

Abstract
(+)-Dihydromethysticin was recently identified as a promising lung cancer chemopreventive agent while (+)-dihydrokavain was completely ineffective. A pilot in vivo structure-activity relationship (SAR) was explored, evaluating the efficacy of its analogs in blocking 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced short-term O6-methylguanine and long-term adenoma formation in the lung tissues in A/J mice. Both results revealed cohesive SARs, demonstrating that the methylenedioxy functional group in DHM is essential while the lactone functional group tolerates modifications.
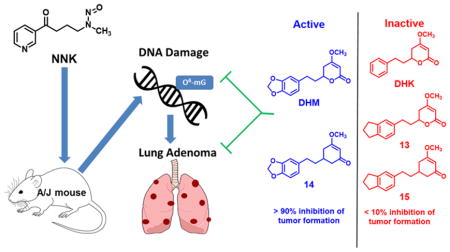
Graphical abstract
INTRODUCTION
Lung cancer causes ~160,000 deaths in the U.S. each year.1,2 Moreover, its five-year survival has been hovering around 15% for decades.2 Given the limited success in early diagnosis and treatment, prevention is of paramount importance. Tobacco cessation is the best strategy to reduce lung cancer risk.3 However, many smokers will not succeed in quitting because of the addictive nature of nicotine in tobacco products. Chemopreventive agents, therefore, are needed for these high-risk populations.
4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK, 1 in Scheme 1) is a tobacco-specific carcinogen that induces primarily lung tumor formation in various species.4 It has been proposed to contribute to lung adenocarcinoma incidence among smokers in the U.S.A.5 Mechanistically NNK needs metabolic activation via cytochrome P450 (CYP450) enzyme-mediated hydroxylation to generate two reactive species, which can react with DNA leading to DNA damages, including methylation and pyridyloxobutylation (POB, Scheme 1).3,6 NNK can also be reduced to 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL, 2), which is likely the dominant metabolic pathway of NNK in humans.7 Upon CYP450-mediated hydroxylation, NNAL generates two reactive species as well, resulting in DNA modifications, including methylation and pyridylhydroxybutylation (PHB). DNA damage induced by NNK and NNAL has been proposed as the underlying mechanism to initiate lung tumorigenesis.4 Inhibiting the formation of such DNA damage is, therefore, a plausible strategy to reduce lung cancer risk. Along the journey of understanding NNK carcinogenesis, Hecht and his colleagues have developed robust methods to quantify different DNA modifications via liquid chromatographyelectrospray ionization/tandem mass spectrometry (LC-ESI-MS/MS) analyses of the hydrolytic products from damaged DNA.
Scheme 1.

Simplified metabolism of NNK and NNAL, focusing on the bioactivation, generation of reactive intermediates, and subsequent formation of different types of DNA damage.
Among lung tumorigenesis models, the NNK-induced A/J mouse model is widely used in identifying lung cancer chemopreventive agents while the tobacco smoke-induced model is less practical.8–10 Because of the predisposed pulmonary adenoma susceptibility 1 (Pas1) gene,9 the A/J mice are highly susceptible to lung tumorigenesis. They can develop lung adenomas in a relatively short time with 100% incidence and high adenoma multiplicity upon appropriate NNK treatment.8,10 Although the contribution of each type of DNA damage to lung tumorigenesis has not been quantified, there has been compelling evidence suggesting that O6-mG is the most carcinogenic in A/J mice.11–16
We have recently identified (+)-dihydromethysticin ((+)-DHM, 3 in Figure 1) from kava as a promising lung cancer chemopreventive agent. (+)-DHM potently and selectively inhibited NNK-induced O6-mG formation in the target lung tissues.17 (+)-DHM also completely blocked lung adenoma formation in A/J female mice at a dose of 0.05 mg/g of diet.17 Interestingly, several structurally similar lactone compounds in kava revealed a clear structure activity relationship (SAR).17 (+)-Dihydrokavain ((+)-DHK, 4), for instance, was completely ineffective in inhibiting O6-mG and lung adenoma formation even at the dose of 0.5 – 1.0 mg/g of diet.17 (+)-Methysticin (5) was able to significantly inhibit NNK-induced O6-mG formation, only slightly less effective than (+)-DHM, while (+)-kavain (6) was not effective at all. These results overall suggest that the methylenedioxy functional group in (+)-DHM and (+)-methysticin is critical for their lung cancer chemopreventive potential.
Figure 1.

Structures of four major natural kavalactones in kava.
