

Abstract
Core-shell structured stem cell microencapsulation in hydrogel has wide applications in tissue engineering, regenerative medicine, and cell-based therapies because it offers an ideal immunoisolative microenvironment for cell delivery and 3D culture and differentiation. Long-term storage of such microcapsules as cell-biomaterial constructs by cryopreservation is an enabling technology for their wide distribution and ready availability for clinical transplantation. However, most of the existing studies focused on cryopreservation of separated cells or cells in microcapsules without a core-shell structure (i.e., hydrogel beads). The goal of this study is to achieve cryopreservation of stem cells encapsulated in core-shell microcapsules as cell-biomaterial constructs or biocomposites. To this end, a capillary microfluidics-based core-shell alginate hydrogel encapsulation technology is developed to facilitate cryopreservation of porcine adipose-derived stem cells (pADSCs) laden microcapsules with very low concentration (2 mol L−1) of cell membrane penetrating cryoprotective agents (CPAs) by suppressing ice formation. This may provide a low-CPA and cost-effective approach for vitreous cryopreservation of “ready-to-use” stem cell-biomaterial constructs, facilitating their off-the-shelf availability and widespread applications.
Keywords: core-shell, microencapsulation, cell-biomaterial constructs, stem cells, cryopreservation
TOC image
G. Zhao, X. Liu, and co-workers report alginate hydrogel encapsulation by capillary microfluidics facilitates rapid-cooling cryopreservation of stem cell-laden large-volume core-shell microcapsules as a cell-biomaterial construct. It enables cryopreservation of encapsulated stem cells with very low concentrations of penetrating cryoprotective agents, providing a cost-effective and high-throughput approach for vitreous cryopreservation of both separated stem cells and stem cell-biomaterial constructs.
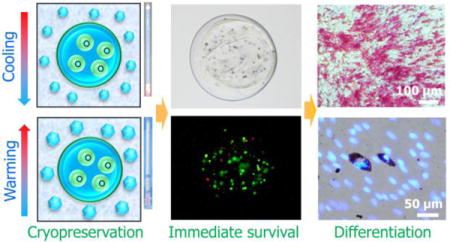
1. Introduction
Cell encapsulation in hydrogels has been widely used in cytotherapy, tissue engineering, regenerative medicine, reproductive medicine and 3D cell culture.[1–4] This is because the microcapsules can protect the encapsulated cells from immune rejection while allowing sufficient diffusion of oxygen, nutrients, secreted molecules, and metabolic wastes for the cells to survive and perform their function.[5, 6] It has shown great promise for treating various diseases including diabetes, hemophilia, cancer, liver and renal failure, as well as cardiovascular diseases.[5–7] Cryopreservation of microencapsulated cells make it possible for their wide distribution to end users so that they are readily available when needed for transplantation.[2, 3, 5, 8] It also offers a powerful tool for the integration of cell expansion and cryopreservation.[9]
Besides, the demand for living cells (especially stem cells) is rapidly increasing for cell-based research and medicine.[10] Although cell culture at 37 °C is a routine method for addressing this demand, it is costly, time-consuming, and labor intensive. Moreover, continuous long-term culture in vitro may induce spontaneous differentiation and/or possible genetic alterations of stem cells.[11] These issues may be resolved by cryopreservation of cells at cryogenic temperature.[10, 12]
Conventional cryopreservation approaches can be divided into two categories: conventional slow (programmable or controlled) freezing and vitrification (amorphous solidification during cooling).[13–15] In slow freezing, the samples are cryopreserved at controlled or programmable slow cooling rates with low concentrations of cryoprotective agents (CPAs, ~ 1.5 mol L−1), while in vitrification, they are transformed into glassy state at ultra-rapid cooling rates with high concentrations of CPAs (e.g., 6–8 mol L−1).[13–15] Both conventional slow freezing and vitrification of microencapsulated cells have been investigated over the past decades.[2, 16–19] It has been reported that a large amount of ice formation in slow/controlled freezing may damage the integrity of microcapsules of ~250 μm.[2, 16, 17] This is due to the fact that the large surface-to-volume ratios of the microcapsule makes it very likely for them to have direct contact with the developing ice crystals during cryopreservation.[2, 18] Besides, the conventional slow freezing approach requires a commercially available programmable freezer or a cryogenic refrigerator with a lengthy (up to hours) cooling process,[16, 20] and after cooling, the samples must be transferred into liquid nitrogen (LN2) for long-term storage.[21] These factors make it uneconomic, time-consuming, and complicated.[5] Vitreous cryopreservation as an emerging strategy, is regarded to be safer and more reliable for cell preservation when compared with the conventional slowing freezing method.[2, 13, 14, 22] This is because no extra- or intracellular ice formation (which may cause injury mechanically) and the resultant imbalance in solute concentrations between extra- and intracellular solutions (which may cause osmotic injuries).[23] However, in conventional vitrification, high concentrations of CPAs (up to ~ 8 mol L−1, which is toxic and may induce metabolic and osmotic injuries[10, 24, 25] and uncontrolled differentiation of stem cells[26]) and/or ultra-rapid cooling/warming rates (even higher than 106 °C/min,[10, 24, 25] which is technically difficult to reach especially for bulk samples), are commonly used to suppress ice formation[27] during cooling and dampen devitrification (the changing of glass from the vitreous state to a crystalline state induced by not-high-enough concentrations of CPAs or not-rapid-enough warming rates) during warming.[28] These requirements may limit the application of vitreous cryopreservation in maintaining stress-sensitive stem cells, immune cells, and oocytes, etc. Nanoliter droplets have been used to confine cells for vitreous cryopreservation with reduced concentrations of CPAs.[10, 29] However, the droplets are exposed to the environment (liquid nitrogen, air, or pre-cooled surfaces) directly,[13, 14, 29, 30] and the cells may suffer from potential contamination.
Alginate hydrogel microencapsulation was recently reported to enable low-CPA cell vitrification by inhibiting devitrification,[10] which marks a significant step towards practical application of vitreous cell cryopreservation. However, most of the encapsulation vitrification studies have been performed with microcapsules of 100 to 250 μm in diameter without a core-shell structure.[10, 31–35] However, core-shell structured microcapsules are needed for various biomedical applications.[32, 33, 35, 36] For example, core-shell structured encapsulation has been reported to better support 3D culture (providing minimized spontaneous differentiation of stem cells encapsulated in the core[4, 32, 34]) and transplantation.[33, 37] In addition, the use of the large microcapsules may allow for rapidly processing a large volume (tens to hundreds of milliliters) of cell suspensions (which is needed for cytotherapy or cell transplantation[38]). However, vitreous cryopreservation of cells encapsulated in large-volume microcapsules (> 500 μm in diameter) with a core-shell structure has not been done. Although one major challenge associated with cryopreservation of encapsulated cells by slow freezing is the loss of integrity of the relatively large microcapsules (> 250 μm) because of ice formation,[31, 39] it may not be an issue for vitreous cryopreservation in the absence of apparent ice formation.
Electrostatic spray and microfluidic channel devices are two commonly used methods for cell encapsulation.[31, 34, 40, 41] However, the size distribution of microcapsules produced by electrospray may be wide. Although microfluidic channel device may be used to produce more homogeneous microcapsules,[42] its fabrication requires specialized facilities including clean room that may not be readily available to many researchers. In addition, plastic (e.g., the most commonly used polydimethylsiloxane or PDMS) microfluidic device may be unsuitable for long-term or repeated use due to the thermal/mechanical aging of plastics.[43] Furthermore, a specially designed nonplanar polydimethylsiloxane (PDMS) microfluidic device may be necessary for fabrication of the core-shell structured microcapsules.[4, 10, 32, 34] Moreover, although core-shell hydrogel microcapsules can be generated with flow-focusing microfluidic devices, most of the approaches involved the use of toxic acid and mineral oil, which may raise a question of biocompatibility.[35, 44]
In this paper, a large-volume (> 500 μm in diameter, at least two times in size of the reported ones[10, 31]) core-shell structured alginate hydrogel encapsulation approach without using toxic acid or oil was developed for vitreous cryopreservation of encapsulated cells. Instead of a nonplanar PDMS microfluidic channel device, we developed a cost-effective, easy-to-make, double emulsion flow-focusing tube-in-tube capillary microfluidic device to encapsulate stem cells in core-shell microcapsules. This device can be used repeatedly in long term without the need of specialized facilities for its fabrication. We demonstrate the feasibility of the approach using porcine adipose-derived stem cells (pADSCs), due to the primary importance of their encapsulation, 3D culture, cryopreservation, and transplantation in cellular therapy.[45]
2. Results
2.1. Tube-In-Tube Capillary Microfluidic Device
A schematic illustration of the experimental setup for fabricating the cell-laden core-shell microcapsules for vitreous cryopreservation is given in Figure 1A. The device has three inlets (I1, I2 and I3) for injecting cell suspension, aqueous sodium alginate solution, and corn oil, respectively. At the exit of the inner and the middle glass capillaries (inset (i) of Figure 1A), the cell suspension (core fluid) and the aqueous sodium alginate solution (shell fluid) were dispersed into droplets as a result of shear force and interfacial tension (step (a), Movie S1). The droplets were immersed into aqueous CaCl2 solution for gelling into hydrogel (step (b), Movie S2), since sodium alginate can be cross-linked into hydrogel of calcium alginate by Ca2+ (step (b), Movie S2).
Figure 1.
