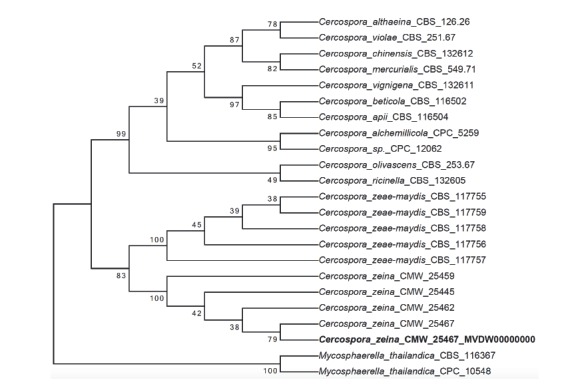
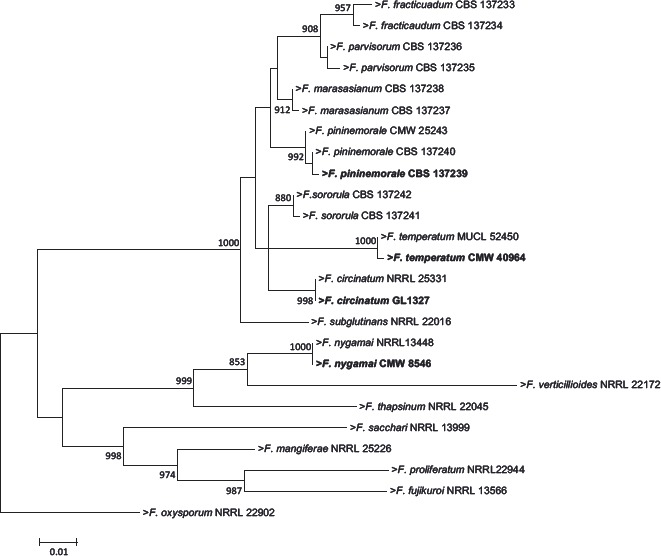
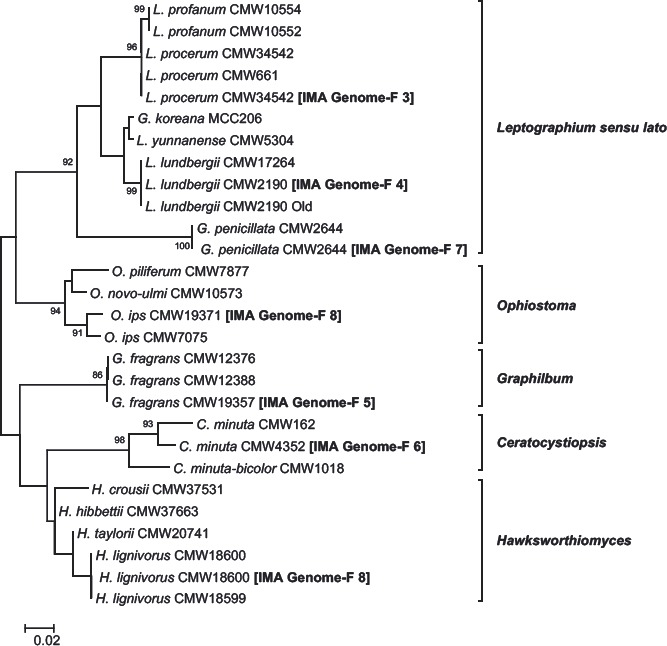
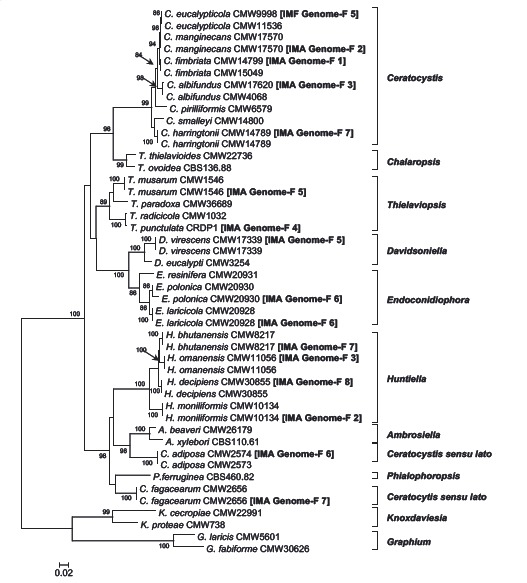
Abstract
The genomes of Cercospora zeina, Fusarium pininemorale, Hawksworthiomyces lignivorus, Huntiella decipiens, and Ophiostoma ips are presented in this genome announcement. Three of these genomes are from plant pathogens and otherwise economically important fungal species. Fusarium pininemorale and H. decipiens are not known to cause significant disease but are closely related to species of economic importance. The genome sizes range from 25.99 Mb in the case of O. ips to 4.82 Mb for H. lignivorus. These genomes include the first reports of a genome from the genus Hawksworthiomyces. The availability of these genome data will allow the resolution of longstanding questions regarding the taxonomy of these species. In addition these genome sequences through comparative studies with closely related organisms will increase our understanding of how these species or close relatives cause disease.