Kava traditionally is an aqueous suspension of the root of “noble” cultivars of piper methysticum and has been consumed as a beverage in the South Pacific Islands to help people relax and improve the quality of sleep.18 The organic (ethanol or acetone) extract of the root had been used as an anxiolytic agent in Europe with some debatable hepatotoxic risk.19,20 The organic extract form is currently available in the US as a dietary supplement.21 Although there has been a long history of kava usage in humans with a number of clinical trials,22 systematic characterization of its pharmacokinetics and metabolism has been limited, particularly with respect to (+)-DHM and (+)-methysticin because they are not believed to be responsible for kava’s relaxing and anxiolytic properties.23 Nevertheless, the methylenedioxy functional group can be metabolized by CYP enzymes via methylene hydroxylation followed by ring opening to generate a hydroquinone intermediate. In the case of DHM, intermediate 7 would be generated, which can be oxidized to 8 (Scheme 2). Although intermediates 7 and 8 have not been detected in humans upon kava exposure,24 a glutathione-conjugated adduct of 8 was detected via LC-MS/MS when (+)-DHM was incubated in human liver microsomes in the presence of glutathione.25 Such a result suggest that the methylenedioxy functional group in (+)-DHM and (+)-methysticin are likely subjected to metabolism in vivo. In addition, molecules of the same molecular weights as intermediates 9 and 10 have been detected via LC-MS/MS by the Xing group when (+)-DHM was incubated in mouse liver microsomes (data not shown), suggesting that the lactone group in (+)-DHM is metabolically labile as well. It therefore remains to be determined whether intact (+)-DHM or any of its potential metabolites (7 – 10) is the active form responsible for its chemopreventive potential.
Scheme 2.

Proposed metabolism of DHM and the corresponding metabolites.
These questions led to our current effort to investigate the role of the methylenedioxy and the lactone of DHM on its lung cancer chemopreventive activity. We have synthesized (±)-DHM (11), (±)-DHK (12) and three analogs (13–15) (Figure 2). By characterizing their effect on NNK-induced O6-mG and adenoma formation in the target lung tissue in A/J mice, a cohesive in vivo SAR of DHM in preventing lung tumorigenesis was observed, which provides key structural insights that will facilitate future mechanistic investigations and structural optimizations.
Figure 2.

Structures of racemic DHM, racemic DHK and three racemic analogs (A) and their effect on NNK-induced O6-mG in the lung tissues in A/J mice (B). Quantification of O6-mG in the target lung tissues in A/J mice (n=3 for 11 – 15 at a dose of 200 ppm and n=6 for NNK control group, O6-mG was not detectable in the negative control group, data not shown). Statistical analysis was performed with ONE-WAY ANOVA followed by Dunnett’s test of each treatment group relative to the NNK control group; *** p<0.001; **** p<0.0001.
RESULTS AND DISCUSSION
Rational design
Since any of the metabolites or intact DHM in Scheme 2 could be the in vivo active form, we proposed three analogs to probe these possibilities (Figure 2A, 13–15). Compound 13 would block methylene hydroxylation, compound 14 would block lactone hydrolysis, while compound 15 would block both pathways of metabolism.
Concise syntheses
We have previously developed a heavy-metal free synthesis to obtain (±)-methysticin (Scheme 3),26 which has been applied to the synthesis of (±)-kavain. We initially envisioned that 11 and 12 could be readily obtained from (±)-methysticin and (±)-kavain via a selective reduction of the 7,8-alkenyl functional group by catalytic hydrogenation (Scheme 3A). 12 indeed was efficiently prepared from (±)-kavain as such. Hydrogenation reaction, however, did not work well for the synthesis of 11 even upon several attempts of optimization; a considerable amount of an undesired hydrogenolytic product, 16, was formed. The formation of 16 not only decreased the yield of 11 but also complicated its purification. The mechanism behind the different hydrogenation outcomes of (±)-methysticin and (±)-kavain requires further investigation. We changed the reaction sequence for the synthesis of 11 to avoid the formation of 16 (Scheme 3B). The Wittig olefination of piperonal with the phosphorous ylide, followed by a quick column filtration, gave the olefinic acetal 17. Compound 17 upon catalytic hydrogenation furnished saturated acetal 18, which upon deprotection with 1 N HCl treatment to afford the saturated aldehyde 19 in a three-step 50% overall yield. Compound 19, without further purification, was subjected to dianion alkylation with ethyl acetoacetate, resulting in the aldol product 20. K2CO3 mediated lactonization of 20 followed by methylation using dimethyl sulfate in acetone afforded 11 in a 64% yield from 19. The synthesis of analog 13 started with indane aldehyde in a 32% overall yield, following the same synthetic route in Scheme 3B. Analogs 14 and 15 were synthesized via a different route (Scheme 3C). Using 14 as the example, aldehyde 19 upon ylide treatment via the Wittig reaction generated olefin ester 21 in a 95% yield. Intermediate 21 was converted to cyclic diketone 22 upon a sequential Michael addition and Dieckmann condensation, which was then methylated to furnish 14 in a 30% overall yield.
