

Abstract
Mammalian organs vary widely in regenerative capacity. Poorly regenerative organs, such as the heart are particularly vulnerable to organ failure. Once established, heart failure (HF) commonly results in mortality1. The Hippo pathway, a kinase cascade that prevents adult cardiomyocyte proliferation and regeneration2, is upregulated in human HF. We show that deletion of the Hippo pathway component Salvador (Salv) in mouse hearts with established ischemic HF after myocardial infarction (MI) induced a reparative genetic program with increased scar border vascularity, reduced fibrosis, and recovery of pumping function compared to controls. Using TRAP (translating ribosomal affinity purification), we isolated cardiomyocyte specific translating mRNA. Hippo deficient cardiomyocytes had increased expression of proliferative genes and stress response genes, such as the mitochondrial quality control (MQC) gene, Park2. Genetic studies indicated that Park2 was essential for heart repair suggesting a requirement for MQC in regenerating myocardium. Gene therapy with a virus encoding Salv shRNA improved heart function when delivered at the time of infarct or after ischemic HF post-MI was established. Our findings indicate that the failing heart has a previously unrecognized reparative capacity involving more than cardiomyocyte renewal.
Graphical abstract

HF, the inability of the heart to pump blood, has no definitive treatment except heart transplantation or Ventricular Assist Device3. In HF, physiologic compensatory mechanisms place strain on cardiomyocytes and promote cardiomyocyte loss and fibrosis in pathologic remodeling4–6. Hippo pathway activation, including Mst and Lats kinases and Salvador (Salv) adaptor, results in phosphorylation and nuclear exclusion of transcriptional cofactors Yap and Taz that cooperatively bind DNA with Tead factors2,7. Concurrent Salv or Lats1 and Lats2 deletion with myocardial infarct (MI) improves heart function8–10. Yap target genes control proliferation, cytoskeletal remodeling, and protect plasma membrane from contractile stress9. Maladaptive activation of Hippo signaling occurs in human HF and mouse ischemia reperfusion11,12. To determine if endogenous cardiomyocyte renewal can be increased in HF, we deleted Salv in a mouse model of ischemic HF (iHF) post-MI. The Hippo pathway represses a reparative genetic program in iHF that includes cardiomyocyte proliferation and survival genes.
Western blots on iHF and nonischemic HF (HF) human samples revealed that pYap and pLats serine 909 levels were higher and Salv levels unchanged in iHF and HF than controls (Fig. 1a-f, Extended Data Fig. 1a-c). We generated αMHC-mcm;Salv f/f (SalvCKO) mice with iHF post-MI (Fig. 2a). Echocardiography three weeks post-MI before tamoxifen, revealed shams had an average ejection fraction (EF) of 64±12% and fractional shortening (FS) of 35±8% (Fig. 2a-c). Post-MI, mice had an average EF of 36±11% and FS of 18±6% (Fig. 2a-c)13. End-systolic variables showed increased systolic left ventricle (LV) chamber diameters (Sham 2.7+/−0.6mm, MI 4.1±0.5mm) and volumes (Sham 29+/−18μL, MI 76±23μL) (Extended Data Fig. 2a-d). Diastolic indices were unchanged indicating iHF was a result of systolic dysfunction (Extended Data Fig. 2b, d). Other iHF signs included pulmonary fluid buildup, hemosiderin-laden macrophages, increased natriuretic peptide B (BNP) serum levels, and increased body weights (Extended Data Fig. 2e-j).
Figure 1. Activated Hippo signaling in human HF.

a, b, Western blots human heart samples. Ctrl: nonfailing nontransplantable, n=6; HF: nonischemic idiopathic end-stage cardiomyopathy, n=6; iHF: ischemic end-stage HF, n=6. Additional samples Extended Data Figure 1. c-f, Quantification Yap (c), pYap (d), pLats (e), Salv (f). Mann-Whitney. Data: mean ± s.e.m, p-values >0.05 nonsignificant (n.s.), *p<0.05, **p<0.01, ***p<0.001.
Figure 2. HF reversal and cardiomyocyte renewal in SalvCKO mice.

a, Experimental timeline. Numbers/group analyzed by echocardiography. b, c, Fractional shortening (FS) (b) ejection fraction (EF) (c), ANOVA Tukey post-test. d, e, Masson’s trichrome serial sections 9 weeks post-MI, n=7/group. scar boundaries (open arrows); ischemic region (solid arrows). Scar categories I, II, III. Scale = 2 mm. f, LV scar size, n=7/group, Mann-Whitney. g, Fibrosis and function: second order polynomial fit. h, Diagram cardiac regions. i, j, PCM-1 IF (i), scale = 100μm; quantification (j), n=3/group, Mann-Whitney. k, l, EdU-labeled cardiomyocytes (arrow) (k) scale = 25μm; quantification (l), SalvCKO MI: 3 weeks (n=4), 9 weeks (n=5), others (n=3), ANOVA Bonferroni post-test. m, n, pHH3 IF (arrow) (m) scale = 25μm; quantification (n), n=3/group, Mann Whiteny. o, p, BZ lineage labeling (o), scale = 25μm; quantification (p), n=3/group, ANOVA Bonferroni post-test. q, Cardiomyocyte cross-sectional area whole heart(WH) and BZ 9 weeks post-MI, Ctrl MI (n=3) SalvCKO MI (n=5), ANOVA Bonferroni post-test. Control: tamoxifen-injected αMHC-mcm; ROSAmT/mG. SalvCKO; tamoxifen-injected αMHC-mcm; ROSAmT/mG; Salvfl/fl. Data: mean ± s.e.m, p-values >0.05 nonsignificant (n.s.), *p<0.05, **p<0.01, ***p<0.001.
3 weeks post-MI, we deleted Salv in cardiomyocytes and performed echocardiography every 2 weeks until 9 weeks post-MI (6 weeks after tamoxifen injection) (Fig. 2a-c, Extended Data Fig. 2a-d, k, l). At 9 weeks post-MI, SalvCKO had improved function (EF; SalvCKO MI 59%±13%, Ctrl MI 38±9%, p=0.001) similar to sham controls (EF; SalvCKO MI 59%±13%, Ctrl and SalvCKO Sham 65%±8%, p=1)(Fig. 2c).