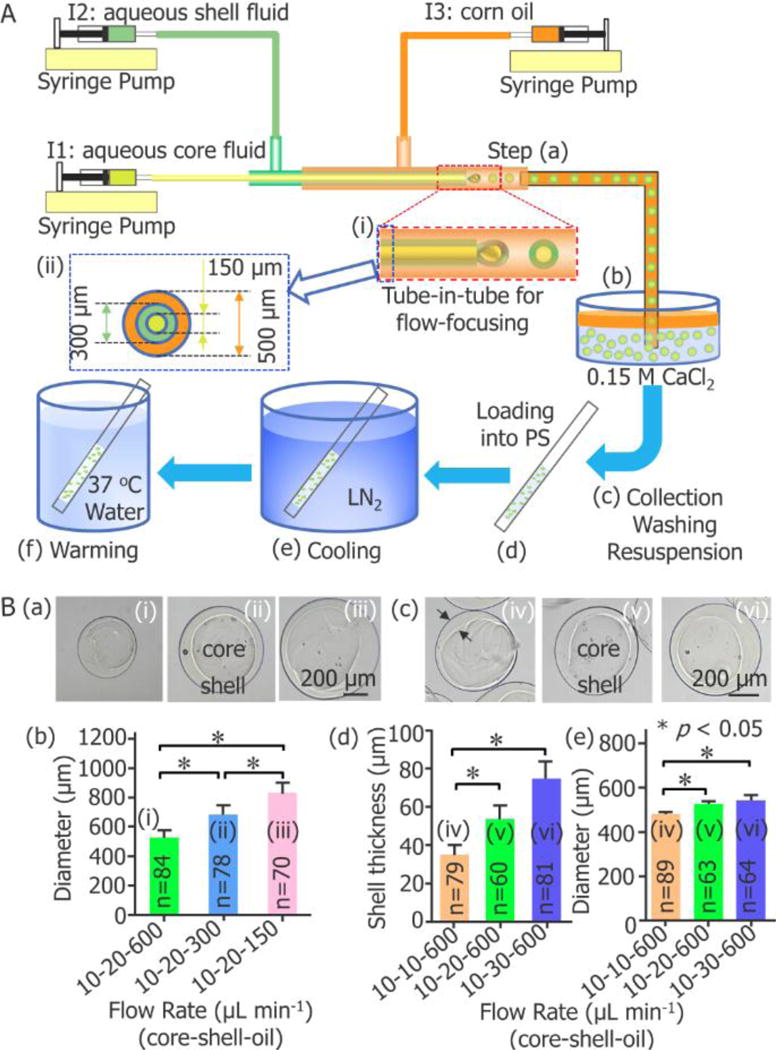
Capillary microfluidic encapsulation of stem cells in core-shell microcapsules for vitreous cryopreservation. (A) A schematic illustration of the major steps: (a) cells are encapsulated in droplets using a double emulsion flow-focusing tube-in-tube capillary microfluidic device, (b) the droplets are immersed into aqueous CaCl2 solution for crosslinking into microcapsules with a calcium alginate hydrogel shell, (c) the microcapsules are collected, washed, and resuspended in 0.9% NaCl solution, (d) after incubated in a cryoprotective agent (CPA) solution, the microcapsules are loaded into a plastic straw (PS), (e) after sealed at the distal end using wax, the PS is plunged into liquid nitrogen (LN2) for cooling, and (f) the PS is plunged into 37 °C waterbath for warming the sample. I1: ice-cold cell suspension with 1% (wt) sodium carboxymethylcellulose, 1% (wt) sodium alginate, and 0.5 mol L−1 trehalose in 0.25 mmol L−1 D-Mannitol. I2: 2% (wt) sodium alginate solution. I3: corn oil. (B) Fabrication of core-shell microcapsules with controllable outer diameter and shell thickness. (a) and (b) show the dependence of the diameter of the microcapsules on the flow rate of the continuous oil phase (n = 70–84, * p < 0.05). (c) - (e) show the dependence of both the shell thickness and diameter of the microcapsules on the flow rate of the aqueous shell fluid (n = 60–89, * p < 0.05).
2.2. Controllable Generation of Core-Shell Microcapsules
Both the size (outer diameter) and the shell thickness of the microcapsules can be well controlled by adjusting the flow rates of the core, shell, and continuous oil phase (Figure 1B). They increase with the decrease in the flow rate of oil phase (Figure 1B (a) and (b)) or the increase in the flow rate of shell fluid (Figure 1B (c) – (e)). Decreasing the flow rate of the oil phase while keeping the other flow rates constant (the flow rates of the core and shell phases at 10 and 20 μL/min, respectively) results in an increase in microcapsule diameter (Figure 1B (a) and (b), Table S1): the microcapsules are 525.3, 682.4 and 832.8 μm in diameter for oil phase flow rates of 600, 300, and 150 μL/min, respectively. In addition, increasing the flow rate of the shell phase while keeping the other flow rates constant (the core and oil flow rates at 10 and 600 μL/min, respectively), results in an increase in shell thickness of the microcapsules (Figure 1B (c) – (e), Table S1). In particular, the flow rates of the shell phase at 600, 300 and 150 μL/min lead to the formation of microcapsules with shell thickness of 35.3, 53.5 and 75.0 μm (480.3, 525.9 and 545.1 μm in diameter), respectively. Finally, the flow rate combination 10-20-600 μL/min (core-shell-oil) was used to produce microcapsules with a diameter of 525.9±8.6 μm (n=63) and thickness of 53.5±6.6 μm (n=60) for further vitreous cryopreservation studies.
2.3. Cryopreservation of Microencapsulated Cells
Cryovials and conventional plastic straw (PS) have been widely used for low-CPA (1–2 mol L−1) slow freezing with controlled rate freezer (or cryogenic refrigerator) [2, 9, 46] and high-CPA (~8 mol L−1) vitrification by plunging into LN2 [8, 22, 47], respectively. Therefore, we used PS for the large-volume-alginate hydrogel-encapsulation based cryopreservation of stem cells by rapid cooling with low concentrations of CPAs in this study. For cryopreservation, the cross-linked hydrogel microcapsules were collected, washed, and re-suspended in CPA solution (step (c), Figure 1A), and then loaded into a plastic straw (PS) (step (d), Figure 1A) that is immediately plunged into liquid nitrogen (LN2) for cooling (step (e), Figure 1A). After 5 minutes when the sample reached thermal equilibrium with LN2 at −196 °C, the PS was shifted into 37 °C water bath for rewarming (step (f), Figure 1A), unloading of the sample, removal of the CPAs and further evaluation.
We found that both CPA #1 and #3 can be vitrified (following previous studies,[10, 48] vitrification is defined as no visible ice formation in the bulk solution) by plunging the PS into LN2, while apparent ice formation can be observed in culture medium (DMEM/F12 with 10% (v/v) FBS) alone, CPA #2, and CPA #4 after plunging into LN2 (Figure 2A, Movie S3). Moreover, the amount of ice formation in CPA #2 is obviously less than that in CPA #4 or culture medium alone (Figure 2A, Movie S3). Therefore, we defined CPA #2 as the partial vitrification group (P-vitri, water of the solution is partially transformed into ice). The specific partial vitrification phenomenon is further confirmed by freezing an additional 10 straws filled with CPA #2 (Figure S1). Furthermore, severe devitrification (from transparent to opaque) and/or recrystallization (increase in opacity) were observable in all the four CPA solutions and the culture medium during warming (Figure 2A, Movie S4).
Figure 2.
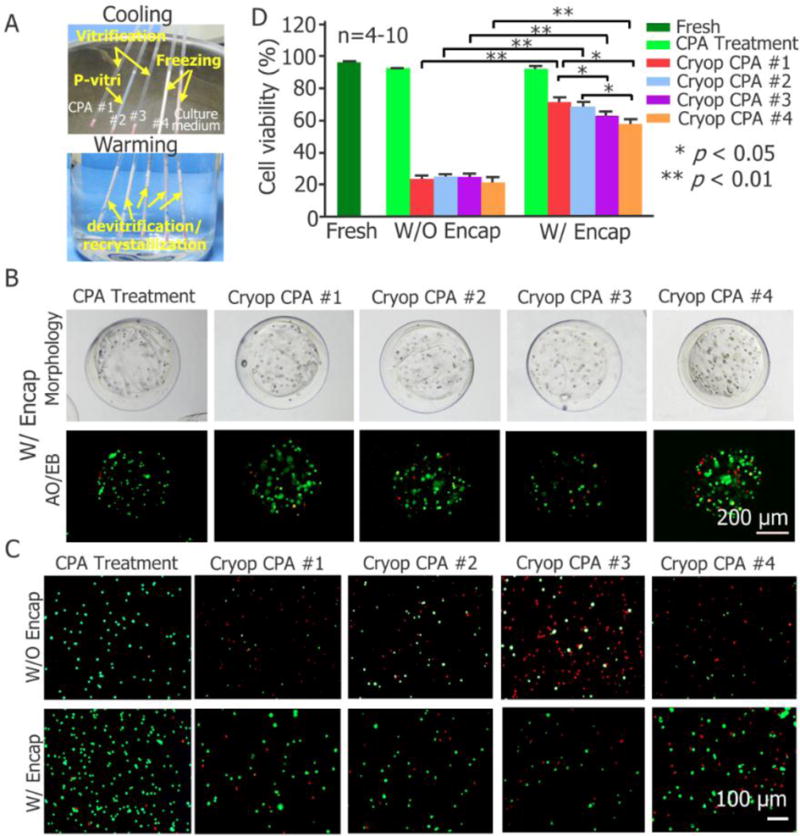
Large-volume-hydrogel-encapsulation vitreous cryopreservation of stem cells using plastic straw (PS). (A) Typical images showing freezing, vitrification, and partial vitrification (p-vitri) of cell culture medium and four cryoprotective agent (CPA) solutions during cooling (top) and devitrification/recrystallization (devitri) during warming (bottom). The red material at the distal end of the straw is sealing wax. (B) Typical phase and live/dead (stained with acridine orange/ethidium bromide or AO/EB as green/red) fluorescence images of pADSCs encapsulated in the microcapsules after treated with CPA #1 (i.e., CPA addition and removal) or cryopreserved with the four different CPAs. (C) Typical fluorescence images of pADSCs with and without encapsulation subjected to CPA #1 treatment and cryopreservation with the four different CPAs. (D) Quantitative cell viability of pADSCs that are fresh, treated with CPA #1 without cryopreservation, or cryopreserved with four different CPAs (n = 4–5. * p < 0.05, ** p < 0.01). Culture medium: Dulbecco’s Modified Eagle’s Medium/Ham’s Nutrient Mixture F-12 (DMEM/F12) with 10% (v/v) fetal bovine serum (FBS). CPA #1: 1 mol L−1 1,2-Propanediol (PROH), 1 mol L−1 ethylene glycol (EG), 10% dextran T50 and 1 mol L−1 trehalose; CPA #2: 1 mol L−1 PROH, 1 mol L−1 EG, 10% dextran T50 and 0.5 mol L−1 Trehalose; CPA #3: 1 mol L−1 PROH, 1 mol L−1 EG and 1 mol L−1 Trehalose; CPA #4: 1 mol L−1 PROH, 1 mol L−1 EG and 10% dextran T50. CPAs #1-#4 were prepared using DMEM/F12 with 80% (v/v) FBS. Fresh: cells kept in culture medium; CPA Treatment: cells subjected to addition and removal of CPA #1; Cryop: cells subjected to cryopreservation. W/O Encap: cells without encapsulation; W/Encap: cells with encapsulation.