Scheme 3.

The synthesis of 11 – 15. (A) The first synthetic route for 11 with 16 being a by-product: (i) (a) (1,3-dioxolan-2-yl)methyl triphenylphosphonium bromide, LiOMe, 70 °C 6h; (b) 1N HCl, THF, RT, 2h; (c) Ethyl acetoacetate, NaH, n-BuLi, THF −78°C to RT; (d) K2CO3/methanol, then dimethylsulfate/acetone; (ii) H2, Pd/C, THF. (B) The second synthetic route of 11: (i) LiOMe, dry THF, 0 °C, piperonal, 70 °C, 6 h; (ii) H2, Pd/C (1 atm), THF, 2 h; (iii) 1N HCl, THF, RT, 2 h; (iv) (a) NaH, n-BuLi, dry THF, 0 °C, 30 min, (b) −55 °C to RT, 6h; (v) (a) K2CO3, MeOH, RT, 6h, (b) Dimethyl sulfate, acetone, RT, 12 h. (C) The synthetic route of 14 and 15: (i) (carbethoxymethylene)triphenylphosphorate, THF, reflux, 12h; (ii) NaH, acetone, THF and toluene, 0 °C-RT, 4h; (iii) Dimethyl sulfate, acetone, RT, 12 h.
Short-term effect of 11–15 on NNK-induced O6-mG in the target lung tissues
Given the carcinogenic importance of O6-mG in NNK-induced lung tumorigenesis in A/J mice and the distinct effects between (+)-DHM and (+)-DHK on O6-mG,17 11–15 were first evaluated for their effect on NNK-induced O6-mG in the target lung tissues in A/J mice at the dose of 200 ppm in diet, following our reported procedures (Figure 2B).17 Consistent with our previous results for (+)-DHM and (+)-DHK, 11 effectively reduced O6-mG adduct formation (72.3% reduction, p < 0.0001) while 12 treatment resulted in a minimal and non-significant reduction (16.4% reduction, p > 0.05). Compounds 13 and 15 also induced minimal and non-significant reductions in O6-mG adducts (29.6% and 11.8% reduction respectively, p > 0.05). Compound 14, on the other hand, resulted in a significant reduction in O6-mG adduct formation (63.8%, p < 0.001) relative to the NNK control group and statistically not different from 11.
Safety of 11 – 14 and their effect on the long-term lung adenoma formation induced by NNK
Based on their short-term effect on O6-mG, 15 was not evaluated for its effect on NNK-induced long-term lung adenoma formation in A/J mice because of its least reduction in O6-mG. 11 – 14 were evaluated at a dose of 25 ppm in diet herein. Such a dose would be equivalent to a dose of ~ 20–30 mg/day for a human of 75 kg bodyweight according to the Body Surface Area Normalization method.27 Based on their impact on the bodyweight and liver weight, all four compounds were well tolerated (Figure 3A–C). Consistent with the short-term O6-mG results and our previous study,17 11 effectively blocked NNK-induced long-term adenoma formation (94.2% reduction, p < 0.0001) while 12 was completely ineffective (6.3% reduction, p > 0.05). Compound 13 also had minimal effect on adenoma multiplicity (10.1% reduction, p > 0.05) while 14 significantly reduced adenoma multiplicity (89.4% reduction, p < 0.0001).
Figure 3.

Preliminary safety data of 11 – 14 (A – C) and their impact on tumor multiplicity (D) (n=5 for 11 – 14 and n=10 for NNK group). Statistical analysis was performed with ONE-WAY ANOVA, followed by Dunnett’s test of each treatment group relative to the NNK control group; **** p<0.0001.