9 week post-MI control hearts had remodeled scars whereas SalvCKO hearts showed less fibrosis and more LV cardiomyocytes (fibrosis: Ctrl 56±12%, SalvCKO 36 ±15%; CM number: Ctrl 1×105±8×104, SalvCKO 6×105±2×105) (Fig. 2d-j). We defined three qualitative categories of damage in SalvCKO hearts post-MI (Fig. 2d, e). Category I, observed in 10% of SalvCKO cohort (N=1 of 10), recovered most LV cardiomyocytes. Category II, observed in 80% of SalvCKO hearts (N=8 of 10), had an equal amount of fibrotic and cardiomyocyte area in LV. Category III, observed in 10% of SalvCKO hearts, had few LV cardiomyocytes with a remodeled scar (N=1 of 10) (Fig. 2d-f). The relationship between fibrosis and function was described by a second order polynomial suggesting that fibrosis is tolerated to maintain cardiac function (Fig. 2g).
3 weeks post-MI, all hearts had LV infarcts, (Extended Data Fig. 3). At two time points, hearts were collected one week prior to echocardiography for histopathology. We discuss those time points as a 2 week range (ie. 4-5 and 6-7 weeks post-MI). There were no differences between groups at 4-5 weeks and 6-7 weeks post-MI (Fig. 2b, c, f, Extended Data Fig. 4,5). At 6 weeks post-MI, Myh7, Nppa, and Nppb, associated with adult cardiomyocyte remodeling, were upregulated in control iHF but not SalvCKO (Extended Data Fig. 6a).
SalvCKO hearts at 6 weeks post-MI had a three fold increase in BZ capillary density and increased endothelial markers, isolectin B4 and CD-31 compared to controls (Extended Data Fig. 6b-e). Cardiomyocyte-enriched TRAP RNA revealed increased vasculogenesis genes encoding angiopoietins, fibroblast growth factors, and vascular endothelial growth factors in cardiomyocytes of SalvCKO MI hearts (Extended Data Fig. 6f).
We found SalvCKO hearts had 2% to 3% cardiomyocyte EdU incorporation at 4 and 6 weeks post-MI and approximately 1% at 9 weeks post-MI (Fig. 2k, l). Phospho-histone H3 IF revealed M-phase cardiomyocytes 9 weeks post-MI in SalvCKO MI mice (Fig. 2m, n). Hence, cardiomyocyte cell cycle entry was diminished in SalvCKO mouse hearts at 9 weeks post-MI as cardiomyocytes repaired the tissue defect.
Lineage tracing by αMHC-mcm transgene revealed mosaic labeling of BZ cardiomyocytes (Fig. 2o). At 4-weeks post-MI, BZ had equivalent numbers of GFP-positive cardiomyocytes in controls (33%±7%) and SalvCKO (22%±2%) (Fig. 2o, p). At 9-weeks post-MI GFP-positive BZ cardiomyocytes were enriched in SalvCKO (Ctrl MI 41%±6%, SalvCKO MI 91%±4%) (Fig. 3o, p) indicating that most new, reparative BZ cardiomyocytes are derived from pre-existing cardiomyocytes rather than Myh6-negative cardiac stem cells that would be GFP-negative. Cardiomyocyte cross-sectional area was reduced, suggesting a more primitive phenotype, in SalvCKO mice at 9 weeks post-MI14 (Fig. 2q).
Figure 3. SalvCKO mice activate reparative molecular response to HF.

a, Total RNA-Seq diagram. b, Volcano plot Total RNA-Seq: SalvCKO MI, Ctrl MI (n=3/group). c, d, Gene ontology (GO) Total RNA-Seq: SalvCKO MI, Ctrl MI (n=3/group). e, TRAP RNA-Seq (TRAP-Seq) diagram. f, Volcano plot TRAP-Seq: SalvCKO MI, Ctrl MI (n=3/group). g, Sample distance matrix: Total RNA-Seq, TRAP-Seq (n=3/group). h, Fold change TRAP/Total RNA (n=3/group) cardiomyocyte, noncardiomyocyte-enriched genes (GSE49906), t-test, ***p<0.001. i, Cardiomyocyte transcripts TRAP-Seq enriched; non-cardiomyocyte and non-coding RNA transcripts Total RNA-Seq enriched (n=3/group). j, PCA Total and TRAP-Seq (n=3/group). k, l, GO TRAP-seq: SalvCKO MI vs Ctrl MI (n=3/group). m, Normalized Park2 read counts, n=3/group. Control: tamoxifen-injected αMHC-mcm; Rpl22HA. SalvCKO: tamoxifen-injected αMHC-mcm; Rpl22HA; Salvfl/fl.
We sequenced Total-RNA and TRAP-RNA from BZ 6 weeks post-MI (Extended Data Fig. 7a-d). Total-RNA revealed upregulated genes (N=932) in SalvCKO BZ were involved in oxidative phosphorylation, RNA processing, stress response, cell cycle, and muscle contraction (Fig. 3a-c, Extended Data Fig. 8a). SalvCKO BZ downregulated genes (N=792) included developmental genes, glycolytic metabolism genes, and connective tissue genes including pro-fibrotic genes like Tgfbr1, Ctgf, and Pdgfrδ (Fig. 3d, Extended Data Fig. 8a)15,16.
Comparison of TRAP-seq, enriched for cardiomyocyte translating RNA (Fig. 3e,f), to total RNA-seq revealed that TRAP-seq was enriched for cardiomyocyte genes and depleted of non-cardiomyocyte genes (Fig. 3g-i, Extended Data Fig. 8b). Non-translated LncRNAs were depleted from TRAP-seq (Fig. 4I, Extended Data Fig. 8b). Comparison of total RNA-seq to TRAP-seq revealed total RNA and TRAP-seq groups clustered separately (Fig. 3j). SalvCKO MI TRAP-seq clustered closely to control sham TRAP-seq data revealing that Hippo deficient cardiomyocytes expressed a genetic program similar to control shams (Fig. 3j).
Figure 4. Park2 in HF and Salv gene therapy.
