

Abstract
Conotruncal defects (CTDs) account for ~30% of all types of congenital heart disease and contribute to increased morbidity and mortality rates. Increasing evidence suggests that genetic risk factors are involved in the pathogenesis of CTDs. Mutations in a number of genes, including the TBX1 gene that codes for a T-box transcription factor essential for normal cardiovascular development, may contribute to the development of CTD. CTDs are genetically heterogeneous and the genetic defects responsible for CTDs in the majority of patients remain unknown. The present study sequenced the coding regions and splicing junction boundaries of TBX1 in 136 patients with CTDs and 300 matched healthy individuals. The disease-causing potential of the identified TBX1 sequence variation was evaluated using MutationTaster, PolyPhen-2, SIFT and PROVEN software. The functional characteristics of the mutant TBX1 gene were defined using a dual-luciferase reporter assay system. A novel heterozygous TBX1 mutation, p.S233Y, was identified in a patient with transposition of the great arteries (TGA) and a ventricular septal defect. This mutation was absent in the 300 controls and altered the amino acid produced, serine, which is evolutionarily conserved across several species, and was predicted to be pathogenic in silico. Luciferase assays conducted in COS-7 cells demonstrated that the newly identified TBX1 mutation was associated with significantly diminished transcriptional activation of the ANF promoter compared with the wild-type TBX1. To the best of our knowledge, the present study is the first to associate a TBX1 loss-of-function mutation with enhanced susceptibility to TGA, which adds significant insight to the molecular mechanism of TGA.
Keywords: congenital heart defect, genetics, transcription factors, TBX1, reporter gene assay
Introduction
Congenital heart disease (CHD) is the most common birth defect in humans. It occurs in ~1% of live births and accounts for ~30% of birth defect-related mortalities (1). Conotruncal heart defect (CTD), a subset of serious and relatively common CHD, is defined as a defect of the cardiac outflow tracts or great arteries and includes tetralogy of Fallot (TOF), transposition of the great arteries (TGA), persistent truncus ateriosus, interrupted aortic arch (IAA), double outlet right ventricle (DORV), ventricular septal defect (VSD) and pulmonary atresia (PA) (2). It is estimated that CTD occurs in 1 out of every 1,000 live births, accounting for ~1/3 of all CHD cases. If surgery to repair CTD is not performed, patients experience a reduced quality of life, decreased performance during exercise and delayed brain development. CTD may also induce pulmonary hypertension, cardiac enlargement, ventricular dysfunction, heart failure, cardiac arrhythmia and even sudden mortality (2–4). Advances in therapeutic strategies to treat patients with CTD have resulted in more neonates with CTD surviving into adulthood; however, morbidity and mortality rates remain high in such survivors (5–8). CTD induces a heavy economic burden on patients' families and health care systems and this socioeconomic burden is anticipated to increase in the future due to increasing survival rates of neonates with CTD and a consequent increase in the number of adults living with CTD (9). Therefore, the etiology underlying CTD requires elucidation.
Previous studies have established the important role genetic risk factors serve in the pathogenesis of CTD (10–15). The 22q11 deletion syndrome (22q11DS), also known as DiGeorge syndrome, is a chromosomal abnormality responsible for ~12% of conotruncal malformations (16–21). Furthermore, mutations in a number of genes, particularly those encoding cardiac transcriptional factors, including NKX2-5, NKX2-6, GATA4, GATA5, GATA6, PITX2, HAND2, TBX5 and TBX20, are associated with CTD (22–44). However, these CTD-associated genes are uncommon causes of CTD (44), and in the majority of patients, the genetic defects underpinning CTD remain unknown.
TBX1 is a member of the T-box family of transcription factors that share an evolutionarily conserved DNA-binding domain. It serves a crucial role in cardiovascular morphogenesis, particularly in elongating the cardiac tube at the anterior pole and maintaining the proliferation of mesenchymal precursor cells for the formation of a myocardialized and septated outflow tract (45). The expression of TBX1 varies spatiotemporally in the tissues that form the pharyngeal apparatus, which develops into the heart during embryogenesis (46). Genetic studies in mice have demonstrated that TBX1 expression during embryogenesis requires specific regulation and in patients with 22q11DS, it has been observed that increases or decreases in TBX1 transcript levels may result in developmental malformations (46–49). Furthermore, in vivo studies have identified that TBX1 is a candidate gene for 22q11DS (48–50). Heterozygous TBX1-null mice exhibit cardiac, craniofacial, thymic and parathyroid deformities, similar to those observed in patients with severe 22q11DS (50–52). In addition to syndromic CTD, TBX1 mutations have been causally linked to non-syndromic CTD in humans, including isolated TOF, VSD, PA, DORV, IAA and aortic arch anomalies (4,53–55). However, the prevalence and spectrum of TBX1 mutations in patients with other forms of non-syndromic CTD remain unknown. The aim of the present study was to evaluate the prevalence and spectrum of TBX1 mutations in a cohort of patients with isolated CTD and characterize the functional effect of an identified TBX1 mutation associated with CTD.