CONCLUSION
In this study, we investigated the importance of the methylenedioxy and lactone functional groups on the chemopreventive activity of DHM using three rationally designed synthetic analogs that can respectively block either methylene hydroxylation, lactone hydrolysis or both routes of metabolism. To achieve this goal, we first developed facile syntheses of these analogs. Their lung cancer chemopreventive properties in comparison to the racemic DHM (active) and the racemic DHK (inactive) were then evaluated under the short-term and long-term conditions using a well-established NNK-induced lung tumorigenesis A/J mouse model. Compounds 13 and 15, devoid of the dioxy functional group of DHM while retaining the five-membered ring, did not show any significant inhibitory activity against NNK-induced O6-mG formation or lung tumor multiplicity in A/J mice. Interestingly compound 14, with the intact methylenedioxy functional group but the mask of the lactone functional group, recapitulated the O6-mG adduct reduction potential and antitumorigenic efficiency of DHM. It is noteworthy to restate that DHK, which lacks the methylenedioxy group, is inactive against NNK-induced DNA adducts and tumorigenesis. From these results, it is clear that the methylenedioxy functional group of DHM is critical for its chemopreventive activity while the lactone functional group tolerates modifications. In addition, the reduction in O6-mG correlates well with the blockage of lung adenoma formation, consistent with the results from Peterson et al. that O6-mG reduction could serve as a short-term surrogate to screen for effective chemopreventive candidates.11 Together, the results of this study reveal a cohesive structure-activity relationship of DHM for its chemopreventive potential. Studies are ongoing to determine whether the intact methylenedioxy moiety of DHM is involved in the direct interactions with the cellular target or the methylenedioxy functional group in DHM is metabolized to reveal the in vivo active form.
EXPERIMENTAL SECTION
Chemistry
All commercial reagents and anhydrous solvents were purchased from vendors and were used without further purification or distillation unless otherwise stated. Analytical thin layer chromatography was performed on Whatman silica gel 60 Å with fluorescent indicator (partisil K6F). Compounds were visualized by UV light and/or stained with potassium permanganate solution followed by heating. Flash column chromatography was performed on Whatman silica gel 60 Å (230–400 mesh). NMR (1H and 13C) spectra were recorded on a Varian 400 MHz or a Bruker 500 MHz spectrometer and calibrated using an internal reference. Chemical shifts (δ values) and coupling constants (J values) were given in ppm and hertz, respectively. ESI mode mass spectra were recorded on a Bruker BiotofII mass spectrometer. All compounds synthesized are racemic mixtures and are more than 95% pure, analyzed using HPLC. Detailed procedures and characterizations see Supporting Information.
A/J mouse in vivo studies
The animal studies performed herein were approved by the University of Minnesota Institutional Animal Care and Use Committee and conducted following the National Institutes of Health guidelines. AIN-G and AIN-M powdered diet was purchased from Harlan Teklad (Madison, WI). Female A/J mice at the age of 6 weeks were purchased from Jackson Laboratory (Bar Harbor, Maine).
Diet preparation and characterization
AIN-G powdered diets supplemented with 11 – 15 respectively were prepared following our reported procedures.8 Briefly, each compound was reconstituted in absolute ethanol (50 mL) and then mixed with the AIN-93 G powdered diet (150 g). Absolute ethanol (50 mL) was mixed with the AIN-93 G powdered diet (150g) for the control diet. The reconstituted diets were dried under vacuum to remove ethanol and then ground into fine powders. All diets were then mixed well with additional AIN-93 G powdered diet to the desired dose. The abundance of the respective compound in the diet was analyzed in triplicate by HPLC and confirmed to be within ± 10% of the specified dose.
Caution: NNK is IACR Group 1 carcinogen. Personnel handlings are expected to have appropriate safety measures. NNK and [CD3]O6-mG were purchased from Toronto Research Chemicals (Toronto, ON, Canada).
Impact of 11 – 15 on NNK-induced O6-mG DNA adduct in the lung tissues
After one week of acclimation, 27 female A/J mice (6–7 weeks of age) were randomized into seven groups each (n=3 except for the negative and NNK control groups n=6) and maintained on the specific diet starting on Day 0 (control diet for negative and NNK control groups, or the corresponding diet supplemented with 11 – 15 respectively at a dose of 200 ppm). Bodyweight of the mice was measured every two or three days and their food intake was monitored twice a week. On Day 7, except for mice in the negative control groups, all mice received a single dose of NNK in saline (100 μL) at the dose of 100 mg/kg of bodyweight via i.p. injection. Mice in the negative control group were given saline. Based on the results from our previous studies,28 four hours after NNK exposure, mice were euthanized with CO2 overdosing. The lung tissues were collected and stored at −80 °C till analysis. DNA isolation from the lung tissues and LC-MS/MS quantification were performed following the standard procedure.17 Briefly, DNA was isolated from half of the whole lung tissue of each individual mouse, following the Puregene DNA isolation protocol (Qiagen Corp). O6-mG was quantified by LC-ESI-MS/MS with [CD3]O6-mG as the internal standard.