Keywords: Gray Leaf Spot, maize, insect vectored fungi, Fusarium fujikuroi species complex, Ophiostomatales, wood decay, Ceratocystidaceae, blue stain
Acknowledgments
Genome sequencing of Cercospora zeina was done by the Phillip SanMiguel, Purdue Genomics Core Facility (USA), and was funded by National Research Foundation (NRF) CPRR, South Africa. The genome sequencing assembly and analyses of the Fusarium pininemorale, Hawksworthiomyces lignivorus, Huntiella decipiens, and Ophiostoma ips genomes were co-funded by the Tree Protection Co-operative Programme (TPCP) and the DST-NRF Centre of Excellence in Tree Health Biotechnology (CTHB) at the Forestry and Agricultural Biotechnology Institute (FABI), and the DST-NRF South African Research Chairs Initiative (SARChI) in Fungal Genomics.
REFERENCES
- Abeel T, Van Parys T, Saeys Y, Galagan J, Van de Peer Y. (2012) GenomeView: a next-generation genome browser. Nucleic Acids Research 40: e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. (1990) Basic local alignment search tool. Journal of Molecular Biology 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, et al. (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology 19: 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes I, Gaur A, Burgess T, Roux J, Wingfield BD, Wingfield MJ. (2001) Microsatellite markers reflect intra-specific relationships between isolates of the vascular wilt pathogen Ceratocystis fimbriata. Molecular Plant Pathology 2: 319–325. [DOI] [PubMed] [Google Scholar]
- Belbahri L. (2015) Genome sequence of Ceratocystis platani, a major pathogen of plane trees. http://www.ncbi.nlm.nih.gov/nuccore/814603118. [Google Scholar]
- Berger DK, Carstens M, Korsman JN, Middleton F, Kloppers FJ, et al. (2014) Mapping QTL conferring resistance in maize to gray leaf spot disease caused by Cercospora zeina. BMC Genetics 15: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetzer M, Pirovano W. (2012) Toward almost closed genomes with GapFiller. Genome Biology 13: R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. (2011) Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27: 578–579. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel BL, Korf I, Robb SM, Parra G, Ross E, et al. (2008) MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Research 18: 188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeau AM, Dufour J, Bouvet GF, Jacobi V, Nigg M, et al. (2015) Functional annotation of the Ophiostoma novo-ulmi genome: insights into the phytopathogenicity of the fungal agent of Dutch elm disease. Genome Biology and Evolution 7: 410–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crous PW, Groenewald JZ, Groenewald M, Caldwell P, Braun U, et al. (2006) Species of Cercospora associated with grey leaf spot of maize. Studies in Mycology 55: 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darribo D, Taboada GL, Doallo R, Posada D. (2012) jModelTest 2: More models, new heuristics and parallel computing. Nature Methods 9: 772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Beer ZW, Duong TA, Barnes I, Wingfield BD, Wingfield MJ. (2014) Redefining Ceratocystis and allied genera. Studies in Mycology 79: 187–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Beer ZW, Wingfield MJ. (2013) Emerging lineages in the Ophiostomatales. In: The Ophiostomatoid Fungi: expanding frontiers (Seifert KA, De Beer ZW, Wingfield MJ, et al., eds): 21–46. [CBS Biodiversity Series no. 12.] Utrecht: CBS-KNAW Fungal Biodiversity Institute. [Google Scholar]
- De Vos L, Steenkamp ET, Martin SH, Santana QC, Fourie G, et al. (2014) Genome-wide macrosynteny among Fusarium species in the Gibberella fujikuroi complex revealed by amplified fragment lengthp-olymorphisms. PloS One 9: e114682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjardins A, Plattner R, Gordon T. (2000) Gibberella fujikuroi mating population A and Fusarium subglutinans from teosinte species and maize from Mexico and Central America. Mycological Research 104: 865–872. [Google Scholar]
