garlick DS, Sluss HK. The ataxia telangiectasia-mutated target site Ser18 is required for p53-mediated tumor suppression. Cancer Res 2007; 67: 11696–11703. [DOI] [PubMed] [Google Scholar]
- Chao C, Herr D, Chun J, Xu Y. Ser18 and 23 phosphorylation is required for p53-dependent apoptosis and tumor suppression. EMBO J 2006; 25: 2615–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Ferguson DO, Xie W, Manis JP, Sekiguchi J, Frank KM et al. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature 2000; 404: 897–900. [DOI] [PubMed] [Google Scholar]
- Li T, Liu X, Jiang L, Manfredi J, Zha S, Gu W. Loss of p53-mediated cell-cycle arrest, senescence and apoptosis promotes genomic instability and premature aging. Oncotarget 2016; 7: 11838–11849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012; 149: 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303: 844–848. [DOI] [PubMed] [Google Scholar]
- Kumamoto K, Spillare EA, Fujita K, Horikawa I, Yamashita T, Appella E et al. Nutlin-3a activates p53 to both down-regulate inhibitor of growth 2 and up-regulate mir-34a, mir-34b, and mir-34c expression, and induce senescence. Cancer Res 2008; 68: 3193–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efeyan A, Ortega-Molina A, Velasco-Miguel S, Herranz D, Vassilev LT, Serrano M. Induction of p53-dependent senescence by the MDM2 antagonist nutlin-3a in mouse cells of fibroblast origin. Cancer Res 2007; 67: 7350–7357. [DOI] [PubMed] [Google Scholar]
- Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M et al. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med 2004; 10: 1321–1328. [DOI] [PubMed] [Google Scholar]
- Di Conza G, Buttarelli M, Monti O, Pellegrino M, Mancini F, Pontecorvi A et al. IGF-1R/MDM2 relationship confers enhanced sensitivity to RITA in Ewing sarcoma cells. Mol Cancer Ther 2012; 11: 1247–1256. [DOI] [PubMed] [Google Scholar]
- Worrall C, Suleymanova N, Crudden C, Trocoli Drakensjo I, Candrea E, Nedelcu D et al. Unbalancing p53/Mdm2/IGF-1 R axis by Mdm2 activation restrains the IGF-1-dependent invasive phenotype of skin melanoma. Oncogene 2017; 36: 3274–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Goodman A, Walavalkar V, Barkauskas DA, Sharabi A, Kurzrock R. Hyperprogressors after immunotherapy: analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res 2017; 23: 4242–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendrysa SM, Perry ME. Tumor suppression by p53 without accelerated aging: just enough of a good thing? Cell Cycle 2006; 5: 714–717. [DOI] [PubMed] [Google Scholar]
- Barzilai N, Huffman DM, Muzumdar RH, Bartke A. The critical role of metabolic pathways in aging. Diabetes 2012; 61: 1315–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasty P, Christy BA. p53 as an intervention target for cancer and aging. Pathobiol Aging Age Relat Dis 2013; 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med 2005; 11: 440–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HT, Park JT, Choi K, Choi HJC, Jung CW, Kim GR et al. Chemical screening identifies ROCK as a target for recovering mitochondrial function in Hutchinson-Gilford progeria syndrome. Aging Cell 2017; 16: 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 2005; 433: 760–764. [DOI] [PubMed] [Google Scholar]
- Sinha M, Jang YC, Oh J, Khong D, Wu EY, Manohar R et al. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science 2014; 344: 649–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loffredo FS, Steinhauser ML, Jay SM, Gannon J, Pancoast JR, Yalamanchi P et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell 2013; 153: 828–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsimpardi L, Litterman NK, Schein PA, Miller CM, Loffredo FS, Wojtkiewicz GR et al. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science 2014; 344: 630–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper SC, Brack A, MacDonnell S, Franti M, Olwin BB, Bailey BA et al. Is growth differentiation factor 11 a realistic therapeutic for aging-dependent muscle defects? Circ Res 2016; 118: 1143–1150 discussion 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egerman MA, Cadena SM, Gilbert JA, Meyer A, Nelson HN, Swalley SE et al. GDF11 increases with age and inhibits skeletal muscle regeneration. Cell Metab 2015; 22: 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber K. No longer going to waste. Nat Biotechnol 2016; 34: 458–461. [DOI] [PubMed] [Google Scholar]
- Freitas-Rodriguez S, Rodriguez F, Folgueras AR. GDF11 administration does not extend lifespan in a mouse model of premature aging. Oncotarget 2016; 7: 55951–55956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HT, Park JT, Choi K, Kim Y, Choi HJC, Jung CW et al. Chemical screening identifies ATM as a target for alleviating senescence. Nat Chem Biol 2017; 13: 616–623. [DOI] [PubMed] [Google Scholar]
- Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 2016; 22: 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baar MP, Brandt RM, Putavet DA, Klein JD, Derks KW, Bourgeois BR et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 2017; 169: 132–47 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer J, Andressoo JO, de Wit J, Huijmans J, Beems RB, van Steeg H et al. Premature aging in mice deficient in DNA repair and transcription. Science 2002; 296: 1276–1279. [DOI] [PubMed] [Google Scholar]
- Serrano M, Blasco MA. Cancer and ageing: convergent and divergent mechanisms. Nat Rev Mol Cell Biol 2007; 8: 715–722. [DOI] [PubMed] [Google Scholar]
- Poyurovsky MV, Prives C. Unleashing the power of p53: lessons from mice and men. Genes Dev 2006; 20: 125–131. [DOI] [PubMed] [Google Scholar]
- Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K et al. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol. 1999; 147: 913–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Lian H, Sharma P, Schreiber-Agus N, Russell RG, Chin L et al. Disruption of the Cockayne syndrome B gene impairs spontaneous tumorigenesis in cancer-predisposed Ink4a/ARF knockout mice. Mol Cell Biol. 2001; 21: 1810–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeda G, Donker I, de Wit J, Morreau H, Janssens R, Vissers CJ et al. Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr Biol. 1997; 7: 427–439. [DOI] [PubMed] [Google Scholar]
- Lebel M, Leder P. A deletion within the murine Werner syndrome helicase induces sensitivity to inhibitors of topoisomerase and loss of cellular proliferative capacity. Proc Natl Acad Sci U S A. 1998; 95: 13097–13102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard DB, Beard C, Johnson B, Marciniak RA, Dausman J, Bronson R et al. Mutations in the WRN gene in mice accelerate mortality in a p53-null background. Mol Cell Biol. 2000; 20: 3286–3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004; 36: 744–749. [DOI] [PubMed] [Google Scholar]
- Tollini LA, Jin A, Park J, Zhang Y. Regulation of p53 by Mdm2 E3 ligase function is dispensable in embryogenesis and development, but essential in response to DNA damage. Cancer Cell. 2014; 26: 235–247. [DOI] [PMC free article] [PubMed] [Google Scholar]