Patients and methods
Study population
Between January 2013 and November 2015, a total of 136 unrelated individuals (80 male, 56 female; median age, 2.3 years old) presenting with isolated CTD were recruited prospectively from the Chinese Han population at the Department of Pediatrics, Shanghai Tenth People's Hospital; Departments of Pediatrics and Cardiology, Tongji Hospital; Department of Pediatrics, Baoshan Branch of Huashan Hospital; Department of Cardiac Surgery, Shanghai Chest Hospital; Department of Cardiovascular Surgery, Renji Hospital (all Shanghai, China). There were 129 patients who underwent cardiac surgery to repair CTD. A total of 30 parents of the patients were also included. All subjects underwent a detailed clinical evaluation that included familial history, medical history, complete physical examination, two-dimensional transthoracic echocardiography with color flow Doppler and a standard 12-lead electrocardiogram. The patients' cardiac phenotypes were confirmed by echocardiography, angiocardiography and/or surgery. Probands with known chromosomal abnormalities or other recognized syndromes were excluded. Proband samples were screened for 22q11.2 deletion as previously described (54,55) and samples with deletions were excluded from further analysis. During a period from September 2014 to January 2016, a total of 300 healthy individuals (178 male, 122 female; median age, 2.4 years old), matched with the patients in age, sex and ethnicity, were enrolled as controls from the above-mentioned institutions where the CTD patients were recruited. The normal cardiac morphology of these controls was confirmed using transthoracic echocardiography.
The present study was performed in accordance with the ethical principles of the Declaration of Helsinki. The study protocol was approved by the Ethics Committee of Tongji Hospital, Tongji University School of Medicine [Shanghai, China; ethical approval no. LL(H)-09-07)] and written informed consent was obtained from the parents of each participant prior to the study.
Genetic analysis
Peripheral venous blood samples were taken from all participants and the genomic DNA was extracted from leukocytes using the Wizard Genomic DNA Purification kit (Promega Corporation, Madison, WI, USA) following the manufacturer's instructions. The referential genomic DNA sequence of TBX1 was derived from GenBank (accession no. NC_000022.11) using the National Center for Biotechnology Information (NCBI; http://www.ncbi.nlm.nih.gov/nucleotide/). The intronic primers used for the amplification of the coding exons and flanking introns of TBX1 in the polymerase chain reaction (PCR) were designed as previously described (4). The primers were as follows: Coding exon 1 forward, 5′-GACGCCATAATCCTCTGGGC-3′ and reverse, 5′-AAGAGCTGCCTCCACCTACT-3′; coding exon 2 forward, 5′-GTCATGATCTCCGCCGTGTC-3′ and reverse, 5′-GAACAGCGAAGGAGGCAGCG-3′; coding exon 3 forward, 5′-AGGGCGAGGCCGAGTTTATG-3′ and reverse, 5′-ACGACCCTTGGAGTTGGGTC-3′; coding exon 4 forward, 5′-GGCACTTTTAGGGTTCGCCC-3′ and reverse, 5′-TCTCCTCATCGGCACACCAG-3′; coding exon 5 forward, 5′-GAGTCCAGGCCAGTGAGGTC-3′ and reverse, 5′-CCGCTTTTCCAGAGGCGTTG-3′; coding exons 6 and 7 forward, 5′-TGGTGCGCTTCTCCTAACACT-3′ and reverse, 5′-CTCCGACGCCCCATGCGAGG-3′; coding exon 8A forward, 5′-CCCTGATCCGACGTCTTTCC-3′ and reverse, 5′-AACACGACAACTCCATGTGC-3′; coding exon 8B forward, 5′-CTGAGTGGGTGCACACTGGA-3′ and reverse, 5′-AGGGCTGGAGGATTCGCTTC-3′; and coding exon 8C forward, 5′-ACTTGGGGTCTCGGGCACGC-3′ and reverse, 5′-CGAACTTCGGGGCTGTGCAG-3′. PCR was performed using HotStar Taq DNA Polymerase (Qiagen GmbH, Hilden, Germany) on a Veriti Thermal Cycler (Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The PCR program used was as follows: Initial pre-denaturation at 95°C for 15 min, followed by 35 cycles of denaturation at 94°C for 30 sec, annealing at 62°C for 30 sec and elongation at 72°C for 1 min, with final elongation at 72°C for 6 min. Samples were subsequently cooled down to −4°C. The amplified products were fractionated on a 2% agarose gel by electrophoresis and purified using the QIAquick Gel Extraction kit (Qiagen GmbH). The amplicons were sequenced on an ABI PRISM 3130 XL DNA Analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc.) using BigDye® Terminator v3.1 Cycle Sequencing kits (Applied Biosystems; Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. To determine any sequence variance, bi-directional re-sequencing of an independent PCR-generated amplicon from the same subject was performed. Additionally, the Human Gene Mutation Database (HGMD; http://www.hgmd.cf.ac.uk/), the NCBI's Single Nucleotide Polymorphism (SNP; http://www.ncbi.nlm.nih.gov/snp) database and the PubMed Database (http://www.ncbi.nlm.nih.gov/pubmed) were used to confirm that any identified sequence variances were novel.
Alignment of multiple TBX1 protein sequences across species
To evaluate evolutionary conservation in the altered amino acid, human TBX1 amino acid sequences were aligned with those in several other species, including dogs, mice, chimpanzees, zebrafish, rats, worms and frogs, using the HomoloGene and Show Multiple Alignment links on the NCBI's web site (http://www.ncbi.nlm.nih.gov/homologene).
Prediction of the disease-causing potential of a TBX1 sequence variation
MutationTaster (http://www.mutationtaster.org), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), SIFT (http://sift.jcvi.org) and PROVEN (http://provean.jcvi.org/index.php) were used to predict disease-causing potential of a TBX1 sequence variation. Each alteration was automatically given a pathogenic or benign probability score.