- Duong TA, De Beer ZW, Wingfield BD, Wingfield MJ. (2013) Characterization of the mating-type genes in Leptographium procerum and Leptographium profanum. Fungal Biology 117: 411–421. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39: 783–791. [DOI] [PubMed] [Google Scholar]
- Forgetta V, Leveque G, Dias J, Grove D, Lyons R, et al. (2013) Sequencing of the Dutch Elm Disease fungus genome using the Roche/454 GS-FLX Titanium system in a comparison of multiple genomics core facilities. Journal of Biomolecular Techniques 24: 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology 52: 696–704. [DOI] [PubMed] [Google Scholar]
- Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, et al. (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Systematic Biology 59: 307–21. [DOI] [PubMed] [Google Scholar]
- Haridas S, Wang Y, Lim L, Alamouti SM, Jackman S, et al. (2013) The genome and transcriptome of the pine saprophyte Ophiostoma piceae, and a comparison with the bark beetle-associated pine pathogen Grosmannia clavigera. BMC Genomics 14: 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS. (2007) Improved pairwise alignment of genomic DNA. PhD thesis, Pennsylvania State University. [Google Scholar]
- Hepting GH, Roth ER. (1946) Pitch canker, a new disease of some southern pines. Journal of Forestry 44: 742–744. [Google Scholar]
- Herron DA, Wingfield MJ, Wingfield BD, Rodas C, Marincowitz S, et al. (2015) Novel taxa in the Fusarium fujikuroi species complex from Pinus spp. Studies in Mycology 80: 131–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff KJ, Stanke M. (2013) WebAUGUSTUS — a web service for training AUGUSTUS and predicting genes in eukaryotes. Nucleic Acids Research 41: W123–W128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshraftar S, Hung S, Khan S, Gong Y, Tyagi V, et al. (2013) Sequencing and annotation of the Ophiostoma ulmi genome. BMC Genomics. 14: 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. (2013) MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Molecular Biology and Evolution 30: 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kile G. (1993) Plant diseases caused by species of Ceratocystis sensu stricto and Chalara. St Paul, MN: American Phytopathological Society Press. [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, et al. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirisits T. (2004) Fungal associates of European bark beetles with special emphasis on the ophiostomatoid fungi. In: Bark and Wood Boring Insects in Living Trees in Europe, a synthesis (Lieutier F, Day KR, Battisti A, Grégoire JC, Evans HF, eds): 181–123. Dordrecht: Kluwer Academic Publishers. [Google Scholar]
- Kirisits T, Konrad H, Wingfield MJ, Chhetri DB. (2013) Ophiostomatoid fungi associated with the Eastern Himalayan spruce bark beetle, Ips schmutzenhoferi, in Bhutan and their pathogenicity to Picea spinulosa and Pinus wallichiana. In: The Ophiostomatoid Fungi: expanding frontiers (Seifert KA, De Beer ZW, Wingfield MJ, eds): 99–112. [CBS Biodiversity Series no. 12.] Utrecht: CBS-KNAW Fungal Biodiversity Institute. [Google Scholar]
- Korf I. (2004) Gene finding in novel genomes. BMC Bioinformatics 5: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Tamura K. (2016) MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for bigger datasets. Molecular Biology and Evolution 33: 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latterell FM, Rossi AE. (1983) Gray leaf spot of corn: a disease on the move. Plant Disease 67: 842–847. [Google Scholar]
- Lieutier F, Yart A, Garcia J, Ham M, Morelet M, et al. (1989) Champignons phytopathogènes associés à deux coléoptères scolytidae du pin sylvestre (Pinus sylvestris L.) et étude préliminaire de leur agressivité envers l’hôte. Annales des Sciences Forestières 46: 201–216. [Google Scholar]