2.4. Cell Viability
Typical images of live/dead (acridine orange/ethidium bromide or AO/EB) staining showing qualitatively the viability of encapsulated cells either without cryopreservation (but treated with CPA #1) or cryopreserved with the four different CPAs are given in Figure 2B. Figure 2C shows the qualitative survival of cells after being released out of the microcapsules together with the non-encapsulated cells treated with CPA #1 or cryopreserved with the four different CPAs. The survival of the released cells under the five different conditions together with fresh cells was further quantified and the results are shown in Figure 2D. Neither the CPA treatment (CPA addition and removal without encapsulation and cryopreservation) nor the combination of CPA treatment and cell encapsulation has significant impact on cell viability (Figure 2B–D and Table S2), while encapsulation does greatly enhance the cell viability post cryopreservation (from 24% to 73%, 25% to 71%, 25% to 63%, and 21% to 56% for CPAs #1, #2, #3, and #4, respectively). Because the cell viability should be >70% for a cell-based product to be clinically usable according to the FDA guidelines,[49] only CPA #1 (vitrification) and #2 (P-vitri) are compliant with the requirement.
An interesting and new finding is that the encapsulated cells still have a high survival (> 70%) post cryopreservation even though apparent (albeit partial) ice formation is observable (Figure 2A, CPA #2, P-vitri). This is probably because the ice formation occurs only outside the microcapsules,[23, 25, 50, 51] These results suggest that alginate hydrogel microencapsulation can effectively protect the cells in the microcapsules by providing an oocyte zona pellucida-like structure against the ice front during rapid cooling[27], in addition to the effect of inhibiting devitrification/recrystallization reported previously.[10] In other words, although the bulk solution outside the microcapsules is partially vitrified, the cell suspension in the core of the microcapsules may still be successfully vitrified. This is also similar to the function of cell membrane during cooling, to block ice propagation into intracellular solution and promote intracellular vitrification.[51]
It is interesting that the partial vitrification group (treated with CPA#2) leads to a slightly higher (but statistically insignificant) survival rate than that of the vitrification group (treated with CPA#3), we attribute it to the cryoprotective effect of dextran T50 in CPA 2#. The main difference between CPA #2 and #3 lies in that 0.5 mol L−1 trehalose in CPA #3 was replaced with 10% (w/v) dextran T50 in CPA #2. The capability of enhancing cell survival post vitrification by dextran could further be demonstrated by comparing CPA #1 and #3 for encapsulation cryopreservation. Where, the presence of 10% (w/v) dextran T50 significantly improves (by > 10%) the cell survival post cryopreservation for the encapsulation group (CPA #1 versus CPA #3, both experience vitrification and devitrification in the presence of 1 mol L−1 trehalose). This may be applicable for explaining the different results between CPA #2 and #3. Even though there is partial ice formation outside the microcapsule for CPA#2, there may be no ice formation inside the microcapsules due to the capability of the hydrogel microcapsule in blocking ice formation as reported by Zhang et al.[31] It is worth noting that the advantage of 10% dextran over 0.5 mol L−1 trehalose for enhancing encapsulation vitrification is marginal because the difference in post-vitrification cell viability between CPA #2 and #3 is not significant. Moreover, the similar viability for CPA #1 (apparent vitrification) and #2 (apparent partial vitrification) may be explained by the same reason. In other words, although the solution outside the microcapsules was partially vitrified with CPA #2, the cell suspension inside the microcapsules may be vitrified to a similar level to CPA #1. Besides, it should be pointed out that only when combined with encapsulation, dextran T50 could enhance the cell survival (Figure 2D). Without trehalose, the presence of 10% (w/v) dextran T50 alone cannot provide enough protection to the encapsulated cells either (survival: 56%, CPA #4) (Figure 2D).
2.5. Cell Attachment and Proliferation
Since the highest cell survival following cryopreservation by full or partial vitrification was achieved using CPAs #1 and #2, further assessment on the cells’ long-term viability and functional properties was focused on these two groups. The long-term viability was assessed by both cell attachment and proliferation. The results indicate that the morphology of pADSCs post encapsulation and vitreous cryopreservation (W/Encap and Cryop) with CPAs #1 and #2 is similar to that of the fresh control group during culture for 3 days (Figure 3A). Furthermore, no significant difference was observed between the proliferation of the fresh, CPA treated, and cryopreserved groups for both CPA #1 (Figure 3B) and #2 (Figure 3C). Consistent with the decrease of cell survival post cryopreservation judged by live/dead staining (Figure 2B), a decrease in attachment efficiency was observed for the cells post cryopreservation compared to fresh cells and cells treated with CPA without cryopreservation (Figure 3D-E).
Figure 3.
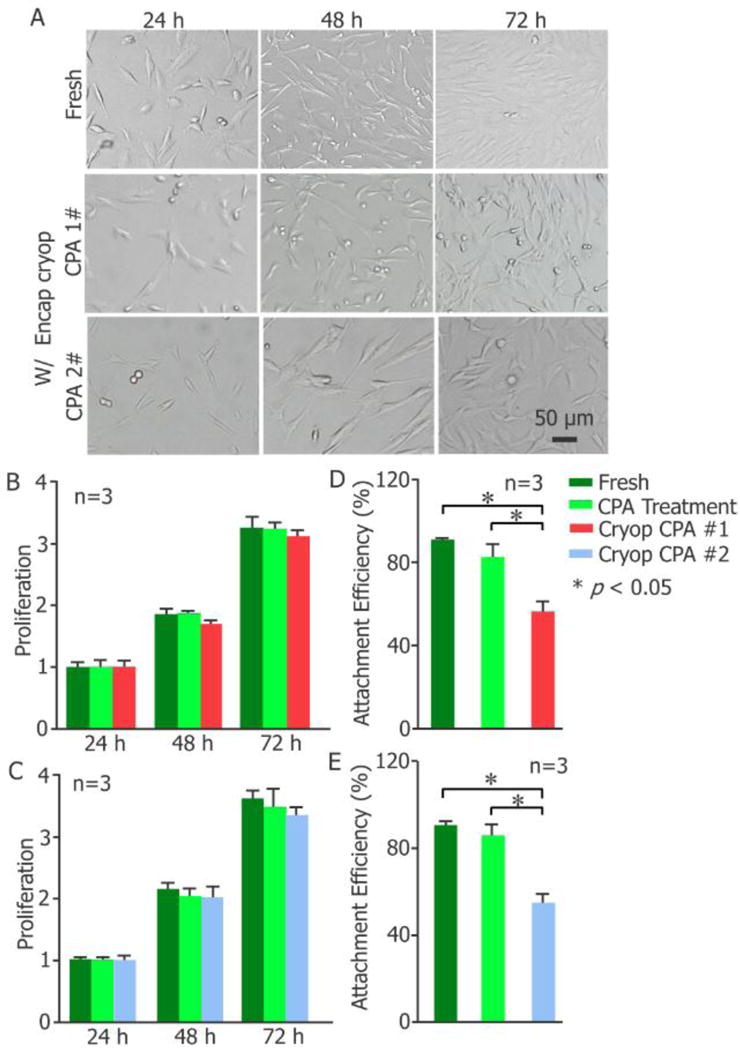
Cell attachment and proliferation. (A) Morphology, (B-C) proliferation, and (D-E) attachment efficiency of fresh pADSCs and the cells post Large-volume-hydrogel-encapsulation vitreous cryopreservation (n = 3, * p < 0.05). (B) and (D) share the same legend in (B) and (C) and (E) share the legend in (C). W/Encap: with encapsulation; Cryop: cryopreservation; CPA Treatment: CPA addition and removal, with encapsulation.
2.6. Stemness and Multi-Lineage Differentiation
The effect of cryopreservation with CPAs #1 and #2 on the pADSC function represented by their stemness and induced multi-lineage differentiation is shown in Figure 4. Immunostaining was conducted to evaluate the expression of CD44, CD29, and CD31 on the cells. For mesenchymal stem cells, both CD44 and CD29 are common surface glycoproteins/receptors, while CD31 is a negative or endothelial differentiation marker. As shown in Figure 4A, encapsulation cryopreservation with CPA #1 or #2 has no significant impact on the expression of the three receptors on the pADSCs. Furthermore, quantitative evaluation on the expressions of four receptors (CD44, CD29, CD31 and CD90 (another positive marker for ADSCs[52])) by flow cytometry indicates that the expressions of CD44, CD29 and CD90 for the cryopreserved cells were similar to the control group, while the expressions of CD31 were low for all groups (Figure 4B). In addition, the relative expression of the four typical stem cell genes, Nanog, Klf4, Sox2, and Oct4, determined by quantitative real-time polymerase chain reaction (qRT-PCR), for the encapsulation cryopreserved cells with CPAs #1 and #2 are not significantly different (p>0.05) from that of fresh cells (Figure 4C). The capability of multi-lineage differentiation, reflecting the functional stemness of the pADSCs, was further tested. The data of induced adipogenic and osteogenic differentiation indicate that there is no significant difference between the differentiation potentials of the fresh and cryopreserved cells (Figure 4D). Both Oil Red O (ORO) staining of adipocyte-like cells with lipid droplets (adipogenic differentiation) and Alizarin Red S (ARS) staining of calcific deposition from osteoblast-like cells (osteogenic differentiation) for the three groups of cells are similar (Figure 4D). Therefore, the pADSCs cryopreserved with CPAs #1 and #2 retain their stemness and capacity of multi-lineage differentiation.
Figure 4.