Impact of 11 – 14 on NNK-induced adenoma formation in the lung tissues and preliminary safety monitoring
After one week of acclimation, 35 female A/J mice (6–7 weeks of age) were randomized into six groups each (n=5 each group except for the NNK-alone group, n=10) and maintained on the specific diet at a dose of 25 ppm, starting on Day 0. NNK was given at a dose of 100 and 67 mg/kg bodyweight via i.p. injection on Day 7 and Day 14 respectively. The dietary treatment stopped 24 hours after the last NNK treatment and mice were maintained on standard AIN-M powdered diet. Food intake was monitored twice a week and bodyweight was monitored once a week. At the end of Day 119, mice were weighed and euthanized via CO2 overdosing. The liver tissues were collected and weighed. The relative liver weight was calculated as the ratio of the liver weight and bodyweight of each mouse. Adenomas on the surface of the lung were counted under blinded conditions by an A.C.V.P board certified pathologist (M.G. O’S.).
Statistical analysis
Data on the quantity of O6-mG and lung adenoma multiplicity were reported as mean ± SD. One-way ANOVA was used to compare means among NNK and NNK + treatment groups. The Dunnett’s test was used for comparisons of the quantity between NNK control and NNK + treatment groups. P value ≤0.05 was considered statistically significant. All analyses were conducted in GraphPad Prism 4 (GraphPad Software, Inc.).
Supplementary Material
Acknowledgments
This work was funded by grants from National Institutes of Health Grant R01-CA193278 (Xing) and in part by the National Cancer Institute Cancer Center Support Grant CA 077598 (Turesky). We thank Dr. Peter Villalta of the Masonic Cancer Center’s Analytical Biochemistry shared resource for direction on the use of LC-MS/MS instrument.
ABBREVIATIONS
- SAR
structure-activity relationship
- DHM
dihydromethysticin
- DHK
dihydrokavain
- NNK
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
- NNAL
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol
- O6-mG
O6-methylguanine
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- ANOVA
analysis of variance
Footnotes
Notes
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information Availability
Synthesis, characterization and molecular formula strings are included, available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 3.Hecht SS. Lung carcinogenesis by tobacco smoke. Int J Cancer. 2012;131:2724–2732. doi: 10.1002/ijc.27816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hecht SS. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem Res Toxicol. 1998;11:559–603. doi: 10.1021/tx980005y. [DOI] [PubMed] [Google Scholar]
- 5.Hecht SS. It is time to regulate carcinogenic tobacco-specific nitrosamines in cigarette tobacco. Cancer Prev Res. 2014;7:639–647. doi: 10.1158/1940-6207.CAPR-14-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hecht SS. Progress and challenges in selected areas of tobacco carcinogenesis. Chem Res Toxicol. 2008;21:160–171. doi: 10.1021/tx7002068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carmella SG, Akerkar S, Hecht SS. Metabolites of the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in smokers’ urine. Cancer Res. 1993;53:721–724. [PubMed] [Google Scholar]
- 8.Hecht SS, Kassie F, Hatsukami DK. Chemoprevention of lung carcinogenesis in addicted smokers and ex-smokers. Nat Rev Cancer. 2009;9:476–488. doi: 10.1038/nrc2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Donnell EP, Zerbe LK, Dwyer-Nield LD, Kisley LR, Malkinson AM. Quantitative analysis of early chemically-induced pulmonary lesions in mice of varying susceptibilities to lung tumorigenesis. Cancer Lett. 2006;241:197–202. doi: 10.1016/j.canlet.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 10.Malkinson AM. Primary lung tumors in mice as an aid for understanding, preventing, and treating human adenocarcinoma of the lung. Lung Cancer. 2001;32:265–279. doi: 10.1016/s0169-5002(00)00232-4. [DOI] [PubMed] [Google Scholar]