- Liew E, Laurence M, Pearce C, Shivas R, Johnson G, et al. (2016) Review of Fusarium species isolated in association with mango malformation in Australia. Australasian Plant Pathology: DOI 10.1007/s13313-016-0454-z1-13. [Google Scholar]
- Ma LJ, Van der Does HC, Borkovich KA, Coleman JJ, Daboussi MJ, et al. (2010) Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464: 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel B, Korsman J, Kloppers F, Berger D. (2009) Cercospora zeina is the causal agent of grey leaf spot disease of maize in southern Africa. European Journal of Plant Pathology 124: 577–583. [Google Scholar]
- Möller E, Bahnweg G, Sandermann H, Geiger H. (1992) A simple and efficient protocol for isolation of high molecular weight DNA from filamentous fungi, fruit bodies, and infected plant tissues. Nucleic Acids Research 20: 6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Kumar S. (2000) Molecular Evolution and Phylogenetics. New York: Oxford University Press. [Google Scholar]
- Niehaus E-M, Münsterkötter M, Proctor RH, Brown DW, Sharon A, et al. (2016) Comparative “omics” of the Fusarium fujikuroi species complex highlights differences in genetic potential and metabolite synthesis. Genome Biology and Evolution 8: 3574–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nkuekam GK, Wingfield MJ, Roux J. (2012) Ceratocystis species, including two new taxa, from Eucalyptus trees in South Africa. Australasian Plant Pathology 42: 283–311. [Google Scholar]
- Ohm RA, Feau N, Henrissat B, Schoch CL, Horwitz BA, et al. (2012) Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes fungi. PLoS Pathogens 8: e1003037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbold CT. (1931) Two blue-staining fungi associated with bark-beetle infestation of pines. Journal of Agricultural Research 43: 847–873. [Google Scholar]
- Seifert K. (1993) Sapstain of commercial lumber by species of Ophiostoma and Ceratocystis. In: Ceratocystis and Ophiostoma: taxonomy, ecology and pathogenicity (Wingfield MJ, et al., eds): 141–151. St Paul, MN: American Phytopathological Society Press. [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31: 3210–3212. [DOI] [PubMed] [Google Scholar]
- Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, et al. (2009) ABySS: a parallel assembler for short read sequence data. Genome Research 19: 1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Six DL, Wingfield MJ. (2011) The role of phytopathogenicity in bark beetle–fungus symbioses: a challenge to the classic paradigm. Annual Review of Entomology. 56: 255–272. [DOI] [PubMed] [Google Scholar]
- Stanke M, Diekhans M, Baertsch R, Haussler D. (2008) Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24: 637–644. [DOI] [PubMed] [Google Scholar]
- Stanke M, Keller O, Gunduz I, Hayes A, Waack S, Morgenstern B. (2006) AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research. 34: W435–W439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavaré S. (1986) Some probabilistic and statistical problems in the analysis of DNA sequences. In: Some Mathematical Questions in Biology: DNA sequence analysis (Muira RM, ed.): 57–86. Providence, RI: American Mathematical Society. [Google Scholar]
- Van der Nest MA, Beirn LA, Crouch JA, Demers JE, De Beer ZW, et al. (2014a) Draft genomes of Amanita jacksonii, Ceratocystis albifundus, Fusarium circinatum, Huntiella omanensis, Leptographium procerum, Rutstroemia sydowiana, and Sclerotinia echinophila. IMA Fungus 5: 473–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Nest MA, Bihon W, De Vos L, Naidoo K, Roodt D, et al. (2014b) Draft genome sequences of Diplodia sapinea, Ceratocystis manginecans, and Ceratocystis moniliformis. IMA Fungus 5: 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wyk M, Roux J, Barnes I, Wingfield BD, Wingfield MJ. (2006) Molecular phylogeny of the Ceratocystis moniliformis complex and description of C. tribiliformis sp. nov. Fungal Diversity 21: 181–201. [Google Scholar]