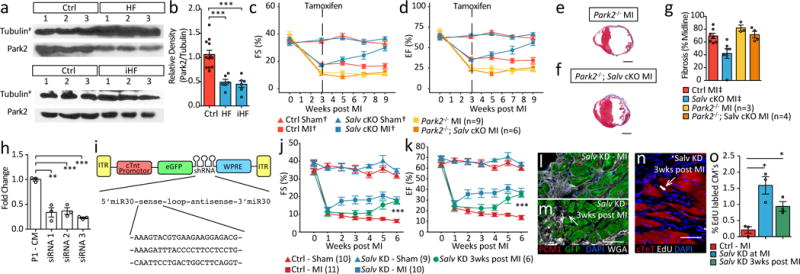
a,b Western blot human heart (a) and quantification(b), Mann-Whitney. (Tubulin# from Figure 1a,b (n=6); additional samples Extended Data Figure 1). c, d, fractional shortening (FS) (c) ejection fraction (EF) (d), ANOVA Tukey post-test, Ctrl Sham†, Ctrl MI†, SalvCKO Sham†, SalvCKO MI† repeated from Figure 2a-c. e, f, Masson’s trichrome: Park2−/− (n=3) (e) Park2−/−; SalvCKO (n=4) (f) 9 weeks post-MI. Scale = 2 mm. g, LV scar size, Ctrl MI‡, SalvCKO MI‡ repeated from Figure 2f (n=7). h, Knockdown (KD) efficiency Salv siRNA, n=3/group. i, AAV9 diagram. j, k, FS (j) and EF (k), ANOVA Tukey post-test. l, m, GFP expression Salv KD BZ (n=3/group), Low GFP (Arrow), scale = 50μm. n, o, EdU-labeled cardiomyocytes (arrow), scale = 50μm; quantification (o), n=3/group, Mann-Whitney. Data: mean ± s.e.m, p-values >0.05 nonsignificant (n.s.), *p<0.05, **p<0.01, ***p<0.001.
We compared TRAP-seq of control post-MI and sham (Ctrl MI vs Ctrl Sham) to determine cardiomyocyte gene expression changes during iHF post-MI (Extended Data Fig. 8c-f). One upregulated category was peptide metabolic processes including Akt1 that exacerbates HF when chronically expressed17(Extended Data Fig. 8d, f). Genes that respond to reactive oxygen species, such as GstP1 and Gpx3, were upregulated as was Clusterin, known to be upregulated in human HF18. Another upregulated gene category, actin filament based process, is consistent with observations that cytoskeletal components are upregulated in human HF19. Downregulated gene categories included chromatin organization and blood vessel development consistent with known gene expression changes in HF20,21(Extended Data Fig. 8e, f). Genes involved in organelle fission, such as mitochondrial fission, were downregulated. Disruption of mitochondrial fission and fusion can lead to HF22 (Extended Data Fig. 8e, f).
We compared TRAP-seq between SalvCKO and control hearts post-MI (SalvCKO MI vs Ctrl MI) (Fig. 3f). Upregulated genes (N=365) included cell cycle genes, consistent with previous studies (Fig. 3k, Extended Data Fig. 8g)8,9. Other upregulated genes included genes involved in heart contraction, heart growth, and cellular response to stress suggesting that SalvCKO cardiomyocytes were recovering the mature cardiomyocyte phenotype with an effective stress response. Downregulated genes (N=261) in SalvCKO cardiomyocytes post-MI included translation, protein metabolism including ubiquitin-proteosome pathway, and inflammation (Fig. 3l, Extended Data Fig. 8g), indicating that inflammation and mis-folded protein response were more effectively resolved in SalvCKO MI.
Park2, a Yap target gene, was upregulated in SalvCKO TRAP-seq (Fig. 3m)9. Park2 encodes a ubiquitin ligase involved in MQC, a surveillance mechanism to recycle damaged mitochondria23. Park2 is recruited to damaged mitochondria to direct mitochondria into mitophagy pathway that salvages damaged mitochondria. Park2 protein levels are reduced in human HF (Fig. 4a,b Extended Data Fig. 9a-c). Moreover, Park2 deletion in adult mice results in dilated cardiomyopathy post-MI24. Similar to Yap, Park2 is required for P1 cardiac regeneration (Extended Data Fig. 9d-i)25.
We tested if Park2 was required for regeneration in P8 SalvCKO8. After P8 MI, SalvCKO had higher mitochondrial DNA content and increased Park2 protein levels (Extended Data Fig. 10a,b), suggesting that MQC is more active in SalvCKO hearts. Park2 was required for recovery of myocardial function in SalvCKO hearts after P8 MI, (Extended Data Fig. 10c-g). Interestingly, although myocardial function was reduced, fibrosis did resolve in SalvCKO;Park2−/− hearts, indicating a myocardial-autonomous requirement for Park2 in SalvCKO neonatal hearts (Extended Data Fig. 10c, g).
Park2 was required for cardiac function recovery in adult iHF (Fig. 4c-g). Compared to SalvCKO mice, double mutant SalvCKO;Park2−/−mice didn’t recover contractile function (Fig. 4c,d). However, unlike SalvCKO adult hearts and SalvCKO;Park2−/− neonatal hearts (Extended Data Fig. 10c), scar failed to resolve in SalvCKO;Park2−/− adult hearts revealing a differential requirement for Park2 in neonatal versus adult fibrosis resolution (Fig. 4e-g).
We engineered a viral vector to knockdown Salv in cardiomyocytes (Fig. 4h, i). Direct myocardial viral injection at the time of MI or systemic viral injection 3 weeks post-MI resulted in improved cardiac function and cell cycle induction (Fig. 4j-o). Hippo pathway inhibition, genetically or with gene therapy, results in an effective stress response in iHF with organ failure reversal.
Since Hippo signaling is upregulated in human iHF, Hippo pathway inhibition may have a therapeutic benefit for iHF patients. We identified cardiomyocyte-directed, non-cell-autonomous injury responses that improve vascular perfusion. New cardiomyocytes, derived from pre-existing cardiomyocytes, direct recovery of a BZ microvasculature by expressing vessel-promoting factors including FGF’s that are protective against cardiac damage26–28. iHF reversal requires a proliferative response and injury resistance since newly generated cardiomyocytes must withstand both mechanical and hypoxic stress. At risk cardiomyocytes also likely benefit from Hippo pathway inhibition.
Our data provide insight into Park2 function in cardiac regeneration. In addition to MQC, Park2 functions in mitochondrial maturation and in fetal-to-adult metabolic transition29. Park2 is essential for neonatal regeneration and for cardiac repair in non-regenerative P8 SalvCKO hearts and in iHF. In P8 SalvCKO hearts, Park2 deletion was detrimental to cardiac function, but dispensable for fibrosis resolution. In contrast, in adult iHF, both cardiac function and fibrosis resolution were impaired in SalvCKO;Park2−/− mice. Park2 requirements in cardiomyocyte function and fibrosis resolution are an area of future study.