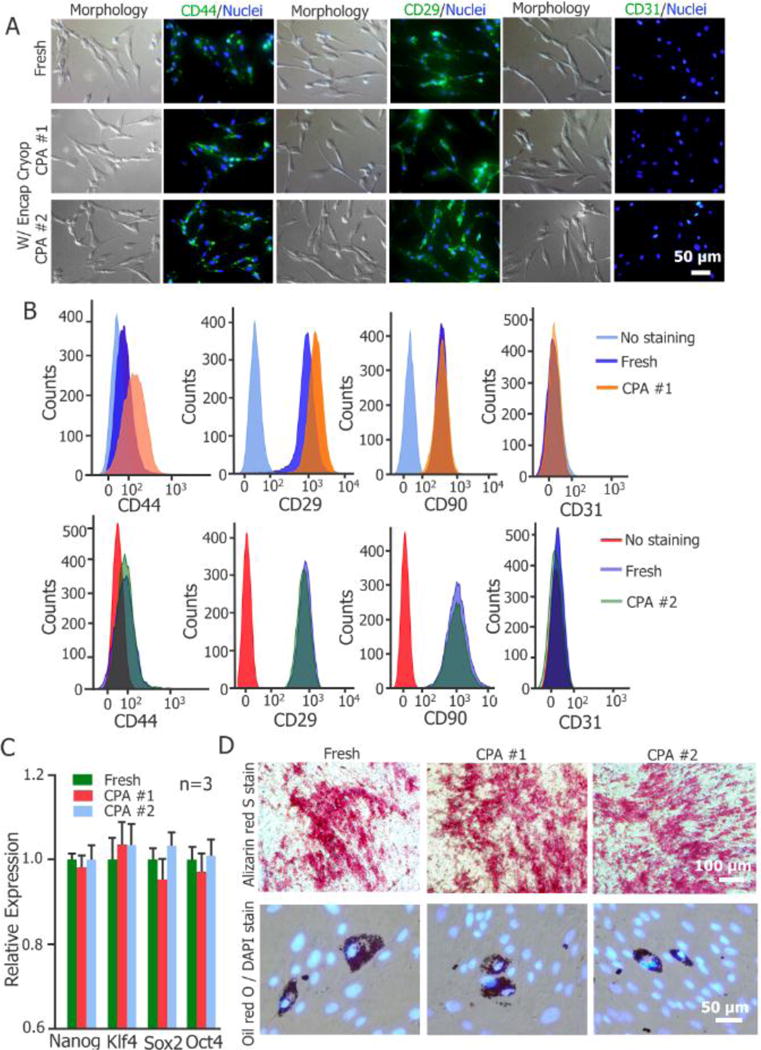
Stemness and function of pADCSs before and after large-volume-hydrogel-encapsulation vitreous cryopreservation. (A) Immunostaining of CD44(+), CD29(+) and CD31(−) expression on the cells. (B) Flow cytometry quantification of the expression of CD44(+), CD29(+), CD90(+) and CD31(−) on the cells. (C) Quantitative RT-PCR analysis of the expression of four stem cell genes in the cells (n = 3). (D) Qualitative evaluation on adipogenic and osteogenic (Alizarin Red S stain of calcific deposition) differentiations of the cells with Oil Red O (ORO) stain of lipid droplets and Alizarin Red S (ARS) stain of calcific deposition, respectively.
3. Discussion
Vitrification of cell suspension of pADSCs in large-volume microcapsules, as a stem cell-hydrogel construct, at a very low concentration of CPAs was achieved. Specifically, 2 mol L−1 cell membrane penetrating (1 mol L−1 PROH and 1 mol L−1 EG) supplemented with 0.5 mol L−1 non-penetrating (0.5 mol L−1 trehalose and 0.002 mol L−1 Dextran T50) CPAs were successfully used to partially vitrify the solution outside the hydrogel capsules with very large volumes (2 times larger in diameter, and accordingly 8 times larger in volume compared to the commonly reported[10, 32, 40] hydrogel capsules for cell encapsulation). It is worth noting that large-volume vitrification with a low concentration of CPAs is the ultimate goal of cryopreservation[53, 54] although it has been challenging because low-CPA vitrification requires an ultrarapid cooling/warming rate[25, 51, 55] that is difficult to achieve with a large sample volume. Moreover, it offers an ideal method for vitrification of bulk volume of cell suspensions with low toxicity because the conventional vitrification requires a very high concentration (up to ~8 mol L−1) of penetrating CPAs that may induce significant metabolic and osmotic injury to cells[10]. We minimized the potential toxic effects by using a low concentration of penetrating CPAs (2 mol L−1), and suppressed the injuries caused by ice formation by using hydrogel encapsulation to facilitate cell vitrification. It was found that the viability of the cells released from the microcapsules post-cryopreservation (with CPA #1 and #2) is higher than 70%, which is comparable to that previously reported for smaller (approximately half in size) microcapsules[10]. Furthermore, the released cells retain normal proliferation, expression of the cell membrane receptors (CD44+, CD29+, CD31-, and CD90+), and expression of the four typical stem cell genes (Nanog, Klf4, Sox2, and Oct4). The capability of multi-lineage differentiation is also well maintained. However, an apparent decrease in attachment efficiency was observed for the released cells post-cryopreservation, which may be due to cryoinjury induced cell apotosis[56] and reduced gene expression of adhesion-related molecules[57], increased degradation of adhesion proteins[57], and loss of some plasma membrane proteins[58] associated with cryopreservation. Further investigation to improve the cell attachment efficiency post cryopreservation is warranted.
The potential mechanism underling the effect of encapsulation on the formation and propagation of ice crystals during both cooling (by LN2 quenching) and rewarming is schematically shown in Figure 5. Specifically, for CPAs (CPA #1 and #3) that are enough to for achieving vitrification of the samples during cooling, the microcapsules could prevent ice recrystallization and/or devitrification of the encapsulated cell suspension in the presence of large amount of external ice crystals during warming (Figure 5A and C). For CPA #2 with only partial vitrification in the sample during cooling, the microcapsules could still prevent ice recrystallization and/or devitrification of the encapsulated cell suspension during warming, although large amount of bigger ice crystals may form outside the microcapsules (Figure 5B). Finally, for CPA #4 that is not sufficient to achieve either vitrification or partial vitrification during cooling, a large amount of ice crystals will form both inside and outside the microcapsules during cooling (Figure 5D). Besides, ice recrystallization may grow or nucleate during warming, resulting in severe injuries to the cells inside the microcapsules (Figure 5D).
Figure 5.
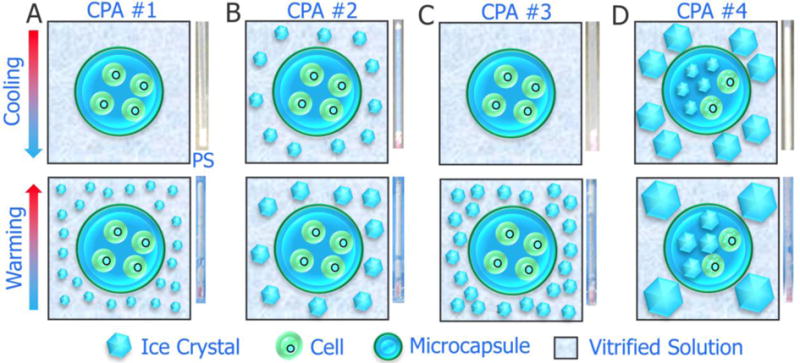
A schematic illustration of the possible mechanisms for enhanced cryopreservation of stem cells by encapsulation of the cells in core-shell microcapsules. (A)–(D) correspond to CPA #1-#4. PS: plastic straw. The appreance of the sample in the PS is shown alongside the sketch for the possible mechanisms.
It is worth noting that no visible ice formation (to the naked eyes) in the bulk solution is only a gross assessment of vitrification.[28, 59] However, it is extremely difficult to achieve true or complete vitrification of aqueous solutions and it is equally difficult to determine true vitrification because contemporary instruments (e.g., x-ray crystallography[60]) for studying crystallization are usually designed for working at room rather than cryogenic temperatures. Differential scanning calorimetry (DSC) has been used to investigate vitrification in aqueous solutions without cells,[59, 61] for which less than 0.2% w/w ice in the solutions is not detectable.[53, 62] Therefore, cell vitrification in the field of cryobiology usually does not refer to true vitrification of the extracellular solution and few studies on cell vitrification claims true vitrification of the extracellular solution either. The absence of visible ice has been practically used as the first-cut judgment of vitrification.[10, 14, 48, 63] Besides, for almost all the theoretical models used for the prediction of vitrification, a small ice crystallization volume fraction (< ~ 0.1% v/v) was used to evaluate the realization of vitrification.[51, 64, 65] This also implies that the vitrification involved is essentially not true or complete vitrification. In this study, we adopted the gross assessment of vitrification based on visible ice formation similar to the previous studies:[10, 48] vitrification refers to no visible ice formation (or more precisely, apparent vitrification), and partial vitrification refers to the formation of a small amount of visible ice. It is also worth noting that the major difference between cell cryopreservation by vitrification and slow freezing in the cryobiology literatures is that ice formation is intentionally induced for the latter while it is not for the former[66].
Scaling up of the microfluidic device is another important topic associated with the approach developed in this study. In fact, a very important issue associated with a single microfluidic unit or channel is the low production rate of emulsion droplets,[67] which is regarded as a major hindrance to the wide application of microfluidic technology at commercial scale.[67] Fortunately, this problem can be solved by parallelizing a large number of microfluidic cell encapsulation units or channels onto a single chip or chipset.[68, 69] Mulligan et al. successfully developed a single microfluidic chip consisting of six microfluidic flow-focusing units operating in parallel to achieve a production rate of hundreds of milliliters per hour.[68] Considering the possibility of parallelizing a large number of microfluidic chips with multiple flow-focusing units, the production rate could even be improved. Since the capillary microfluidic devices are easier than microchannel-based devices for integrating into an array for high-throughout,[70] scaling-up of the capillary microfluidic encapsulation approach developed by this study is technically feasible. Moreover, automation of the complex procedures (except for cooling and rewarming) for the encapsulation cryopreservation approach developed in this study is possible, which may further increase the production rate. Although direct ejection of the microcapsules into LN2 or onto a precooled surface could greatly enhance the cooling rates and it may be integrated into an automatic system[15, 65], we preferred to load the cells into the conventional plastic straws for the successive quench cooling in LN2. This is because a high contamination risk is associated with direct contact of the microcapsules with non-sterile liquid nitrogen or a surface in an open environment.[71] Finally, bundles of conventional plastic straws could be used to load the microcapsules containing tens to hundreds of milliliters of cell suspensions for cooling and thawing at one time, to make this approach practically applicable.