- 11.Peterson LA, Hecht SS. O6-Methylguanine Is a Critical Determinant of 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone Tumorigenesis in A/J Mouse Lung. Cancer Res. 1991;51:5557–5564. [PubMed] [Google Scholar]
- 12.Liu L, Qin X, Gerson SL. Reduced lung tumorigenesis in human methylguanine DNA--methyltransferase transgenic mice achieved by expression of transgene within the target cell. Carcinogenesis. 1999;20:279–284. doi: 10.1093/carcin/20.2.279. [DOI] [PubMed] [Google Scholar]
- 13.Loechler EL, Green CL, Essigmann JM. In vivo mutagenesis by O6-methylguanine built into a unique site in a viral genome. Proc Natl Acad Sci U S A. 1984;81:6271–6275. doi: 10.1073/pnas.81.20.6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Essigmann JM, Loechler EL, Green CL. Mutagenesis and repair of O6-substituted guanines. IARC Sci Publ. 1986:393–399. [PubMed] [Google Scholar]
- 15.Ronai ZA, Gradia S, Peterson LA, Hecht SS. G to A transitions and G to T transversions in codon 12 of the Ki-ras oncogene isolated from mouse lung tumors induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and related DNA methylating and pyridyloxobutylating agents. Carcinogenesis. 1993;14:2419–2422. doi: 10.1093/carcin/14.11.2419. [DOI] [PubMed] [Google Scholar]
- 16.Malkinson AM. Primary lung tumors in mice: an experimentally manipulable model of human adenocarcinoma. Cancer Res. 1992;52:2670s–2676s. [PubMed] [Google Scholar]
- 17.Narayanapillai SC, Balbo S, Leitzman P, Grill AE, Upadhyaya P, Shaik AA, Zhou B, O’Sullivan MG, Peterson LA, Lu J, Hecht SS, Xing C. Dihydromethysticin from kava blocks tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced lung tumorigenesis and differentially reduces DNA damage in A/J mice. Carcinogenesis. 2014;35:2365–2372. doi: 10.1093/carcin/bgu149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teschke R, Sarris J, Lebot V. Kava hepatotoxicity solution: A six-point plan for new kava standardization. Phytomedicine. 2011;18:96–103. doi: 10.1016/j.phymed.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 19.Organization, W. H. WHO Document Production Service. 2007. Assessments of the risk of hepatotoxicity with kava products. [Google Scholar]
- 20.Teschke R, Schwarzenboeck A, Hennermann KH. Kava hepatotoxicity: a clinical survey and critical analysis of 26 suspected cases. Eur J Gastroenterol Hepatol. 2008;20:1182–1193. doi: 10.1097/MEG.0b013e3283036768. [DOI] [PubMed] [Google Scholar]
- 21.River ZXC, Narayanapillai S. Kava as a pharmacotherapy of anxiety disorders: promises and concerns. Medicinal Chemistry. 2016;6:81–87. [Google Scholar]
- 22.Sarris J, Teschke R, Stough C, Scholey A, Schweitzer I. Re-introduction of kava (Piper methysticum) to the EU: is there a way forward? Planta Med. 2011;77:107–110. doi: 10.1055/s-0030-1250290. [DOI] [PubMed] [Google Scholar]
- 23.Lindenberg D, Pitule-Schodel H. D,L-kavain in comparison with oxazepam in anxiety disorders. A double-blind study of clinical effectiveness. Fortschr Med. 1990;108:49–50. [PubMed] [Google Scholar]
- 24.Duffield AM, Jamieson DD, Lidgard RO, Duffield PH, Bourne DJ. Identification of Some Human Urinary Metabolites of the Intoxicating Beverage Kava. J Chromatogr. 1989;475:273–281. doi: 10.1016/s0021-9673(01)89682-5. [DOI] [PubMed] [Google Scholar]
- 25.Johnson BM, Qiu SX, Zhang S, Zhang F, Burdette JE, Yu L, Bolton JL, van Breemen RB. Identification of novel electrophilic metabolites of piper methysticum Forst (Kava) Chem Res Toxicol. 2003;16:733–740. doi: 10.1021/tx020113r. [DOI] [PubMed] [Google Scholar]
- 26.Shaik AATJ, Lu J, Xing C. Economically viable efficient synthesis of (±)-Methysticin - A component in kava potential responsible for its cancer chemopreventive activity. Arkivoc. 2012;viii:137–145. [Google Scholar]
- 27.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- 28.Leitzman P, Narayanapillai SC, Balbo S, Zhou B, Upadhyaya P, Shaik AA, O’Sullivan MG, Hecht SS, Lu J, Xing C. Kava blocks 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced lung tumorigenesis in association with reducing O6-methylguanine DNA adduct in A/J mice. Cancer Prev Res. 2014;7:86–96. doi: 10.1158/1940-6207.CAPR-13-0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.