- Van Wyk M, Roux J, Barnes I, Wingfield BD, Chhetri DB, et al. (2004) Ceratocystis bhutanensis sp. nov., associated with the bark beetle Ips schmutzenhoferi on Picea spinulosa in Bhutan. Studies in Mycology 50: 365–379. [Google Scholar]
- Wang J, Levy M, Dunkle LD. (1998) Sibling species of Cercospora associated with gray leaf spot of maize. Phytopathology 88: 1269–1275. [DOI] [PubMed] [Google Scholar]
- Ward JMJ, Stromberg EL, Nowell DC, Nutter FW. (1999) Gray leaf spot: a disease of global importance in maize production. Plant Disease 83: 884–895. [DOI] [PubMed] [Google Scholar]
- Wiemann P, Sieber CM, Von Bargen KW, Studt L, Niehaus E-M, et al. (2013) Deciphering the cryptic genome: genome-wide analyses of the rice pathogen Fusarium fujikuroi reveal complex regulation of secondary metabolism and novel metabolites. PLoS Pathogens 9: e1003475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilken PM, Steenkamp ET, Wingfield MJ, De Beer ZW, Wingfield BD. (2013) IMA Genome-F 1: Draft nuclear genome sequence for the plant pathogen, Ceratocystis fimbriata. IMA Fungus 4: 357–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield BD, Barnes I, De Beer ZW, De Vos L, Duong TA, et al. (2015a) IMA Genome-F 5: Draft genome sequences of Ceratocystis eucalypticola, Chrysoporthe cubensis, C. deuterocubensis, Davidsoniella virescens, Fusarium temperatum, Graphilbum fragrans, Penicillium nordicum, and Thielaviopsis musarum. IMA Fungus 6: 493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield BD, Ades P, Al-Naemi F, Beirn L, Bihon W, et al. (2015b) IMA Genome-F 4: Draft genome sequences of Chrysoporthe austroafricana, Diplodia scrobiculata, Fusarium nygamai, Leptographium lundbergii, Limonomyces culmigenus, Stagonosporopsis tanaceti, and Thielaviopsis punctulata. IMA Fungus 6: 233–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield BD, Ambler JM, Coetzee MPA, De Beer ZW, Duong TA, et al. (2016a) IMA Genome-F5: Draft genome sequences of Armillaria fuscipes, Ceratocystiopsis minuta, Ceratocystis adiposa, Endoconidiophora laricicola, E. polonica and Penicillium freii DAOMC 242723. IMA Fungus 7: 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield BD, Duong TA, Hammerbacher A, Van der Nest MA, Wilson A, et al. (2016b) IMA Genome-F 7: Draft genome sequences for Ceratocystis fagacearum, C. harringtonii, Grosmannia penicillata, and Huntiella bhutanensis. IMA Fungus 7: 317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield BD, Steenkamp ET, Santana QC, Coetzee MPA, Bam S, et al. (2012) First fungal genome sequence from Africa: a preliminary analysis. South African Journal of Science 108: 01–09. [Google Scholar]
- Wingfield MJ, Marasas WFO. (1980) Ceratocystis ips associated with Orthotomicus erosus (Coleoptera: Scolytidae) on Pinus spp. in the Cape province of South Africa. Phytophylactica. 12: 65–68. [Google Scholar]
- Yuan Z-Q, Mohammed C. (2002) Ceratocystis moniliformopsis sp. nov., an early coloniser of Eucalyptus obliqua logs in Tasmania, Australia. Australian Systematic Botany 15: 125–133. [Google Scholar]
- Zerbino DR, Birney E. (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Research 18: 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XD, Burgess TI, De Beer ZW, Lieutier F, Yart A, et al. (2007) High intercontinental migration rates and population admixture in the sapstain fungus Ophiostoma ips. Molecular Ecology 16: 89–99. [DOI] [PubMed] [Google Scholar]
- Zhou XD, De Beer ZW, Wingfield BD, Wingfield MJ. (2001) Ophiostomatoid fungi associated with three pine-infesting bark beetles in South Africa. Sydowia 53: 290–300. [Google Scholar]
- Zhou XD, De Beer ZW, Wingfield BD, Wingfield MJ. (2002) Infection sequence and pathogenicity of Ophiostoma ips, Leptographium serpens and L. lundbergii to pines in South Africa. Fungal Diversity 10: 229–240. [Google Scholar]
- Zhou XD, De Beer ZW, Ahumada R, Wingfield BD, Wingfield MJ. (2004) Ophiostoma and Ceratocystiopsis spp. associated with two pine-infesting bark beetles in Chile. Fungal Diversity 15: 253–266. [Google Scholar]