Materials and Methods
Patient Samples
Three types of human heart tissue samples were used for this study: control samples (nonfailing, nontransplantable hearts, n=6), HF samples (hearts with nonischemic idiopathic cardiomyopathy in end-stage failure, n=3), and iHF samples (hearts with ischemic heart disease in end-stage failure, n=3). For Control and both types of HF, samples were obtained from the left ventricular apex. The HF samples were collected at the time of left ventricular assist device implantation, the area from which the tissue core is obtained. The tissue samples were provided by the Texas Heart Institute (THI) Center for Cardiac Support Cardiovascular Surgery Research and Cardiovascular Pathology Research in accordance with human research protocol approved by the Texas Heart Institute Institutional Review Board under which patient informed consent was obtained. Sample analysis was conducted in accordance with human research protocol approved by the Baylor College of Medicine Institutional Review Board for which additional patient consent was waived. The tissue samples were digested to obtain protein lysates with the following buffer: 50mM Tris-Cl (pH7.5), 150mM NaCl, 1% Triton X-100, 0.25% sodium deoxycholate, and protease/phosphatase inhibitors. Samples were homogenized with a Dounce tissue homogenizer, passed through a 22-gauge needle, and sonicated for 10 cycles (30 seconds on, 30 seconds off) in a Diagenode Bioruptor Pico Plus NGS. For western blot analysis, we used antibodies against total YAP (Novus # NB110-58358), pYAP (Cell Signaling # 4911S), pLats1 (Cell Signaling # 9157), Sav1 (Novus # NBP2-13282), Park2 (Cell Signaling # 2132S), and alpha-tubulin (Sigma # T5168).
Mice
Adult C57/BL6×129/S mice, 8 to 10 weeks old were used for the heart failure studies, and underwent either a Sham procedure or an MI procedure. C57/BL6 and 129/S strains are weakly regenerative strains30. For the AAV9 experiments, adult (8-10 week old) ICR (CD1) mice were used. The regenerative characteristics of outbred ICR (CD1) compared to inbred strains has not been systematically investigated. At 3 weeks post-MI, we injected tamoxifen into mice to induce Cre activity in cardiomyocytes. Control (Ctrl) mice included tamoxifen-injected αMHC-mcm; ROSAmT/mG(mTmG) mice, tamoxifen-injected Salvfl/fl mice, and oil-injected αMHC-mcm; Salvfl/fl mice to control for Tamoxifen and cre recombinase cardiotoxicity31–33. SalvCKO mice included tamoxifen-injected αMHC-mcm; ROSAmT/mG; Salvfl/fl mice and tamoxifen-injected αMHC-mcm; Salvfl/fl mice. The ROSAmT/mG reporter was used to linage trace αMHC-mcm cardiomyocytes. Tamoxifen-injected αMHC-mcm; Rpl22HA mice were used as controls for TRAP experiments. For experiments involving TRAP SalvCKO mice, we used tamoxifen-injected αMHC-mcm; Salvfl/fl; Rpl22HA mice. For the P1 Park2 experiments, we used C57BL/6J and Park2tm1shn/j mice. For the P8 and adult double mutant experiments, we used αMHC-mcm; Salvfl/fl; Park2tm1shn/j mice injected with tamoxifen.
To achieve recombination of floxed alleles in the adult mice, tamoxifen dissolved in peanut oil was administered via intraperitoneal injection at a dose of 1 mg/day for 4 days beginning 3 weeks post-MI. No tamoxifen was administered to the mice in the P1 experiments. To achieve recombination of the floxed alleles in the P8 mouse experiments, tamoxifen was administered at P7, P8, P9, and P10, at 0.5 mg/day dissolved in peanut oil, as previously described8.
All mouse procedures were performed in accordance with institutional and governmental guidelines, and approved by the Baylor College of Medicine Institutional Animal Care and Use Committee.
Surgical Procedures
Model of heart failure in adult mice
To induce MI in 8- to 10-week-old mice, we permanently ligated the LAD artery as previously described8. Briefly, mice were anesthetized with 2% isoflurane and then intubated. We exposed the heart by performing a thoracotomy through the fourth or fifth intercostal space and tied an 8-0 nylon suture around the LAD artery. The mice were allowed to recover for 3 weeks, and then heart function was analyzed by using echocardiography. Because the clinical definition of heart failure is a 20% reduction in left ventricular EF as indicated on echocardiography (i.e., from >50% EF to <40% EF in humans),34 only mice with a >20% decrease in left ventricular ejection fraction were included in the study13. Our observed rates of survival (64%) and inclusion (70%) were consistent with those previously reported (50%-60% and 54%-92%, respectively)13,34,35.
Model of myocardial infarction in early and late neonatal mice
To induce MI in neonatal mice, we permanently ligated the LAD artery on postnatal day 1 (P1) or postnatal day 8 (P8), as previously described8. Briefly, mice were anesthetized with hypothermia, the heart was exposed via thoracotomy through the fourth or fifth intercostal space, and an 8-0 nylon suture was tied around the LAD coronary artery.
Echocardiography
Cardiac function was determined by echocardiography (VisualSonics, Vevo 2100, 40Mhz-550S probe). After alignment in the transverse B-mode with the papillary muscles, cardiac function was measured on M-mode images.
Injury regions
Cardiac tissue regions used for RNA, or image characterization described as; whole heart (WH), ischemic zone (IZ, LV free wall), border zone (BZ, LV anterior and posterior wall), or distal zone (DZ, interventricular septum).
Histological analysis
Whole hearts were fixed with 10% formalin, embedded in paraffin, and sectioned at 7-μm intervals. Each slide had 10 sections, which started at the apex and ended at the suture ligation site (approximately 50 slides). Every 4th slide was stained with Masson’s trichrome to identify areas of fibrosis. To determine scar size we examined serial sections from the apex to the ligation suture site and calculated the average percent fibrosis of the midline circumference around the LV36. To quantify cardiomyocytes we used PCM-1 immunostaining and methods as described37.