Expression plasmids and site-directed mutagenesis
The recombinant expression plasmid TBX1-pcDNA3.1, which contains the full-length complementary DNA of the human TBX1 isoform C and the 4XT-luc reporter vector, which contains four conserved T-half sites ‘ATTTCACACCT’ and expresses Firefly luciferase, were constructed as previously described (4). The mutant TBX1-pcDNA3.1 plasmid was produced with PCR-based site-directed mutagenesis using a QuickChange II XL Site-Directed Mutagenesis kit (Stratagene; Agilent Technologies, Inc., Santa Clara, CA, USA) according to the manufacturer's protocol and a complementary pair of primers (forward, 5′-ACATTATTCTGAATTACATGCACAGATACCA-3′ and reverse, 5′-TGGTATCTGTGCATGTAATTCAGAATAATGT-3′), using the wild-type TBX1-pcDNA3.1 as a template. The PCR thermal cycling conditions used were as follows: Initial denaturation at 95°C for 1 min, followed by 18 cycles of denaturation at 95°C for 50 sec, annealing at 60°C for 50 sec and extension at 68°C for 7 min, with final extension at 58°C for 10 min. The mutant TBX1 was selected using restriction enzyme DpnI and confirmed by direct PCR-sequencing.
Cell culture and reporter gene analysis
The COS-7 cells (Cardiovascular Research Laboratory, Shanghai Chest Hospital, Shanghai, China) were maintained in Dulbecco's modified Eagle's medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum (Invitrogen; Thermo Fisher Scientific, Inc.) in 5% CO2 at 37°C. A total of 1.5×105 cells/well were plated in a 6-well plate. Cell transfections were performed using the Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher Scientific, Inc.), following the manufacturer's protocol. All transfections were performed in triplicate. pGL4.75 (hRluc/CMV; Promega Corporation), which expresses Renilla luciferase, was considered to be the internal control vector and was used to normalize transfection efficiency. COS-7 cells at ~90% confluence were transfected with 0.4 µg wild-type or mutant TBX1-pcDNA3.1, in combination with 1.0 µg 4XT-luc and 0.04 µg pGL4.75. For co-transfection experiments, 0.2 µg wild-type TBX1-pcDNA3.1 together with 0.2 µg mutant TBX1-pcDNA3.1 or 0.2 µg empty pcDNA3.1 vector was used in the presence of 1.0 µg 4XT-luc and 0.04 µg pGL4.75. A total of 48 h following transfection, cells were harvested and lysed. Subsequently, Firefly luciferase and Renilla luciferase activities were measured with the lysates using the Dual-Glow luciferase assay system (Promega Corporation). The activity of the 4XT promoter was presented as the fold activation of Firefly luciferase relative to Renilla luciferase.
Statistical analysis
The SPSS version 17.0 software package (SPSS, Inc., Chicago, IL, USA) was used for statistical analyses. Data were expressed as the mean ± standard deviation, unless otherwise indicated. Continuous variables were tested for normal distribution and the Unpaired Student's t-test was used to compare numerical variables between the two groups. Comparison of the categorical variables between two groups was made using Pearson's χ2 test or Fisher's exact test when appropriate. P<0.05 was considered to indicate a statistically significantly difference.
Results
Clinical features of the patients
In the present study, 136 children with isolated CTD (80 male, 56 female; median age, 2.3 years old) were clinically evaluated in comparison with 300 sex-age- and ethnically-matched, unrelated healthy individuals (178 male, 122 female; median age, 2.4 years old). All subjects were negative for the 22q11.2 microdeletion and no individuals exhibited any extracardiac anomalies or features suggestive of 22q11.2DS. Of the 136 patients with CTD, 15 (~11%) had a family history of CHD. There was no recorded family history of CHD in any of the controls. Echocardiograms indicated that the control subjects were physically healthy and did not have any structural cardiac diseases. Furthermore, there were no significant differences between the patient and control groups in demographic characteristics including age, sex and ethnicity (data not shown). The proportion of various types of congenital CTD in the patients is indicated in Table I.
Table I.
Distribution of various types of congenital conotruncal defects in patients with CTD.
| Congenital conotruncal defect | Number of patients | Percentage |
|---|---|---|
| Tetralogy of fallot | 52 | 38 |
| Double outlet right ventricle | 25 | 18 |
| Pulmonary atresia | 18 | 13 |
| Transposition of great arteries | 16 | 12 |
| Single ventricle | 11 | 8 |
| Interrupted aortic arch | 6 | 4 |
| Persistent truncus arteriosus | 4 | 3 |
| Hypoplastic right heart syndrome | 2 | 2 |
| Co-arctation of aorta | 2 | 2 |
| Total | 136 | 100 |
Detection of a TBX1 variation in CTD
Direct DNA sequencing led to identification of a heterozygous mutation in TBX1 in 1 of the 136 patients with CTD. Specifically, a substitution of adenine (A) for cytosine (C) in the second nucleotide of codon 233 (c.698C>A) was detected in a 6-month-old boy affected by congenital TGA and VSD with no family history of CHD. This was equivalent to the transversion of serine (S) into tyrosine (Y) at amino acid position 233 (p.S233Y). Physical examination of the mutation carrier was normal and the subject presented without apparent extracardiac abnormalities. The missense mutation was not detected in either of the boy's parents, who did not possess any physical abnormalities, implying this was a de novo mutation. The sequence chromatograms indicating the heterozygous TBX1 mutation of c.698C>A and its control sequence are presented in Fig. 1A. Fig. 1B presents a schematic drawing of TBX1, revealing the structural domains and the location of the detected mutation. The identified TBX1 variation c.698C>A was absent in the 300 controls and other 135 CTD patients, and was not found in the HGMD, SNP and PubMed databases (last accessed on March 11, 2016), suggesting that this was a novel mutation.
Figure 1.

A novel TBX1 mutation causally linked to congenital contruncal defects. (A) Sequence electropherograms indicated the TBX1 mutation compared with its wild-type form. Arrow indicated the heterozygous nucleotides of C/A in the mutation carrier (mutant) or the homozygous nucleotides of C/C in the corresponding control individual (wild-type). The rectangle marks the nucleotides comprising a codon of TBX1. (B) Schematic presentation of TBX1 protein structures with the identified mutation indicated in the T-box domain. The mutation identified in a patient with transposition of the great arteries and ventricular septal defect was indicated above the structural domains. NH2, amino-terminus; NLS, nuclear location signal; TAD, transcriptional activation domain; COOH, carboxyl-terminus.