Cryopreservation of encapsulated stem cells (cell-biomaterials systems) are of primary importance for clinical applications, while such investigations are very few. Therefore, pADSCs were used to demonstrate the feasibility of the approach developed in this study. It is worth noting that we intend to cryopreserve “ready-to-use” cell-biomaterial constructs instead of the separated cells in this study, because cryopreservation of microencapsulated cell systems is an enabling technology for their clinical translation.[2, 8] In view of the fact that i) most of the microcapsules used for cell encapsulation range from ~100 to 500 μm in diameter,[33, 72] ii) cryopreservation of large-volume microcapsules is desired,[2, 8] iii) the size of the microcapsules should not be too large to compromise sufficient diffusion of oxygen, nutrients, secreted molecules, and metabolic wastes for the transplanted encapsulated cells to survive, and iv) the size should also be appropriate for smooth loading and unloading from the plastic straws used for cryopreservation, we chose the microcapsules with a diameter of ~500 μm In this study. Importantly, our data show that cell-laden large-size (~500 μm in diameter) core-shell microcapsules can be successfully cryopreserved with very low concentrations of cryoprotective agents (only 2 mol L−1 penetrating CPAs supplemented with 0.5 mol L−1 non-penetrating CPAs).
4. Conclusion
A capillary microfluidics-based large-volume core-shell alginate hydrogel encapsulation technology is developed to facilitate vitreous cryopreservation of encapsulated stem cells with very low concentrations of cryoprotective agents (only 2 mol L−1 penetrating CPAs is used), by suppressing ice formation to minimize cryoinjury to cells. This may provide a low-CPA, cost-effective and high-throughput approach for cryopreservation of both cells and cell-biomaterial constructs. Compared to previous approaches, the reported cryopreservation approach is innovative in several aspects including a core-shell structure for better 3D culture and better immunoisolation post-transplantation, a large construct volume allowing for high throughput capacity for fast processing of mass production, and a very low concentration of penetrating CPAs for minimized CPA toxicity. As an important supporting and enabling technology, this approach may find its important applications in the fields of clinical medicine and fundamental research, since generation and cryopreservation of “ready-to-use” stem cell-biomaterial constructs is attracting ever-increasing attention for 3D cell culture, 3D bio-printing, precise modulation of stem cell differentiation, cell delivery, and cellular therapy. Meanwhile, the self-designed and fabricated tube-in-tube capillary microfluidic device for generation of the microcapsules in this study is both durable and reusable, which is one of its featured advantages compared with PDMS microfluidic devices. It has the potential of scaling-up and can be easily incorporated into an automatic system for high-throughput. Therefore, the encapsulation-based vitreous cryopreservation approach with a low concentration of CPAs is valuable for fast processing of large volumes of stem cell-hydrogel biocomposites for clinical and practical use. Collectively, this study opens up an attractive avenue for banking stem cell-hydrogel constructs and offers the potential for their “off-the-shelf” availability in clinical applications.
5. Experimental Section
Materials and reagents
All chemicals were purchased from Sigma, USA, unless otherwise stated. We developed four different CPA solutions: CPA #1: 1 mol L−1 1,2-Propanediol (PROH), 1 mol L−1 ethylene glycol (EG), 10% (w/v) dextran T50 (≈ 0.002 mol L−1) and 1 mol L−1 trehalose; CPA #2: 1 mol L−1 PROH, 1 mol L−1 EG, 10% (w/v) dextran T50 and 0.5 mol L−1 trehalose; CPA #3: 1 mol L−1 PROH, 1 mol L−1 EG and 1 mol L−1 trehalose; CPA #4: 1 mol L−1 PROH, 1 mol L−1 EG and 10% (w/v) dextran T50. They were made by dissolving the different combinations of CPAs in Dulbecco’s Modified Eagle’s Medium/Ham’s Nutrient Mixture F-12 (DMEM/F12) with 80% (v/v) fetal bovine serum (FBS).
Cell culture
Porcine adipose derived stem cells (pADSCs) obtained as a gift from Prof. Zhang’s lab[73] were cultured with DMEM/F12 containing 10% FBS, 50 μg/ml vitamin C (Sigma, USA), 10 ng/ml basic fibroblast growth factor (bFGF, Pepro Tech, USA), and 0.002 mol L−1 GlutaMAXTM-100x (Life Technologies, USA) at 37°C in a 5% CO2 humidified incubator. Culture medium was changed every three days till cells reached 80%–90% confluency, and the cells were washed with phosphate-buffered saline (PBS), digested with 0.25% w/v trypsin-EDTA (Sigma, USA) for 3 min, and centrifuged at 94 g for 5 min. Then, they were re-suspended and kept at 4 °C until further use.
Fabrication of tube-in-tube capillary microfluidic device
The microfluidic device for cell microencapsulation was fabricated by using three tube-in-tube patterned glass capillaries with different diameters (150, 300, and 500 μm in outer diameter) (Figure 1A). The relative and concentric position of the three glass capillaries was fixed by using hot melt glue, and the outermost capillary was further adhered to a rectangular glass (9×5×0.29 cm) for convenient observation under a microscope.
Microencapsulation of pADSCs
Syringes, holders, capillaries, and all other accessories were washed with 75% (v/v) alcohol, and further sterilized using UV light in a laminar flow hood (SW-CJ-1FD, Suzhou Purification Equipment Co., ltd, China) for 30 min before use. All solutions were filtered with 0.22 μm of sterilizing-grade filters before use. The core fluid consisted of 1% (w/v) sodium alginate, 1% (w/v) sodium carboxymethylcellulose, and 0.5 mol L−1 trehalose with pADSCs (inlet I1, Figure 1A), while the shell fluid consisted of 2% (w/v) sodium alginate (inlet I2, Figure 1A). Both the core and the shell fluids were prepared by dissolving the solutes in 0.25 mol L−1 aqueous D-mannitol solution to make the shell fluid isotonic, while the core fluid hypertonic (for partial dehydration of the cells to suppress intracellular ice formation during cryopreservation[10, 74]). Food grade corn oil was used as the outer carrier oil emulsion (inlet I3). The flow rates of inlets I1, I2, and I3 were precisely controlled using programmable syringe pumps (Legato 110P, KD Scientific, USA). Under appropriate combinations of the flow rates, microcapsules could be stably and continuously fabricated (Figure 1A, step (a)). Sodium alginate in the shell layer of the droplets was further gelled to form calcium alginate hydrogel when immersed into 0.15 mol L−1 aqueous CaCl2 solution (buffered at pH=7.2 using 0.01 mol L−1 HEPES) for 20 min (Figure 1A, step (b)). The excess oil on top was aspirated off after the crosslinked microcapsules settled down, and then 0.9% NaCl aqueous solution was added to stir up the remaining oil that was aspirated off for collecting the microcapsules from the bottom of the solution (Figure 1A, step (c)). The collected microcapsules were kept at 4 °C for further use.
Size and shell thickness controllable microcapsules can be fabricated (Figure 1B) by adjusting the flow rates of inlets I1, I2 and I3. Both the production rate of the microcapsules and the number of the cells encapsulated per microcapsule are controllable by adjusting the combination of the flow rates and the cell concentration. The microcapsules with an outer diameter and a shell thickness of 525.9±8.6 μm and 53.5±6.6 μm at the flow rate combination 10-20-600 μL min−1 (core-shell-oil) were used for all cell encapsulation and vitrification experiments. The cell encapsulation rate was around 500 cells s−1 at the cell density of 2.47×107 cells mL−1.
Cryopreservation of encapsulated pADSCs
The above mentioned four CPAs (#1 - #4) were tested in this study, and in which Dulbecco’s modified Eagle medium/Ham’s Nutrient Mixture F-12 (DMEM/F12) with 80% fetal bovine serum (FBS) was used as the base solution. To minimize the toxicity while loading enough CPAs into the cells, the nonencapsulated cells were incubated in CPAs #1 - #4 for 10 min, while the encapsulated cells were incubated in 2 mol L−1 penetrating CPAs (1 mol L−1 PROH + 1 mol L−1 EG) for 30 min first and then incubated in CPAs #1 - #4 for another 10 min at 4°C (to obtain a rebalance for nonpenetrating CPAs, trehalose and/or dextran T10). After CPA addition, the cells (either nonencapsulated or encapsulated) were loaded into 0.25 ml conventional plastic straws (PS) (FHK, Japan) (step (d), Figure 1A), and the cell-laden PS was plunged into liquid nitrogen and held there for 5 min to achieve thermal equilibrium between the sample and liquid nitrogen (LN2) (step (e), Figure 1A). After that, the sample was rapidly immersed into 37 °C water bath for fast warming (step (f), Figure 1A). Once warmed, CPA removal was conducted in a two-step procedure: the cells (nonencapsulated group) or the microcapsules (encapsulated group) were incubated with 0.5 mol L−1 PROH and 0.5 mol L−1 EG at room temperature for 5 min, and then equal volume of DMEM/F12 was added for another 5 min of incubation. After that, the cells (for the encapsulated group, the cells were released from the microcapsules by liquefying the alginate hydrogel using 0.075 mol L−1 trisodium citrate) were centrifuged at 1100 RPM (~104 g) for 5 min at 4 °C using an Eppendorf 5424R centrifuge (Eppendorf, Hamburg, Germany), and resuspended in phosphate buffer solution (PBS) for viability assays after removal of the supernatant.
Cell viability assays
The viability of the cells post cryopreservation was evaluated using an acridine orange/ethidium bromide (AO/EB) staining kit (KeyGen BioTECH Co., Ltd., China). Equal volumes of AO (0.5 g mL−1) and EB (0.5 g mL−1) were mixed to make the fluorescence staining solution. Green fluorescence (AO) is an indicator of live cells (excitation: 420–485 nm, emission: ~515 nm), while red fluorescence is an indicator of dead cells (excitation: 460–550 nm, emission: ~590 nm). The fluorescence staining solution (1 μL) was added into the cell suspension (25 μL), incubated for 3 min at room temperature, and then the live and dead cells (stained green and red, respectively) were checked under an inverted fluorescence microscope (Ti-U, Nikon, Japan) with a 10× objective lens. Fluorescence images of the cells were taken using a CCD camera (DS-Ri1, Nikon, Japan), and then the live and dead cells were counted using the software Image J (NIH, Bethesda, USA).
Assessment of attachment efficiency and proliferation
To evaluate cell attachment efficiency, the pADSCs were incubated with fresh DMEM/F12 supplemented with 10% FBS in 12-well plates for 1 d, and then both the attached and unattached cells were counted using the Muse™ cell Analyzer (Merck Millipore, Germany) to calculate the attachment efficiency.