Immuno-fluorescence Experiments
To assess cell cycle entry, we added thymidine analogs to the drinking water for a duration of 4 days prior to dissection. The analog 5-iodo-2′-deoxyuridine (IdU; 0.2 g/L, MP Biomedical, catalog #0210035701) was used at the 3 week post-MI time point prior to tamoxifen delivery. The analog 5-ethynyl-2′-deoxyuridine (EdU; 0.2 g/L, Santa Cruz, catalog #sc-284628A) was used for the 4, 6, and 9 weeks post-MI time points. IF experiments were performed on FFPE (formalin-fixed and paraffin-embedded sections) or on fresh frozen optimum cutting temperature-embedded sections. The stains and antibodies used for these experiments included; Click-iT EdU kit, IdU staining (IdU cross-reactive BrdU antibody, BD Biosciences, 347580), mTmG lineage trace (endogenous mTomato and mGFP fluorescence), isolectin B4 (Vector Labs, FL-1201), CD31 antibody (Pecam, BD Pharmingen, 550274), PCM1 (Sigma, HPA023370), cTnT (ThermoFisher, MS295), wheat germ agglutinin (WGA, Vector Labs, RL-1022), and pHH3 (cell signaling, #9701).
Linage tracing was done using the Rosa26mT/mG reporter at 4 and 9 weeks post-MI in the BZ. A full dose of tamoxifen was administered, and mosaicism of the reporter was observed in the BZ. Mosacism was not observed in the DZ. We assume this is due to the variability of drug-included cre activity because of the poor BZ vascularity. The 4 weeks post-MI time point was used as a baseline to determine cre activity in the BZ. The.
RNA
Total RNA
Using trizol reagent, we isolated total RNA from the non-fibrotic myocardium (interventricular septum, anterior and posterior free wall) bellow the ligation suture.
Translating ribosomal affinity purification (TRAP RNA)
The TRAP method utilizes an inducible HA epitope-tagged ribosomal allele for the Cre-mediated cell-specific isolation of RNA38. By crossing this allele into the SalvCKO background and then performing TRAP-seq, we enriched for mRNAs that were loaded onto ribosomes in cardiomyocytes and determined the cardiomyocyte translating RNA profile. We quickly excised the whole heart from each mouse and obtained the LV anterior and posterior free-walls (the myocardial border zone) below the ligation suture to isolate ribosomal-associated cardiomyocyte-specific RNA. The tissue was homogenized on ice in 1 mL of supplemented homogenization buffer (1% NP-40, 0.1M KCl, 0.05M Tris, 0.012M MgCl2, 0.1mg/mL cyclohexamide, protease inhibitors, RNAsin, 0.01M DTT). The samples were centrifuged, and the supernatants were incubated at 4°C for 4 hours with the primary HA-antibody (Cell Signaling, catalog #3724) and then overnight with protein-G magnetic beads (Pierce, catalog #88847). Bead-antibody RNA isolation was performed on the homogenate, which was washed with a high-salt buffer (1% NP-40, 0.3M KCl, 0.05M Tris, 0.012M MgCl2, 0.1mg/mL cyclohexamide, 0.01M DTT). In samples with no expression of the Rpl22HA transgene, we recovered less than 1% of the total input RNA; therefore, we assumed that the RNA isolation was highly specific.
Real Time - quantitative PCR (RT-qPCR)
Transcript levels were quantified by using RT-qPCR. First-strand synthesis was performed by using iScript reverse transcription super mix for RT-qPCR (BioRad, catalog #1708841); then, qPCR was carried out by using iTaq Universal SYBR Green super mix (BioRad, catalog #172-5121). All qPCR primers are listed in Supplementary Information Table 1.
RNA-seq library preparation and data analysis
To isolate mRNA from Total RNA and TRAP RNA samples, we used the Dynabeads mRNA DIRECT Micro kit. ERCC Ex Fold RNA Spike-In control mixes were added before mRNA purification. Ion Torrent RNA-seq libraries were prepared with the Ion Total RNA-Seq kit v2 for whole transcriptome libraries. RNA sequencing was performed by using the Ion Proton system for next-generation sequencing. Reads were mapped to the mouse genome (mm10) and read counts quantified using STAR (version 2.5). Differentially expressed genes were detected using R package DESeq2 with the following parameters: threshold p ≤ 0.05, fold change ≥ 0.5, and FDR ≤ 10%. GO analysis was performed on differentially expressed genes (p<0.05) with the Metascape online platform, and annotation clusters with terms that had a p-value ≤ 0.05 were included.
We compared our TRAP vs Total RNA analysis to the cardiomyocyte and non-cardiomyocyte isolation data set (GSE49906)39. When sorted by fold change, the top 400 cardiomyocyte-enriched genes extracted from the GSE49906 P60 mouse hearts, were on average 1.5 fold more highly expressed in TRAP-seq samples than in Total RNA-seq samples. Likewise, the top 400 non-cardiomyocyte enriched genes were on average 1.5 fold more highly expressed in the Total RNA-Seq data.
AAV9 targeting Salvador
First, three siRNA constructs targeting Salv were tested in vitro in primary isolated neonatal mouse cardiomyocytes. Then, a triple short hairpin (sh)RNA construct with flanking miR30 sequences was cloned into the pENN.AAV.cTNT, p1967-Q vector downstream of GFP. Expression of both the GFP and the shRNA’s is driven by the cardiac troponin T promotor. The miR30-based shRNA design allows for efficient shRNA expression from an RNA Pol II promotor40. The resulting construct was packaged into the muscle-trophic serotype AAV9 by the IDDRC Neuroconnectivity Core at Baylor College of Medicine. Two methods of viral delivery were used: direct myocardial injection at the time of MI (Salv KD-MI), or systemic injection by retro-orbital injection 3 weeks post-MI (Salv KD 3 weeks post-MI). Viral titer for efficient knockdown was done as described previously and shown to effectively knock down Salv41. For direct myocardial injection at the time of MI, the mice were given 2 or 3 intra-myocardial injections by using a Hamilton syringe (50μL capacity) with a 33-gauge needle to deliver a total of 2×10^11 viral genomes (10 μl total volume delivered) into the ischemic risk area. In a separate cohort of mice, AAV9 was injected systemically via retro-orbital injection to deliver a total of 1×10^12 viral genomes (50μL total volume delivered).