Conservative assessment of the altered amino acid
Alignment of multiple TBX1 protein sequences across species revealed that the altered serine at amino acid position 233 (p.S233) was completely evolutionarily conserved (Fig. 2). This assessment suggests that TBX1 may serve an important functional role in CTD.
Figure 2.

Multiple alignments of TBX1 protein sequences across species. The altered serine at amino acid 233 was completely evolutionarily conserved among various species.
Disease-causing potential of the detected TBX1 sequence variation
The TBX1 sequence variation c.698C>A was predicted to be disease-causing by MutationTaster, (P=1.000). Furthermore, the resultant amino acid substitution p.S233Y was predicted to be damaging by PolyPhen-2 (with a score of 1.000; sensitivity, 0.00; specificity, 1.00), damaging by SIFT (with a SIFT score of 0) and deleterious by PROVEN (with a PROVEN score of −5.70). These findings support that the mutation may predispose the development of CTD.
Diminished transcriptional activity of the mutant TBX1 protein
As indicated in Fig. 3, dual-luciferase assays revealed that the same quantity (0.4 µg) of wild-type and S233Y-mutant TBX1 activated the 4XT promoter by ~4- and ~1-fold, respectively (0.4 µg wild-type TBX1 vs. 0.4 µg S233Y-mutant TBX1: t=6.51501, P<0.003; Fig. 3), whereas 0.2 µg wild-type TBX1 in combination with 0.2 µg S233Y-mutant TBX1 only activated the 4XT promoter by ~2-fold (0.4 µg wild-type TBX1 vs. 0.2 µg wild-type TBX1 plus 0.2 µg S233Y-mutant TBX1: t=6.04274, P<0.004; Fig. 3). These results suggest that the S233Y-mutant TBX1 significantly reduces transcriptional activity. However, 0.2 µg wild-type TBX1 alone induced the activation of the 4XT promoter by ~2-fold, similar to that of TBX1 plus S233Y (0.4 µg wild-type TBX1 vs. 0.2 µg wild-type TBX1: t=4.22682, P=0.01340; 0.2 µg wild-type TBX1 vs. 0.2 µg wild-type TBX1 plus 0.2 µg of S233Y-mutant TBX1: t=−1.50844, P=0.20590). The present findings suggest the activity of TBX1 is dose-dependent.
Figure 3.
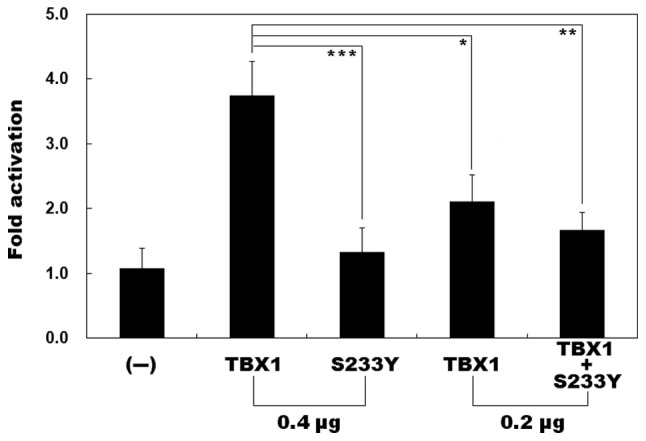
Functional impairment of the mutant TBX1 protein. Activation of 4XT promoter-driven luciferase reporter in COS-7 cells by wild-type TBX1 or S233Y-mutant TBX1, alone or in combination. The S233Y-mutant TBX1 significantly decreased transcriptional activity. Experiments were performed in triplicate. Data are presented as the mean ± standard deviation. -, empty pcDNA3.1 plasmid. ***P<0.003; **P<0.004; *P<0.014.
Discussion
In the present study, a cohort of patients with CTD that did not possess 22q11.2 deletions were screened for rare genetic variants in all coding exons and splicing junctions of TBX1 by sequencing. A novel heterozygous mutation, p.S233Y, was identified in a patient with isolated TGA and VSD. This missense mutation, which was absent in 600 reference chromosomes, altered an amino acid that was evolutionarily conserved across several species. The mutation was predicted to be disease-causing by MutationTaster, PolyPhen-2, SIFT and PROVEN. Biological analyses revealed that the S233Y-mutant TBX1 protein was associated with significantly diminished transcriptional activity. Therefore, it is likely that the identified TBX1 mutation may have induced TGA and VSD in the carrier.
The T-box genes code for a large family of transcription factors that are characteristic of a highly conserved DNA-binding motif referred to as the T-box (45). The T-box recognizes and binds to specific DNA elements in the promoters of target genes and mediates the transcriptional activation or inhibition of downstream genes (45). The T-box domain is also a conserved protein-protein interaction domain for other transcription factors, chromatin remodeling complexes and histone-modifying enzymes (45). As a core member of the T-box gene family, TBX1 is mapped on human chromosome 22q11.21, the center of the 22q11DS chromosomal region and codes for a protein that is made up of 495 amino acids (4). Based on experimental evidence from animal models, TBX1 is expressed in the secondary heart field during embryogenesis and functions in a combinatorial or hierarchical fashion to regulate the proliferation of mesenchymal precursor cells, which is required for normal cardiovascular development, particularly for the development of a myocardialized and septated outflow tract (45). In the present study, the TBX1 mutation was identified in a patient with CTD was located within the T-box and was thus expected to decrease transcriptional activity, potentially by affecting TBX1-DNA interaction. This expectation was in accordance with the experimental data, which indicated that the mutant exhibited significantly reduced transactivation activity. This diminished transcriptional activation supports a previous hypothesis that normal cardiovascular development is particularly susceptible to differences in TBX1 activity and a hypomorphic allele may predispose non-syndromic CHD (54).