To quantify cell proliferation, the cells were seeded in 96-well plates for culturing. After 1, 2, and 3 d, the medium was removed and the cells were washed using PBS twice, and then the cell number in each well was determined using the CCK-8 kit according to the instruction of the manufacturer (Dojindo, Kumamoto, Japan). Proliferation was calculated as the relative cell number on day 2 and day 3 to that on day 1.
Immunostaining
For immunofluorescent staining of three surface markers CD44(+), CD29(+) and CD31(-), the pADSCs were seeded on glass coverslips placed in a 6-well plate and incubated overnight. Cells attached onto the coverslips were washed with PBS for three times before being fixed with Immunol Staining Fix Solution (Beyotime, Haimen, China) at room temperature for 15 min. The fixed cells were washed with PBS for three times, and incubated in Immunol Staining Blocking Buffer (Beyotime, Haimen, China) for 1 h to block potential nonspecific binding, followed by overnight incubation at 4 °C with the primary antibody (1:10 dilution, according to the instruction of the manufacturer) of mouse anti-human CD44 monoclonal antibody (Proteintech, Wuhan, China), purified mouse anti-pig CD29 (BD Pharmingen, USA) or purified mouse anti-human CD31 (BD Pharmingen, USA). After that, the samples were incubated at 37 °C for 30 min, washed with PBS for three times, and incubated in dark at room temperature for 1 h with the secondary antibody Goat anti-mouse secondary coupled to Antibody Alexa Fluor 488 (Thermo Fisher Scientific, USA) (1:50 dilution). The samples were then washed with PBS for three times and stained for nuclei using DAPI (Beyotime, Haimen, China) at room temperature for 10 min. Then, Antifade Mounting Medium (Beyotime, Haimen, China) was added for further analysis under an inverted fluorescent microscope (Nikon Eclipse Ti-U, Tokyo, Japan).
Assessment of cell surface marker expression
To quantify the expression of CD44(+), CD29(+), CD90(+), and CD31(-) surface markers with flow cytometry, pADSCs at 90% confluence were digested with trypsin/EDTA and washed three times with PBS, followed by incubation at 4 °C with primary antibody (1:1000 dilution, according to the instruction of the manufacturer) of CD44-FITC (Invitrogen, USA), CD31-FITC (Abcam, USA), CD29-FITC (BD Pharmingen, USA), and CD90-FITC (BD Pharmingen, USA) for 1 h. The cells were then washed with PBS for three times for further analysis using a flow cytometer (BD FACSVerse, NJ, USA) together with the FACS suite software.
Assessment of gene marker expression
Quantitative real-time polymerase chain reaction (qRT-PCR) analysis was used to quantify the expressions of the four stem cell pluripotency gene markers: Oct4, Sox2, Klf4, and Nanog. After 4–5 days of culture in 60 mm petri dish, the cells were digested with trypsin/EDTA and washed three times with PBS. Total RNA was isolated using Trizol (Invitrogen, Carlsbad, CA) followed by DNase (Ambion, Houston, TX, USA) treatment and reverse transcription with iScript cDNA Synthesis Kit (Bio-rad, Hercules, CA). The qRT-PCR was performed using SYBR Green master mix (Vazyme, Nanjing, China) on a Bio-Rad iCycler. Primer sequences used were given in Table S3. All samples were normalized to the 18S rRNA. The fold change of target mRNA expression was calculated based on threshold cycle (Ct), where ΔCt = Cttarget – Ct18S and Δ(ΔCt) =ΔCtControl – ΔCtIndicated condition.
Assessment of induced multi-lineage differentiation
The multi-lineage differentiation potential of pADSCs was tested by induced adipogenic and osteogenic differentiation. The cells were seeded in 12-well plates for culture and the medium was changed every other day till the cell confluency reached 80–90%. The adipogenic (or osteogenic) differentiation medium (ThermoScientific, USA) was added for further culture for 14 d (or 21 d) with the medium being changed every 3 d. The differentiated cells were harvested, washed with PBS, and fixed for 30 min with 4% paraformaldehyde. The fixed cells were washed with PBS and stained with Oil red O (Sigma, USA) for 60 min or Alizarin red S stain (Sigma, USA) for 10 min. The stained cells were washed three times with PBS for further examination under an inverted fluorescence microscope (Nikon Eclipse Ti-U, Nikon, Japan).
Statistical analysis
For the studies on the size controllability of the microcapsules, 60–89 microcapsules were measured for each combination of the core, shell, and oil flow rates. The diameter of the microcapsules was measured from their projected areas on the micrographs using ImageJ (Version 1.51q, NIH) with manually circled boundaries. The shell thickness of the microcapsules was calculated by subtracting the diameter of the core from that of the microcapsules, and then dividing by 2. The assessments on cell viability were repeated 4–5 times independently, while the evaluations on proliferation and relative gene expression were repeated 3 times independently. All the data were presented as mean ± standard deviation (SD). The statistical analysis was performed using the Student’s two-tailed t-test in Microsoft Excel with the p-value less than 0.05 being considered statistically significant.
Supplementary Material
Acknowledgments
X.L. and K.Z. contributed equally to this work. This work was partially supported by grants from NSFC (Nos. 51476160 and 51528601) to G.Z. and NIH (R01EB012108 and R01EB023632) to X.H.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Prof. Gang Zhao, Department of Electronic Science and Technology, University of Science and Technology of China, Hefei, Anhui 230027, China
Xiaoli Liu, Department of Electronic Science and Technology, University of Science and Technology of China, Hefei, Anhui 230027, China.
Kaixuan Zhu, Department of Electronic Science and Technology, University of Science and Technology of China, Hefei, Anhui 230027, China.
Xiaoming He, Department of Biomedical Engineering, The Ohio State University, Columbus, Ohio 43210, USA.
References
- 1.Wilson JL, McDevitt TC. Biotechnology and bioengineering. 2013;110:667. doi: 10.1002/bit.24802. [DOI] [PMC free article] [PubMed] [Google Scholar]; Li YH, Huang GY, Zhang XH, Li BQ, Chen YM, Lu TL, Lu TJ, Xu F. Advanced Functional Materials. 2013;23:660. [Google Scholar]
- 2.Murua A, Orive G, Hernandez RM, Pedraz JL. Biomaterials. 2009;30:3495. doi: 10.1016/j.biomaterials.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 3.Bhakta G, Lee KH, Magalhaes R, Wen F, Gouk SS, Hutmacher DW, Kuleshova LL. Biomaterials. 2009;30:336. doi: 10.1016/j.biomaterials.2008.09.030. [DOI] [PubMed] [Google Scholar]
- 4.Agarwal P, Choi JK, Huang H, Zhao S, Dumbleton J, Li J, He X. Part Part Syst Charact. 2015;32:809. doi: 10.1002/ppsc.201500025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang WJ, He XM. J Healthc Eng. 2011;2:427. doi: 10.1260/2040-2295.2.4.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orive G, Hernandez RM, Gascon AR, Calafiore R, Chang TMS, De Vos P, Hortelano G, Hunkeler D, Lacik I, Shapiro AMJ, Pedraz JL. Nature medicine. 2003;9:104. doi: 10.1038/nm0103-104. [DOI] [PubMed] [Google Scholar]
- 7.Vegas AJ, Veiseh O, Doloff JC, Ma M, Tam HH, Bratlie K, Li J, Bader AR, Langan E, Olejnik K, Fenton P, Kang JW, Hollister-Locke J, Bochenek MA, Chiu A, Siebert S, Tang K, Jhunjhunwala S, Aresta-Dasilva S, Dholakia N, Thakrar R, Vietti T, Chen M, Cohen J, Siniakowicz K, Qi M, McGarrigle J, Lyle S, Harlan DM, Greiner DL, Oberholzer J, Weir GC, Langer R, Anderson DG. Nat Biotechnol. 2016;34:345. doi: 10.1038/nbt.3462. [DOI] [PMC free article] [PubMed] [Google Scholar]; Vegas AJ, Veiseh O, Gurtler M, Millman JR, Pagliuca FW, Bader AR, Doloff JC, Li J, Chen M, Olejnik K, Tam HH, Jhunjhunwala S, Langan E, Aresta-Dasilva S, Gandham S, McGarrigle JJ, Bochenek MA, Hollister-Lock J, Oberholzer J, Greiner DL, Weir GC, Melton DA, Langer R, Anderson DG. Nature medicine. 2016;22:306. doi: 10.1038/nm.4030. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhao S, Xu Z, Wang H, Reese BE, Gushchina LV, Jiang M, Agarwal P, Xu J, Zhang M, Shen R, Liu Z, Weisleder N, He X. Nature communications. 2016;7:13306. doi: 10.1038/ncomms13306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmad HF, Sambanis A. Acta Biomater. 2013;9:6814. doi: 10.1016/j.actbio.