Statistical analysis
Throughout this study, we used either the Mann-Whitney U test or analysis of variance (ANOVA) with Tukey’ pairwise post-hoc test or Bonferroni’s post-hoc test to compare means and the F test to compare variance. All error bars show the standard error of the mean. Mice were assigned unique identifiers to blind experimenters to genotypes. A block randomization scheme was used to assign animals to groups on a rolling admissions basis to obtain adequate samples for each time point and each experiment. For echocardiography sample sizes were estimated using power analysis determined from our previously described experiments.
Data Availability
The sequencing data in this manuscript is available through the accession number on GEO: GSE100532.
Extended Data
Extended Data Fig. 1. Activated Hippo signaling in Human HF.
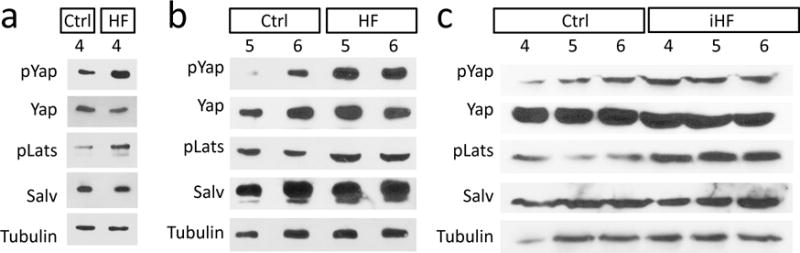
a-c, Western blots human heart samples. Ctrl: nonfailing nontransplantable, n=6 (a-c). HF: nonischemic idiopathic cardiomyopathy in end-stage heart failure, n=6 (a, b). iHF: ischemic heart in end-stage heart failure, n=6 (c). Quantification presented in Figure 1.
Extended Data Fig. 2. Mouse model of systolic heart failure.

a-d, Systolic diameter (a), diastolic diameter (b), systolic volume (c) and diastolic volume (d), n values indicated in Fig. 1a, ANOVA Tukey post-test. e, f, H&E edema liquid (pink transudate fluid) in lung tissue three weeks post-MI, sham (n=3) (e) and MI (n=5) (f), scale = 50 μm. g, h, Prussian blue hemosiderin (blue) in lung tissue three weeks post-MI, sham (n=3) (g) and MI (n=5) (h), scale = 50μm. i, BNP (Natriuretic Peptide B) in blood serum three weeks post-MI, Mann Whitney (i). j, Weight gain three weeks post-MI (j), t-test. k, l, Longitudinal echocardiography beginning 3 weeks post-MI, data are a subset of Fig. 2c. Ctrl Sham and Ctrl MI samples were split by Cre genotype or by injection type, indicated in parenthesis (k). No significant effect of Cre or tamoxifen (Tam) was observed, ANOVA Tukey post-test (k). Data: mean ± s.e.m, p-values >0.05 nonsignificant (n.s.), *p<0.05, **p<0.01, ***p<0.001.
Extended Data Fig. 3. Histologic analysis at 3 weeks post-MI.
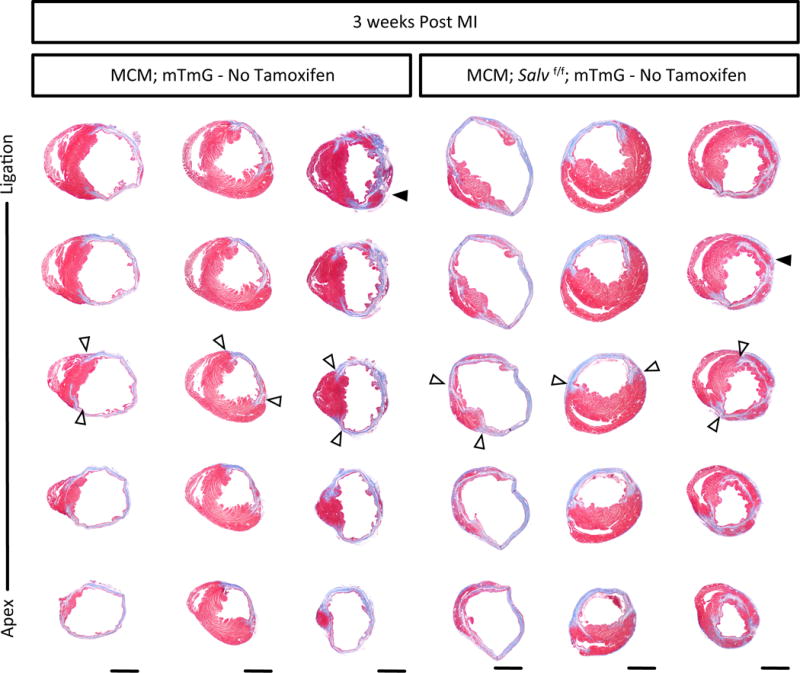
Masson’s Trichrome of serial sagittal sections 3 weeks post-MI, no tamoxifen was delivered, genotype indicated, n=3/group, scar boundaries (open arrows), cardiomyocytes in the ischemic region (solid arrows), scale = 2 mm.
Extended Data Fig. 4. Histologic analysis at 4 weeks post-MI.

Masson’s Trichrome of serial sagittal sections 4 weeks post-MI, 1 week post-tamoxifen, Ctrl (αMHC-mcm; mTmG) and SalvCKO (αMHC-mcm; mTmG; Salvfl/fl), n=3/group, scar boundaries (open arrows), cardiomyocytes in the ischemic region (solid arrows), scale = 2 mm.
Extended Data Fig. 5. Histologic analysis at 6 weeks post-MI.

Masson’s Trichrome of serial sagittal sections 6 weeks post-MI, 3 weeks post-tamoxifen, Ctrl (αMHC-mcm; mTmG) and SalvCKO (αMHC-mcm; mTmG; Salvfl/fl), n=3/group, scar boundaries (open arrows), cardiomyocytes in the ischemic region (solid arrows), scale = 2 mm.
Extended Data Fig. 6. Vessel growth in the border zone of Hippo deficient mouse hearts.