In humans, TBX1 haploinsufficiency is the etiology responsible for 22q11DS, a clinical syndrome with a varied phenotypic spectrum, including thymic hypoplasia, congenital cardiac defects, facial dysmorphisms, velopharyngeal insufficiency with or without cleft palate and immune deficiency (20). However, an increasing number of TBX1 mutations have been identified in patients with the clinical phenotypes of 22q11DS, but no detectable XXIIq11 deletion (56–58). Previous studies have associated TBX1 mutations with isolated CTD (4,53–55). Gong et al (53) performed a mutation analysis of TBX1 in 65 patients with isolated CTD who did not have deletions of 22q11.2, and identified three mutations, including two missense mutations (p.G350D and p.P396L) and one duplication mutation (p.A466_A476dup) in 3 patients with aortic arch anomalies, with a mutation prevalence of ~4.62%. However, the functional effect of these mutations was not characterized. Griffin et al (54) screened TBX1 in 93 TOF probands with had no known chromosomal abnormalities or other recognized syndromes. Two non-synonymous mutations in two probands, including the amino acid substitution p.G39S in a proband with TOF and the in-frame amino acid deletion p.P43_P61del in a proband with TOF as well as a right-sided aortic arch were identified, with a mutational prevalence of ~2.15%. In the dual-luciferase assay, the G39S-mutant TBX1 protein indicated no significant difference in transcriptional activity compared with wild-type TBX1, whereas the P43_P61-deletious TBX1 protein exhibited a significant reduction in transcriptional activity compared with wild-type TBX1 (54). By sequencing the TBX1 gene in 199 non-22q11.2 deletion patients with CTD, Xu et al (55) identified the de novo missense mutation p.E129K in a patient with PA, VSD and patent ductus arteriosus, with a mutational prevalence of ~0.50%.
In vitro experiments have demonstrated that the E129K-mutant TBX1 protein exhibits severely decreased transactivation activity. Pan et al (4) sequenced the coding exons and flanking introns of the TBX1 gene in 230 unrelated children with CHD and detected a novel heterozygous mutation, p.Q277X, in an index patient with DORV and VSD, with a mutational prevalence of ~0.43%. The index patient had a family history of CHD and genetic analysis indicated that the mutation co-segregated with VSD transmitted in an autosomal dominant pattern with complete penetrance. Biochemical analysis revealed that Q277X-mutant TBX1 lost transcriptional activating function when compared with its wild-type counterpart (4). Taken together, these results, together with those of the present study, support that genetically compromised TBX1 confers an enhanced susceptibility of CTD in a minority of cases.
There were certain limitations in the present study. Firstly, only the coding exons and exon-intron boundaries of the TBX1 gene were sequenced. Due to the important role of non-coding regions (promoter regions and regulatory elements) in gene expression, further studies are required to examine the association between sequence variants in non-coding regions and the pathogenesis of CTD. Secondly, non-syndromic CTD has a complex etiology that likely includes multiple genes. Therefore, in-depth exome sequencing studies are required identify novel culprit genes that may increase embryonic susceptibility to the development of CTD. Furthermore, not all aspects of the functional characteristics of the identified TBX1 mutation have been delineated. Further investigations are required to more comprehensively define the functional roles of the TBX1 mutation, including functional analyses of the ability of the mutant TBX1 protein to bind to target DNA, nuclear localization of the mutant TBX1 protein and the synergistic activation of target genes by the mutant TBX1 protein and its transcriptionally cooperative partners.
In conclusion, to the best of our knowledge, the present study was the first to identify an association between the TBX1 loss-of-function mutation and increased vulnerability to TGA. Furthermore, it provided an insight into the underlying molecular mechanism involved in TGA. The results of the present study suggest potential implications for molecular diagnosis and genetic counseling of patients with TGA.
Acknowledgements
The present study was supported, in part, by grants from the National Natural Science Fund of China (grant nos. 81470372 and 81641014), the Key Program for Basic Research of Shanghai, China (grant no. 14JC1405500) and the Natural Science Fund of Baoshan, Shanghai, China (grant no. 12-E-25).