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Serra M, Correia C, Malpique R, Brito C, Jensen J, Bjorquist P, Carrondo MJ, Alves PM. PLoS One. 2011;6:e23212. doi: 10.1371/journal.pone.0023212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang HS, Choi JK, Rao W, Zhao ST, Agarwal P, Zhao G, He XM. Advanced Functional Materials. 2015;25:6839. doi: 10.1002/adfm.201503047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosland GV, Svendsen A, Torsvik A, Sobala E, McCormack E, Immervoll H, Mysliwietz J, Tonn JC, Goldbrunner R, Lonning PE, Bjerkvig R, Schichor C. Cancer research. 2009;69:5331. doi: 10.1158/0008-5472.CAN-08-4630. [DOI] [PubMed] [Google Scholar]; Izadpanah R, Kaushal D, Kriedt C, Tsien F, Patel B, Dufour J, Bunnell BA. Cancer research. 2008;68:4229. doi: 10.1158/0008-5472.CAN-07-5272. [DOI] [PMC free article] [PubMed] [Google Scholar]; Froelich K, Mickler J, Steusloff G, Technau A, Ramos Tirado M, Scherzed A, Hackenberg S, Radeloff A, Hagen R, Kleinsasser N. Cytotherapy. 2013;15:767. doi: 10.1016/j.jcyt.2012.12.009. [DOI] [PubMed] [Google Scholar]; Capelli C, Pedrini O, Cassina G, Spinelli O, Salmoiraghi S, Golay J, Rambaldi A, Giussani U, Introna M. Haematologica. 2014;99:e94. doi: 10.3324/haematol.2014.104711. [DOI] [PMC free article] [PubMed] [Google Scholar]; Pan Q, Fouraschen SM, de Ruiter PE, Dinjens WN, Kwekkeboom J, Tilanus HW, van der Laan LJ. Experimental biology and medicine. 2014;239:105. doi: 10.1177/1535370213506802. [DOI] [PubMed] [Google Scholar]; Liu H, Lin J, Roy K. Biomaterials. 2006;27:5978. doi: 10.1016/j.biomaterials.2006.05.053. [DOI] [PubMed] [Google Scholar]; Scadden DT. Nature. 2006;441:1075. doi: 10.1038/nature04957. [DOI] [PubMed] [Google Scholar]; McDevitt TC, Palecek SP. Current opinion in biotechnology. 2008;19:527. doi: 10.1016/j.copbio.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kondo E, Wada KI, Hosokawa K, Maeda M. Biotechnology and bioengineering. 2016;113:237. doi: 10.1002/bit.25704. [DOI] [PubMed] [Google Scholar]; Fang C, Ji F, Shu Z, Gao D. Lab Chip. 2017;17:951. doi: 10.1039/c6lc01523a. [DOI] [PubMed] [Google Scholar]; Zhao G, Zhang Z, Zhang Y, Chen Z, Niu D, Cao Y, He X. Lab Chip. 2017;17:1297. doi: 10.1039/c6lc01532h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tasoglu S, Gurkan UA, Wang S, Demirci U. Chemical Society reviews. 2013;42:5788. doi: 10.1039/c3cs60042d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demirci U, Montesano G. Lab Chip. 2007;7:1428. doi: 10.1039/b705809h. [DOI] [PubMed] [Google Scholar]
- 15.Dou R, Saunders RE, Mohamet L, Ward CM, Derby B. Lab Chip. 2015;15:3503. doi: 10.1039/c5lc00674k. [DOI] [PubMed] [Google Scholar]
- 16.Canaple L, Nurdin N, Angelova N, Saugy D, Hunkeler D, Desvergne B. J Hepatol. 2001;34:11. doi: 10.1016/s0168-8278(00)00086-6. [DOI] [PubMed] [Google Scholar]
- 17.Heng BC, Yu YJH, Ng SC. J Microencapsul. 2004;21:455. doi: 10.1080/02652040410001729250. [DOI] [PubMed] [Google Scholar]
- 18.Heng BC, Yu H, Ng SC. Biotechnology and bioengineering. 2004;85:202. doi: 10.1002/bit.10837. [DOI] [PubMed] [Google Scholar]
- 19.Stensvaag V, Furmanek T, Lonning K, Terzis AJA, Bjerkvig R, Visted T. Cell transplantation. 2004;13:35. doi: 10.3727/000000004772664879. [DOI] [PubMed] [Google Scholar]; Wu YN, Yu HR, Chang S, Magalhaes R, Kuleshova LL. Tissue Eng. 2007;13:649. doi: 10.1089/ten.2006.0075. [DOI] [PubMed] [Google Scholar]
- 20.Zhou DB, Vacek I, Sun AM. Transplantation. 1997;64:1112. doi: 10.1097/00007890-199710270-00005. [DOI] [PubMed] [Google Scholar]
- 21.Zhou XH, Zhang D, Shi J, Wu YJ. Medicine. 2016:95. doi: 10.1097/MD.0000000000004095. [DOI] [PMC free article] [PubMed] [Google Scholar]; Meintjes M, Purcell S, Chantilis SJ, Navarrete GR, Tilley B, Lee KL, Thomas MR, Qin Y, Davidson K, Mehta R, Guerami A. Fertility and sterility. 2015;104:E188. [Google Scholar]; Naik BR, Rao BS, Vagdevi R, Gnanprakash M, Amarnath D, Rao VH. Animal reproduction science. 2005;86:329. doi: 10.1016/j.anireprosci.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 22.Wang JY, Zhao G, Zhang ZL, Xu XL, He XM. Acta Biomaterialia. 2016;33:264. doi: 10.1016/j.actbio.2016.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mazur P. J Gen Physiol. 1963;47:347. doi: 10.1085/jgp.47.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fahy GM, Wowk B, Wu J, Paynter S. Cryobiology. 2004;48:22. doi: 10.1016/j.cryobiol.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 25.Zhao G, Takamatsu H, He X. J Appl Phys. 2014;115:144701. doi: 10.1063/1.4870826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ji L, de Pablo JJ, Palecek SP. Biotechnology and bioengineering. 2004;88:299. doi: 10.1002/bit.20243. [DOI] [PubMed] [Google Scholar]
- 27.Choi JK, Yue T, Huang H, Zhao G, Zhang M, He X. Cryobiology. 2015;71:350. doi: 10.1016/j.cryobiol.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fahy GM, MacFarlane DR, Angell CA, Meryman HT. Cryobiology. 1984;21:407. doi: 10.1016/0011-2240(84)90079-8. [DOI] [PubMed] [Google Scholar]
- 29.El Assal R, Guven S, Gurkan UA, Gozen I, Shafiee H, Dalbeyler S, Abdalla N, Thomas G, Fuld W, Illigens BMW, Estanislau J, Khoory J, Kaufman R, Zylberberg C, Lindeman N, Wen Q, Ghiran I, Demirci U. Adv Mater. 2014;26:5815. doi: 10.1002/adma.201400941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang XH, Khimji I, Shao L, Safaee H, Desai K, Keles HO, Gurkan UA, Kayaalp E, Nurreddin A, Anchan RM, Maas RL, Demirci U. Nanomedicine. 2012;7:553. doi: 10.2217/nnm.11.145. [DOI] [PMC free article] [PubMed] [Google Scholar]; Samot J, Moon S, Shao L, Zhang X, Xu F, Song Y, Keles HO, Matloff L, Markel J, Demirci U. PLoS One. 2011;6:e17530. doi: 10.1371/journal.pone.0017530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang W, Yang G, Zhang A, Xu LX, He X. Biomed Microdevices. 2010;12:89. doi: 10.1007/s10544-009-9363-z. [DOI] [PubMed] [Google Scholar]
- 32.Zhang WJ, Zhao ST, Rao W, Snyder J, Choi JK, Wang JF, Khan IA, Saleh NB, Mohler PJ, Yu JH, Hund TJ, Tang CB, He XM. J Mater Chem B. 2013;1:1002. doi: 10.1039/C2TB00058J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma ML, Chiu A, Sahay G, Doloff JC, Dholakia N, Thakrar R, Cohen J, Vegas A, Chen DL, Bratlie KM, Dang T, York RL, Hollister-Lock J, Weir GC, Anderson DG. Advanced healthcare materials. 2013;2:667. doi: 10.1002/adhm.201200341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Agarwal P, Zhao ST, Bielecki P, Rao W, Choi JK, Zhao Y, Yu JH, Zhang WJ, He XM. Lab Chip. 2013;13:4525. doi: 10.1039/c3lc50678a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim C, Chung S, Kim YE, Lee KS, Lee SH, Oh KW, Kang JY. Lab Chip. 2011;11:246. doi: 10.1039/c0lc00036a. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen DK, Son YM, Lee NE. Advanced healthcare materials. 2015;4:1537. doi: 10.1002/adhm.201500133. [DOI] [PubMed] [Google Scholar]
- 37.Choi YH, Lee JH, Yuk SH, Suh SH, Yoon KH. J Bioact Compat Pol. 2006;21:71. [Google Scholar]; Zhao ST, Xu ZB, Wang H, Reese BE, Gushchina LV, Jiang M, Agarwal P, Xu JS, Zhang MJ, Shen RL, Liu ZG, Weisleder N, He XM. Nature communications. 2016;7 doi: 10.1038/ncomms13306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shu Z, Kang X, Chen H, Zhou X, Purtteman J, Yadock D, Heimfeld S, Gao D. Cytotherapy. 2010;12:161. doi: 10.3109/14653240903377037. [DOI] [PMC free article] [PubMed] [Google Scholar]; Rubinstein P, Dobrila L, Rosenfield RE, Adamson JW, Migliaccio G, Migliaccio AR, Taylor PE, Stevens CE. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:10119. doi: 10.1073/pnas.92.22.10119. [DOI] [PMC free article] [PubMed] [Google Scholar]; Areman EM, Simonis TB, Carter CS, Read EJ, Klein HG. Transfusion. 1988;28:151. doi: 10.1046/j.1537-2995.1988.28288179020.x. [DOI] [PubMed] [Google Scholar]; Zhou X, Kang X, Shu Z, Chen H, Ding W, Du P, Yadock D, Chi Liu C, Chung JH, Heimfeld S, Gao D. Biopreserv Biobank. 2009;7:107. doi: 10.1089/bio.2009.0005. [DOI] [PubMed] [Google Scholar]
- 39.Wu Y, Yu H, Chang S, Magalhaes R, Kuleshova LL. Tissue Eng. 2007;13:649. doi: 10.1089/ten.2006.0075. [DOI] [PubMed] [Google Scholar]; Stensvaag V, Furmanek T, Lonning K, Terzis AJ, Bjerkvig R, Visted T. Cell transplantation. 2004;13:35. doi: 10.3727/000000004772664879. [DOI] [PubMed] [Google Scholar]; Heng BC, Yu H, Chye Ng S. Biotechnology and bioengineering. 2004;85:202. doi: 10.1002/bit.10837. [DOI] [PubMed] [Google Scholar]
- 40.Zhao ST, Agarwal P, Rao W, Huang HS, Zhang RL, Liu ZG, Yu JH, Weisleder N, Zhang WJ, He XM. Integr Biol-Uk. 2014;6:874. doi: 10.1039/c4ib00100a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang W, He X. Journal of biomechanical engineering. 2009;131:074515. doi: 10.1115/1.3153326. [DOI] [PubMed] [Google Scholar]