a, Quantitative real-time PCR (qPCR) of known markers of heart Failure at 6 weeks post-MI, Myosin Heavy chain 6 (Myh6), Myosin heavy Chain 7 (Myh7), Natriuretic Peptide A (Nppa), Natriuretic Peptide B (Nppb), n=3/group, ANOVA Bonferroni post-test. b, Masson’s Trichrome (scale=100μm) and IF staining for isolectin B4 (scale = 25μm) and CD31 (scale = 25μm), Ctrl (n=3) SalvCKO (n=5). c-e, Quantification 9 weeks post-MI in the BZ for capillary density (c), isolectin+ vessels (d), and CD31+ cells (e), Ctrl (n=3) SalvCKO (n=5), Mann-Whitney. f, qPCR of angiogenic growth factors, cardiomyocyte-specific TRAP RNA, 6 weeks post-MI, Angiopoietin 1 (Angpt1) and 2 (Angpt2), fibroblast growth factor 14 (Fgf14) & 18 (Fgf18), Vascular endothelial growth factor b (Vegfb) & c (Vegfc), n=3/group, ANOVA Bonferroni post-test. Data: mean ± s.e.m, p-values >0.05 nonsignificant (n.s.), *p<0.05, **p<0.01, ***p<0.001.
Extended Data fig. 7. TRAP RNA sequencing reproducibility.

a, Reproducibility correlation matrices of the RNAseq read count, linear regression, n=3/group. b, c, Plot of the per-gene standard deviation (sd) across samples, against the rank (mean) and read count, variance stabilizing transformation, Total RNAseq (b), Trap RNAseq (c). d, Log2FoldChange values between Trap and Total RNA-Seq for Ctrl-MI and SalvCKO-MI were highly correlated.
Extended Data Fig. 8. A reparative molecular response to heart failure in Hippo-deficient hearts.

a, Gene lists of the top 10 genes with the highest fold change in each GO category for Total RNA: SalvCKO MI vs Ctrl MI. b, Boxplot of the normalized read count for Tnnt2, Cdh5, and Malat1. c, Volcano plot TRAP-Seq: Ctrl MI vs Ctrl Sham. d, e, Gene ontology (GO) upregulated (d) and downregulated (e) genes. f, g, Gene lists of the top 10 genes with the highest fold change in each GO category for Trap RNA: Ctrl MI vs Ctrl Sham (f), Trap RNA: SalvCKO MI vs Ctrl MI (g).
Extended Data Fig. 9. Requirement of Park 2 in the regenerating mouse heart.

a-c, Human heart western blots, quantification presented in Figure 4, Tubulin blot is repeated from Extended Data Fig. 1. d, Scar size 21 days post-MI in P1 Park2 wild-type (+/+) and null (−/−) mice, Mann-Whitney. e, f, Echocardiography: ejection fraction (EF) (e), and fractional shortening (FS) (f), ANOVA Bonferroni post-test. g, Cardiomyocyte cell size measured by cross-sectional area, mean (red dashed line), t-test. h, Masson’s trichrome, 21 days post-MI in P1 Park2 wild type (+/+) (n=8) and null (−/−) (n=4) mice, scale = 2mm. i, Summary of results indicating Park2 is necessary for cardiac regeneration. Data: mean ± s.e.m, p-values >0.05 nonsignificant (n.s.), *p<0.05, **p<0.01, ***p<0.001.
Extended Data Fig. 10. Requirement of Park 2 in the P8 Hippo-deficient regenerating mouse heart.
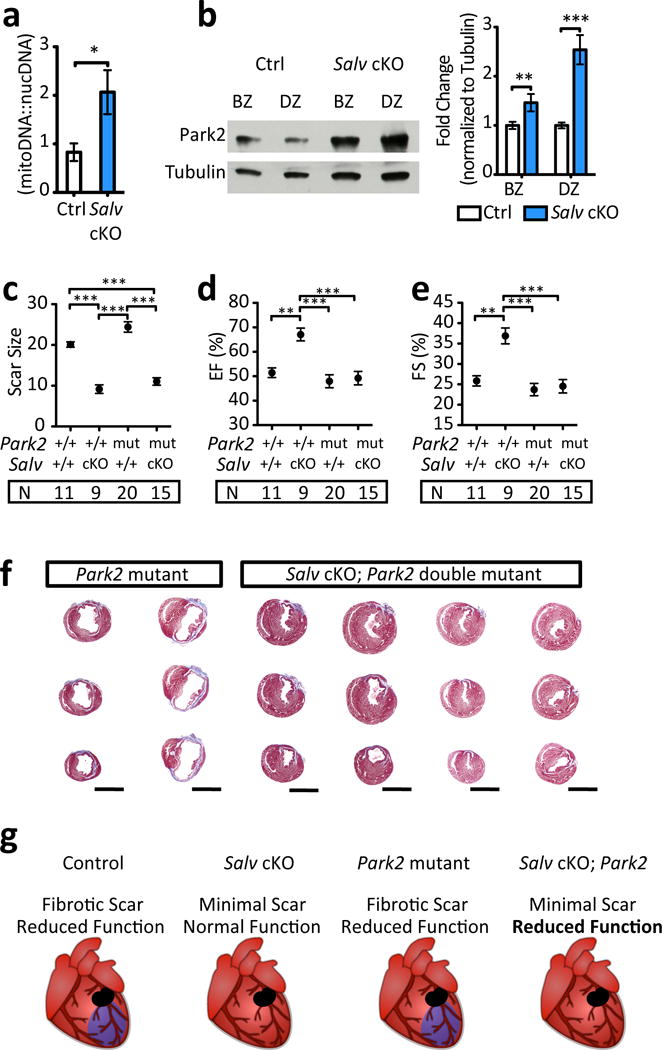
a, Mitochondrial DNA content 4 days post-MI in P8 Ctrl and SalvCKO (n=3/group). b, Park2 protein levels in border zone (BZ) and distal zone (DZ) myocardium at 4 days post-MI in P8 Ctrl and SalvCKO (n=3/group). c, Scar size 21 days post-MI in P8 Park2 wild type (+/+) and mutant (mut; −/− or +/−) mice, in combination with Salv cKO, ANOVA Bonferroni post-test. d, e, Echocardiograpy: ejection fraction (EF) (d), and fractional shortening (FS) (e), ANOVA Bonferroni post-test. f, Masson’s trichrome, 21 days post-MI in P8 Park2 mutant (n=20) and Salv cKO; Park2 double mutant mice (n=15), scale = 2mm. g, Summary of results indicating Park2 is necessary for regeneration. Data: mean ± s.e.m, p-values >0.05 nonsignificant (n.s.), *p<0.05, **p<0.01, ***p<0.001.