References
- 1.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Després JP, Fullerton HJ, Howard VJ, et al. Heart disease and stroke statistics-2015 update: A report from the American heart association. Circulation. 2015;131:e29–e32. doi: 10.1161/CIR.0000000000000157. [DOI] [PubMed] [Google Scholar]
- 2.Agopian AJ, Mitchell LE, Glessner J, Bhalla AD, Sewda A, Hakonarson H, Goldmuntz E. Genome-wide association study of maternal and inherited loci for conotruncal heart defects. PLoS One. 2014;9:e96057. doi: 10.1371/journal.pone.0096057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang W, Shen L, Deng Z, Ding Y, Mo X, Xu Z, Gao Q, Yi L. Novel missense variants of ZFPM2/FOG2 identified in conotruncal heart defect patients do not impair interaction with GATA4. PLoS One. 2014;9:e102379. doi: 10.1371/journal.pone.0102379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pan Y, Wang ZG, Liu XY, Zhao H, Zhou N, Zheng GF, Qiu XB, Li RG, Yuan F, Shi HY, et al. A novel TBX1 loss-of-function mutation associated with congenital heart disease. Pediatr Cardiol. 2015;36:1400–1410. doi: 10.1007/s00246-015-1173-x. [DOI] [PubMed] [Google Scholar]
- 5.van der Bom T, Zomer AC, Zwinderman AH, Meijboom FJ, Bouma BJ, Mulder BJ. The changing epidemiology of congenital heart disease. Nat Rev Cardiol. 2011;8:50–60. doi: 10.1038/nrcardio.2010.166. [DOI] [PubMed] [Google Scholar]
- 6.Müller J, Engelhardt A, Fratz S, Eicken A, Ewert P, Hager A. Improved exercise performance and quality of life after percutaneous pulmonary valve implantation. Int J Cardiol. 2014;173:388–392. doi: 10.1016/j.ijcard.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Valente AM, Gauvreau K, Assenza GE, Babu-Narayan SV, Evans SP, Gatzoulis M, Groenink M, Inuzuka R, Kilner PJ, Koyak Z, et al. Rationale and design of an International Multicenter Registry of patients with repaired tetralogy of Fallot to define risk factors for late adverse outcomes: The INDICATOR cohort. Pediatr Cardiol. 2013;34:95–104. doi: 10.1007/s00246-012-0394-5. [DOI] [PubMed] [Google Scholar]
- 8.Tutarel O, Kempny A, Alonso-Gonzalez R, Jabbour R, Li W, Uebing A, Dimopoulos K, Swan L, Gatzoulis MA, Diller GP. Congenital heart disease beyond the age of 60: Emergence of a new population with high resource utilization, high morbidity, and high mortality. Eur Heart J. 2014;35:725–732. doi: 10.1093/eurheartj/eht257. [DOI] [PubMed] [Google Scholar]
- 9.Verheugt CL, Uiterwaal CS, van der Velde ET, Meijboom FJ, Pieper PG, Sieswerda GT, Plokker HW, Grobbee DE, Mulder BJ. The emerging burden of hospital admissions of adults with congenital heart disease. Heart. 2010;96:872–878. doi: 10.1136/hrt.2009.185595. [DOI] [PubMed] [Google Scholar]
- 10.Peyvandi S, Ingall E, Woyciechowski S, Garbarini J, Mitchell LE, Goldmuntz E. Risk of congenital heart disease in relatives of probands with conotruncal cardiac defects: An evaluation of 1,620 families. Am J Med Genet A. 2014;164A:1–1495. doi: 10.1002/ajmg.a.36500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Racedo SE, McDonald-McGinn DM, Chung JH, Goldmuntz E, Zackai E, Emanuel BS, Zhou B, Funke B, Morrow BE. Mouse and human CRKL is dosage sensitive for cardiac outflow tract formation. Am J Hum Genet. 2015;96:235–244. doi: 10.1016/j.ajhg.2014.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, DePalma SR, McKean D, Wakimoto H, Gorham J, et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science. 2015;350:1262–1266. doi: 10.1126/science.aac9396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Werner P, Latney B, Deardorff MA, Goldmuntz E. MESP1 mutations in patients with congenital heart defects. Hum Mutat. 2016;37:308–314. doi: 10.1002/humu.22947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andersen TA, Kde L Troelsen, Larsen LA. Of mice and men: Molecular genetics of congenital heart disease. Cell Mol Life Sci. 2014;71:1327–1352. doi: 10.1007/s00018-013-1430-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lalani SR, Belmont JW. Genetic basis of congenital cardiovascular malformations. Eur J Med Genet. 2014;57:402–413. doi: 10.1016/j.ejmg.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Warburton D, Ronemus M, Kline J, Jobanputra V, Williams I, Anyane-Yeboa K, Chung W, Yu L, Wong N, Awad D, et al. The contribution of de novo and rare inherited copy number changes to congenital heart disease in an unselected sample of children with conotruncal defects or hypoplastic left heart disease. Hum Genet. 2014;133:11–27. doi: 10.1007/s00439-013-1353-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu AP, Chow PC, Lee PP, Mok GT, Tang WF, Lau ET, Lam ST, Chan KY, Kan AS, Chau AK, et al. Under-recognition of 22q11.2 deletion in adult Chinese patients with conotruncal anomalies: Implications in transitional care. Eur J Med Genet. 2014;57:306–311. doi: 10.1016/j.ejmg.2014.03.014. [DOI] [PubMed] [Google Scholar]
- 18.Chen M, Yang YS, Shih JC, Lin WH, Lee DJ, Lin YS, Chou CH, Cameron AD, Ginsberg NA, Chen CA, et al. Microdeletions/duplications involving TBX1 gene in fetuses with conotruncal heart defects which are negative for 22q11.2 deletion on fluorescence in-situ hybridization. Ultrasound Obstet Gynecol. 2014;43:396–403. doi: 10.1002/uog.12550. [DOI] [PubMed] [Google Scholar]