- 42.Morimoto Y, Tan WH, Tsuda Y, Takeuchi S. Lab Chip. 2009;9:2217. doi: 10.1039/b900035f. [DOI] [PubMed] [Google Scholar]
- 43.Suchy T, Sedlacek R, Sucharda Z, Balik K, Bouda T. Comput Method Biomec. 2013;16:255. doi: 10.1080/10255842.2013.815960. [DOI] [PubMed] [Google Scholar]; Kim M, Moon BU, Hidrovo CH. J Micromech Microeng. 2013:23. [Google Scholar]; Xiang KW, Huang GS, Zheng J, Wang XA, Li GX, Huang JY. J Polym Res. 2012:19. [Google Scholar]; McCarthy DW, Mark JE. Rubber Chemistry and Technology. 1998;71:906. [Google Scholar]
- 44.Velasco D, Tumarkin E, Kumacheva E. Small. 2012;8:1633. doi: 10.1002/smll.201102464. [DOI] [PubMed] [Google Scholar]
- 45.Dovgan B, Barlic A, Knezevic M, Miklavcic D. J Membr Biol. 2017;250:1. doi: 10.1007/s00232-016-9916-z. [DOI] [PubMed] [Google Scholar]; Swioklo S, Constantinescu A, Connon CJ. Stem cells translational medicine. 2016;5:339. doi: 10.5966/sctm.2015-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ray SS, Pramanik K, Sarangi SK, Jain N. Biotechnol Lett. 2016;38:1397. doi: 10.1007/s10529-016-2111-6. [DOI] [PubMed] [Google Scholar]; Hassan W, Dong Y, Wang W. Stem Cell Res Ther. 2013;4:32. doi: 10.1186/scrt182. [DOI] [PMC free article] [PubMed] [Google Scholar]; Rubin JP, DeFail A, Rajendran N, Marra KG. J Drug Target. 2009;17:207. doi: 10.1080/10611860802669231. [DOI] [PubMed] [Google Scholar]
- 46.Chen WY, Shu ZQ, Gao DY, Shen AQ. Advanced healthcare materials. 2016;5:223. doi: 10.1002/adhm.201500515. [DOI] [PubMed] [Google Scholar]
- 47.Zheng Y, Zhao G, Fazil Panhwar F, He X. Tissue engineering. Part C, Methods. 2016;22:964. doi: 10.1089/ten.tec.2016.0335. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lai D, Ding J, Smith GW, Smith GD, Takayama S. Human reproduction. 2015;30:37. doi: 10.1093/humrep/deu284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choi JK, Huang HS, He XM. Cryobiology. 2015;70:269. doi: 10.1016/j.cryobiol.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; Fahy GM, Wowk B, Pagotan R, Chang A, Phan J, Thomson B, Phan L. Organogenesis. 2009;5:167. doi: 10.4161/org.5.3.9974. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wowk B, Leitl E, Rasch CM, Mesbah-Karimi N, Harris SB, Fahy GM. Cryobiology. 2000;40:228. doi: 10.1006/cryo.2000.2243. [DOI] [PubMed] [Google Scholar]; Fahy GM. Cryobiology. 1987;24:580. doi: 10.1016/0011-2240(87)90023-x. [DOI] [PubMed] [Google Scholar]; Fahy GM. Progress in clinical and biological research. 1986;224:305. [PubMed] [Google Scholar]; Fahy GM, Macfarlane DR. Cryobiology. 1982;19:668. [Google Scholar]; Fahy GM. Cryobiology. 1981;18:617. doi: 10.1016/0011-2240(81)90206-6. [DOI] [PubMed] [Google Scholar]
- 49.Matsubara H. J Card Fail. 2008;14:S144. [Google Scholar]
- 50.Zhao G, Fu JP. Biotechnology Advances. 2017;35:323. doi: 10.1016/j.biotechadv.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]; Karlsson JOM, Cravalho EG, Rinkes IHMB, Tompkins RG, Yarmush ML, Toner M. Biophys J. 1993;65:2524. doi: 10.1016/S0006-3495(93)81319-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; Mazur P, Rall WF, Leibo SP. Cell Biophys. 1984;6:197. doi: 10.1007/BF02788619. [DOI] [PubMed] [Google Scholar]; Mazur P. Am J Physiol. 1984;247:C125. doi: 10.1152/ajpcell.1984.247.3.C125. [DOI] [PubMed] [Google Scholar]; Mazur P, Leibo SP, Chu EH. Exp Cell Res. 1972;71:345. doi: 10.1016/0014-4827(72)90303-5. [DOI] [PubMed] [Google Scholar]
- 51.Karlsson JOM, Cravalho EG, Toner M. J Appl Phys. 1994;75:4442. [Google Scholar]
- 52.Bourin P, Bunnell BA, Casteilla L, Dominici M, Katz AJ, March KL, Redl H, Rubin JP, Yoshimura K, Gimble JM. Cytotherapy. 2013;15:641. doi: 10.1016/j.jcyt.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fahy GM, Wowk B. Methods in molecular biology. 2015;1257:21. doi: 10.1007/978-1-4939-2193-5_2. [DOI] [PubMed] [Google Scholar]
- 54.Fahy GM, Wowk B, Wu J, Phan J, Rasch C, Chang A, Zendejas E. Cryobiology. 2004;48:157. doi: 10.1016/j.cryobiol.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 55.Zhao G, Xu Y, Ding WP, Hu MB. Cryoletters. 2013;34:40. [PubMed] [Google Scholar]
- 56.Zvibel I, Smets F, Soriano H. Cell transplantation. 2002;11:621. doi: 10.3727/000000002783985404. [DOI] [PubMed] [Google Scholar]
- 57.Terry C, Hughes RD, Mitry RR, Lehec SC, Dhawan A. Cell transplantation. 2007;16:639. doi: 10.3727/000000007783465000. [DOI] [PubMed] [Google Scholar]
- 58.Ollero M, Bescos O, Cebrian-Perez JA, Muino-Blanco T. Theriogenology. 1998;49:547. doi: 10.1016/s0093-691x(98)00006-5. [DOI] [PubMed] [Google Scholar]
- 59.Wowk B. Cryobiology. 2010;60:11. doi: 10.1016/j.cryobiol.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 60.Takahashi H, Katagiri C, Ueno S, Inoue K. Cryobiology. 2008;57:75. doi: 10.1016/j.cryobiol.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 61.Matsumura K, Bae JY, Kim HH, Hyon SH. Cryobiology. 2011;63:76. doi: 10.1016/j.cryobiol.2011.05.003. [DOI] [PubMed] [Google Scholar]; Aksan A, Toner M. Langmuir. 2004;20:5521. doi: 10.1021/la0355186. [DOI] [PubMed] [Google Scholar]; Shaw JM, Kuleshova LL, MacFarlane DR, Trounson AO. Cryobiology. 1997;35:219. doi: 10.1006/cryo.1997.2043. [DOI] [PubMed] [Google Scholar]; Fahy GM, Saur J, Williams RJ. Cryobiology. 1990;27:492. doi: 10.1016/0011-2240(90)90038-6. [DOI] [PubMed] [Google Scholar]; Boutron P, Kaufmann A. Cryobiology. 1978;15:93. doi: 10.1016/0011-2240(78)90012-3. [DOI] [PubMed] [Google Scholar]
- 62.Boutron P. Cryobiology. 1988;25:569. doi: 10.1016/0011-2240(88)90019-3. [DOI] [PubMed] [Google Scholar]
- 63.Zhang XH, Catalano PN, Gurkan UA, Khimji I, Demirci U. Nanomedicine. 2011;6:1115. doi: 10.2217/nnm.11.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Song YS, Adler D, Xu F, Kayaalp E, Nureddin A, Anchan RM, Maas RL, Demirci U. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:4596. doi: 10.1073/pnas.0914059107. [DOI] [PMC free article] [PubMed] [Google Scholar]; Karlsson JOM. Cryobiology. 2010;60:43. doi: 10.1016/j.cryobiol.2009.07.004. [DOI] [PubMed] [Google Scholar]; Karlsson JOM. Science. 2002;296:655. doi: 10.1126/science.296.5568.655d. [DOI] [PubMed] [Google Scholar]; Karlsson JOM. Cryobiology. 2001;42:154. doi: 10.1006/cryo.2001.2318. [DOI] [PubMed] [Google Scholar]
- 65.Shi M, Ling K, Yong KW, Li Y, Feng S, Zhang X, Pingguan-Murphy B, Lu TJ, Xu F. Sci Rep. 2015;5:17928. doi: 10.1038/srep17928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang H, Zhao G, Zhang Y, Xu J, Toth TL, He X. Acs Biomater Sci Eng. 2017;3:1758. doi: 10.1021/acsbiomaterials.7b00201. [DOI] [PMC free article] [PubMed] [Google Scholar]; Yang G, Veres M, Szalai G, Zhang AL, Xu LX, He XM. Annals of biomedical engineering. 2011;39:580. doi: 10.1007/s10439-010-0158-4. [DOI] [PubMed] [Google Scholar]
- 67.Singh A, Kim W, Kim Y, Jeong K, Kang CS, Kim Y, Koh J, Mahajan SD, Prasad PN, Kim S. Advanced Functional Materials. 2016;26:7057. doi: 10.1002/adfm.201602808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mulligan MK, Rothstein JP. Microfluid Nanofluid. 2012;13:65. [Google Scholar]
- 69.Korczyk PM, Dolega ME, Jakiela S, Jankowski P, Makulska S, Garstecki P. J Flow Chem. 2015;5:110. [Google Scholar]; Tendulkar S, Mirmalek-Sani SH, Childers C, Saul J, Opara EC, Ramasubramanian MK. Biomed Microdevices. 2012;14:461. doi: 10.1007/s10544-011-9623-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shang LR, Cheng Y, Wang J, Ding HB, Rong F, Zhao YJ, Gu ZZ. Lab Chip. 2014;14:3489. doi: 10.1039/c4lc00698d. [DOI] [PubMed] [Google Scholar]; Wang W, Xie R, Ju XJ, Luo T, Liu L, Weitz DA, Chu LY. Lab Chip. 2011;11:1587. doi: 10.1039/c1lc20065h. [DOI] [PubMed] [Google Scholar]
- 71.Mirabet V, Alvarez M, Solves P, Ocete D, Gimeno C. Cryobiology. 2012;64:121. doi: 10.1016/j.cryobiol.2011.12.005. [DOI] [PubMed] [Google Scholar]; Harris DT, Mapother ME, Goodman C. Transfusion. 2000;40:111s. [Google Scholar]
- 72.Zhang WJ, Zhao XT, He XM. Archives of Stem Cell Research. 2015;1004(6) [Google Scholar]
- 73.Zhang Y, Wei C, Zhang PF, Li X, Liu T, Pu Y, Li YS, Cao ZB, Cao HG, Liu Y, Zhang XR, Zhang YH. Plos One. 2014;9 doi: 10.1371/journal.pone.0085089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rao W, Huang H, Wang H, Zhao S, Dumbleton J, Zhao G, He X. ACS applied materials & interfaces. 2015;7:5017. doi: 10.1021/acsami.5b00655. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.