- 19.Guo T, Chung JH, Wang T, McDonald-McGinn DM, Kates WR, Hawuła W, Coleman K, Zackai E, Emanuel BS, Morrow BE. Histone modifier genes alter conotruncal heart phenotypes in 22q11.2 deletion syndrome. Am J Hum Genet. 2015;97:869–877. doi: 10.1016/j.ajhg.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mlynarski EE, Sheridan MB, Xie M, Guo T, Racedo SE, McDonald-McGinn DM, Gai X, Chow EW, Vorstman J, Swillen A, et al. Copy-number variation of the glucose transporter gene SLC2A3 and congenital heart defects in the 22q11.2 deletion syndrome. Am J Hum Genet. 2015;96:753–764. doi: 10.1016/j.ajhg.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hacıhamdioğlu B, Hacıhamdioğlu D, Delil K. 22q11 deletion syndrome: Current perspective. Appl Clin Genet. 2015;8:123–132. doi: 10.2147/TACG.S82105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qu XK, Qiu XB, Yuan F, Wang J, Zhao CM, Liu XY, Zhang XL, Li RG, Xu YJ, Hou XM, et al. A novel NKX2.5 loss-of-function mutation associated with congenital bicuspid aortic valve. Am J Cardiol. 2014;114:1891–1895. doi: 10.1016/j.amjcard.2014.09.028. [DOI] [PubMed] [Google Scholar]
- 23.Zheng J, Li F, Liu J, Xu Z, Zhang H, Fu Q, Wang J, Sun K. Investigation of somatic NKX2-5 mutations in Chinese children with congenital heart disease. Int J Med Sci. 2015;12:538–543. doi: 10.7150/ijms.11700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao Y, Wang J, Wei C, Hou Z, Li Y, Zou H, Meng M, Wang W, Jiang L. Genetic variations of NKX2-5 in sporadic atrial septal defect and ventricular septal defect in Chinese Yunnan population. Gene. 2016;575:29–33. doi: 10.1016/j.gene.2015.08.033. [DOI] [PubMed] [Google Scholar]
- 25.Ta-Shma A, El-lahham N, Edvardson S, Stepensky P, Nir A, Perles Z, Gavri S, Golender J, Yaakobi-Simhayoff N, Shaag A, et al. Conotruncal malformations and absent thymus due to a deleterious NKX2-6 mutation. J Med Genet. 2014;51:268–270. doi: 10.1136/jmedgenet-2013-102100. [DOI] [PubMed] [Google Scholar]
- 26.Zhao L, Ni SH, Liu XY, Wei D, Yuan F, Xu L, Xin Li, Li RG, Qu XK, Xu YJ, et al. Prevalence and spectrum of Nkx2.6 mutations in patients with congenital heart disease. Eur J Med Genet. 2014;57:579–586. doi: 10.1016/j.ejmg.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Mao JH, Ding KK, Xu WJ, Liu XY, Qiu XB, Li RG, Qu XK, Xu YJ, Huang RT, et al. A novel NKX2.6 mutation associated with congenital ventricular septal defect. Pediatr Cardiol. 2015;36:646–656. doi: 10.1007/s00246-014-1060-x. [DOI] [PubMed] [Google Scholar]
- 28.Xiang R, Fan LL, Huang H, Cao BB, Li XP, Peng DQ, Xia K. A novel mutation of GATA4 (K319E) is responsible for familial atrial septal defect and pulmonary valve stenosis. Gene. 2014;534:320–323. doi: 10.1016/j.gene.2013.10.028. [DOI] [PubMed] [Google Scholar]
- 29.Han H, Chen Y, Liu G, Han Z, Zhao Z, Tang Y. GATA4 transgenic mice as an in vivo model of congenital heart disease. Int J Mol Med. 2015;35:1545–1553. doi: 10.3892/ijmm.2015.2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen J, Qi B, Zhao J, Liu W, Duan R, Zhang M. A novel mutation of GATA4 (K300T) associated with familial atrial septal defect. Gene. 2016;575:473–477. doi: 10.1016/j.gene.2015.09.021. [DOI] [PubMed] [Google Scholar]
- 31.Huang RT, Xue S, Xu YJ, Zhou M, Yang YQ. Somatic GATA5 mutations in sporadic tetralogy of Fallot. Int J Mol Med. 2014;33:1227–1235. doi: 10.3892/ijmm.2014.1674. [DOI] [PubMed] [Google Scholar]
- 32.Shi LM, Tao JW, Qiu XB, Wang J, Yuan F, Xu L, Liu H, Li RG, Xu YJ, Wang Q, et al. GATA5 loss-of-function mutations associated with congenital bicuspid aortic valve. Int J Mol Med. 2014;33:1219–1226. doi: 10.3892/ijmm.2014.1700. [DOI] [PubMed] [Google Scholar]
- 33.Huang RT, Xue S, Xu YJ, Yang YQ. Somatic mutations in the GATA6 gene underlie sporadic tetralogy of Fallot. Int J Mol Med. 2013;31:51–58. doi: 10.3892/ijmm.2012.1188. [DOI] [PubMed] [Google Scholar]
- 34.Wang X, Ji W, Wang J, Zhao P, Guo Y, Xu R, Chen S, Sun K. Identification of two novel GATA6 mutations in patients with nonsyndromic conotruncal heart defects. Mol Med Rep. 2014;10:743–748. doi: 10.3892/mmr.2014.2247. [DOI] [PubMed] [Google Scholar]
- 35.Wei D, Gong XH, Qiu G, Wang J, Yang YQ. Novel PITX2c loss-of-function mutations associated with complex congenital heart disease. Int J Mol Med. 2014;33:1201–1208. doi: 10.3892/ijmm.2014.1689. [DOI] [PubMed] [Google Scholar]
- 36.Sun YM, Wang J, Qiu XB, Yuan F, Xu YJ, Li RG, Qu XK, Huang RT, Xue S, Yang YQ. PITX2 loss-of-function mutation contributes to tetralogy of Fallot. Gene. 2016;577:258–264. doi: 10.1016/j.gene.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 37.Lu CX, Gong HR, Liu XY, Wang J, Zhao CM, Huang RT, Xue S, Yang YQ. A novel HAND2 loss-of-function mutation responsible for tetralogy of Fallot. Int J Mol Med. 2016;37:445–451. doi: 10.3892/ijmm.2015.2436. [DOI] [PubMed] [Google Scholar]
- 38.Sun YM, Wang J, Qiu XB, Yuan F, Li RG, Xu YJ, Qu XK, Shi HY, Hou XM, Huang RT, et al. A HAND2 loss-of-function mutation causes familial ventricular septal defect and pulmonary stenosis. G3 (Bethesda) 2016;6:987–992. doi: 10.1534/g3.115.026518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baban A, Postma AV, Marini M, Trocchio G, Santilli A, Pelegrini M, Sirleto P, Lerone M, Albanese SB, Barnett P, et al. Identification of TBX5 mutations in a series of 94 patients with tetralogy of Fallot. Am J Med Genet A. 2014;164A:1–3107. doi: 10.1002/ajmg.a.36783. [DOI] [PubMed] [Google Scholar]
- 40.Al-Qattan MM, Al-Shaar H Abou. Molecular basis of the clinical features of Holt-Oram syndrome resulting from missense and extended protein mutations of the TBX5 gene as well as TBX5 intragenic duplications. Gene. 2015;560:129–136. doi: 10.1016/j.gene.2015.02.017. [DOI] [PubMed] [Google Scholar]
- 41.Pan Y, Geng R, Zhou N, Zheng GF, Zhao H, Wang J, Zhao CM, Qiu XB, Yang YQ, Liu XY. TBX20 loss-of-function mutation contributes to double outlet right ventricle. Int J Mol Med. 2015;35:1058–1066. doi: 10.3892/ijmm.2015.2077. [DOI] [PubMed] [Google Scholar]
- 42.Chen J, Sun F, Fu J, Zhang H. Association of TBX20 gene polymorphism with congenital heart disease in Han Chinese neonates. Pediatr Cardiol. 2015;36:737–742. doi: 10.1007/s00246-014-1073-5. [DOI] [PubMed] [Google Scholar]
- 43.McCulley DJ, Black BL. Transcription factor pathways and congenital heart disease. Curr Top Dev Biol. 2012;100:253–277. doi: 10.1016/B978-0-12-387786-4.00008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fahed AC, Gelb BD, Seidman JG, Seidman CE. Genetics of congenital heart disease: The glass half empty. Circ Res. 2013;112:707–720. doi: 10.1161/CIRCRESAHA.112.300853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Greulich F, Rudat C, Kispert A. Mechanisms of T-box gene function in the developing heart. Cardiovasc Res. 2011;91:212–222. doi: 10.1093/cvr/cvr112. [DOI] [PubMed] [Google Scholar]
- 46.Gao S, Li X, Amendt BA. Understanding the role of Tbx1 as a candidate gene for 22q11.2 deletion syndrome. Curr Allergy Asthma Rep. 2013;13:613–621. doi: 10.1007/s11882-013-0384-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Z, Baldini A. In vivo response to high-resolution variation of Tbx1 mRNA dosage. Hum Mol Genet. 2008;17:150–157. doi: 10.1093/hmg/ddm291. [DOI] [PubMed] [Google Scholar]
- 48.Vitelli F, Huynh T, Baldini A. Gain of function of Tbx1 affects pharyngeal and heart development in the mouse. Genesis. 2009;47:188–195. doi: 10.1002/dvg.20476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liao J, Kochilas L, Nowotschin S, Arnold JS, Aggarwal VS, Epstein JA, Brown MC, Adams J, Morrow BE. Full spectrum of malformations in velo-cardio-facial syndrome/DiGeorge syndrome mouse models by altering Tbx1 dosage. Hum Mol Genet. 2004;13:1577–1585. doi: 10.1093/hmg/ddh176. [DOI] [PubMed] [Google Scholar]
- 50.Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, Jurecic V, Ogunrinu G, Sutherland HF, Scambler PJ, et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410:97–101. doi: 10.1038/35065105. [DOI] [PubMed] [Google Scholar]
- 51.Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 2001;27:286–291. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- 52.Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001;104:619–629. doi: 10.1016/S0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- 53.Gong W, Gottlieb S, Collins J, Blescia A, Dietz H, Goldmuntz E, McDonald-McGinn DM, Zackai EH, Emanuel BS, Driscoll DA, Budarf ML. Mutation analysis of TBX1 in non-deleted patients with features of DGS/VCFS or isolated cardiovascular defects. J Med Genet. 2001;38:E45. doi: 10.1136/jmg.38.12.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Griffin HR, Töpf A, Glen E, Zweier C, Stuart AG, Parsons J, Peart I, Deanfield J, O'Sullivan J, Rauch A, et al. Systematic survey of variants in TBX1 in non-syndromic tetralogy of Fallot identifies a novel 57 base pair deletion that reduces transcriptional activity but finds no evidence for association with common variants. Heart. 2010;96:1651–1655. doi: 10.1136/hrt.2010.200121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu YJ, Chen S, Zhang J, Fang SH, Guo QQ, Wang J, Fu QH, Li F, Xu R, Sun K. Novel TBX1 loss-of-function mutation causes isolated conotruncal heart defects in Chinese patients without 22q11.2 deletion. BMC Med Genet. 2014;15:78. doi: 10.1186/1471-2350-15-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1366–1373. doi: 10.1016/S0140-6736(03)14632-6. [DOI] [PubMed] [Google Scholar]
- 57.Paylor R, Glaser B, Mupo A, Ataliotis P, Spencer C, Sobotka A, Sparks C, Choi CH, Oghalai J, Curran S, et al. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: Implications for 22q11 deletion syndrome; Proc Natl Acad Sci USA; 2006; pp. 7729–7734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Torres-Juan L, Rosell J, Morla M, Vidal-Pou C, Garcia-Algas F, de la Fuente MA, Juan M, Tuban A, Bachiller D, Bemues M, et al. Mutations in TBX1 genocopy the 22q11.2 deletion and duplication syndromes: A new susceptibility factor for mental retardation. Eur J Hum Genet. 2007;15:658–663. doi: 10.1038/sj.ejhg.5201819. [DOI] [PubMed] [Google Scholar]