Supplementary Material
Acknowledgments
Supported by grants from the National Institutes of Health (DE023177, HL127717, HL130804, HL118761 [J.F.M.]; and 5T32HL007676-23 [J.P.L.]), Vivian L. Smith Foundation (J.F.M.), State of Texas funding (J.F.M. & J.T.W), LeDucq Foundation Transatlantic Networks of Excellence in Cardiovascular Research (14CVD01) “Defining the genomic topology of atrial fibrillation.” (J.F.M.). Supported by IDDRC grant number 1U54 HD083092 from the Eunice Kennedy Shriver National Institute of Child Health & Human Development and the Mouse Phenotyping Core at Baylor College of Medicine (U54 HG006348). T.H. was supported by American Heart Association Scientist Development Grant (16SDG26460001). Also supported in part by Neuroconnectivity core and Optical Imaging and Vital Microscopy core at Baylor College of Medicine. Nicole Stancel, PhD. ELS provided editorial assistance.
Footnotes
Author Contributions:
J.P.L, J.T.W., and J.F.M conceived and designed experiments and interpreted data. J.P.L, T.H, M.Z, M.C.H, Y.M. and M.R. performed experiments. J.P.L and J.F.M. analyzed data and compiled figures. A.S. provided human tissue samples J.P.L., J.T.W., and J.F.M wrote and edited the manuscript.
The authors have no competing financial interest to declare.
References
- 1.Loehr LR, Rosamond WD, Chang PP, Folsom AR, Chambless LE. Heart failure incidence and survival (from the Atherosclerosis Risk in Communities study) Am J Cardiol. 2008;101:1016–1022. doi: 10.1016/j.amjcard.2007.11.061. [DOI] [PubMed] [Google Scholar]
- 2.Halder G, Johnson RL. Hippo signaling: growth control and beyond. Development (Cambridge, England) 2011;138:9–22. doi: 10.1242/dev.045500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJ. Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet. 2006;367(06):1747–1757. 68770–9. doi: 10.1016/s0140-6736. [DOI] [PubMed] [Google Scholar]
- 4.Lenneman AJ, Birks EJ. Treatment strategies for myocardial recovery in heart failure. Curr Treat Options Cardiovasc Med. 2014;16:287. doi: 10.1007/s11936-013-0287-9. [DOI] [PubMed] [Google Scholar]
- 5.Birks EJ. Molecular changes after left ventricular assist device support for heart failure. Circ Res. 2013;113:777–791. doi: 10.1161/CIRCRESAHA.113.301413. [DOI] [PubMed] [Google Scholar]
- 6.Braunwald E. Heart failure. JACC Heart Fail. 2013;1:1–20. doi: 10.1016/j.jchf.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Halder G, Dupont S, Piccolo S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nature reviews Molecular cell biology. 2012;13:591–600. doi: 10.1038/nrm3416. [DOI] [PubMed] [Google Scholar]
- 8.Heallen T, et al. Hippo signaling impedes adult heart regeneration. Development (Cambridge, England) 2013;140:4683–4690. doi: 10.1242/dev.102798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morikawa Y, et al. Actin cytoskeletal remodeling with protrusion formation is essential for heart regeneration in Hippo-deficient mice. Science signaling. 2015;8 doi: 10.1126/scisignal.2005781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tao G, et al. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature. 2016 doi: 10.1038/nature17959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsuda T, et al. NF2 Activates Hippo Signaling and Promotes Ischemia/Reperfusion Injury in the Heart. Circ Res. 2016;119:596–606. doi: 10.1161/CIRCRESAHA.116.308586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Del Re DP, et al. Mst1 promotes cardiac myocyte apoptosis through phosphorylation and inhibition of Bcl-xL. Mol Cell. 2014;54:639–650. doi: 10.1016/j.molcel.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao XM, Dart AM, Dewar E, Jennings G, Du XJ. Serial echocardiographic assessment of left ventricular dimensions and function after myocardial infarction in mice. Cardiovascular research. 2000;45:330–338. doi: 10.1016/s0008-6363(99)00274-6. [DOI] [PubMed] [Google Scholar]
- 14.Doupe DP, et al. A single progenitor population switches behavior to maintain and repair esophageal epithelium. Science. 2012;337:1091–1093. doi: 10.1126/science.1218835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leask A. Getting to the heart of the matter: new insights into cardiac fibrosis. Circ Res. 2015;116:1269–1276. doi: 10.1161/circresaha.116.305381. [DOI] [PubMed] [Google Scholar]
- 16.Ponten A, Folestad EB, Pietras K, Eriksson U. Platelet-derived growth factor D induces cardiac fibrosis and proliferation of vascular smooth muscle cells in heart-specific transgenic mice. Circ Res. 2005;97:1036–1045. doi: 10.1161/01.RES.0000190590.31545.d4. [DOI] [PubMed] [Google Scholar]
- 17.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 18.Meredith AJ, et al. Circulating biomarker responses to medical management vs. mechanical circulatory support in severe inotrope-dependent acute heart failure. ESC heart failure. 2016;3:86–96. doi: 10.1002/ehf2.12076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kostin S, Hein S, Arnon E, Scholz D, Schaper J. The cytoskeleton and related proteins in the human failing heart. Heart failure reviews. 2000;5:271–280. doi: 10.1023/a:1009813621103. [DOI] [PubMed] [Google Scholar]
- 20.Kim SY, Morales CR, Gillette TG, Hill JA. Epigenetic regulation in heart failure. Current opinion in cardiology. 2016;31:255–265. doi: 10.1097/hco.0000000000000276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jessup M, Brozena S. Heart failure. The New England journal of medicine. 2003;348:2007–2018. doi: 10.1056/NEJMra021498. [DOI] [PubMed] [Google Scholar]
- 22.Marin-Garcia J, Akhmedov AT. Mitochondrial dynamics and cell death in heart failure. Heart failure reviews. 2016;21:123–136. doi: 10.1007/s10741-016-9530-2. [DOI] [PubMed] [Google Scholar]
- 23.Dorn GW., 2nd Parkin-dependent mitophagy in the heart. Journal of molecular and cellular cardiology. 2016;95:42–49. doi: 10.1016/j.yjmcc.2015.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kubli DA, et al. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem. 2013;288:915–926. doi: 10.1074/jbc.M112.411363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xin M, et al. Hippo pathway effector Yap promotes cardiac regeneration. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:13839–13844. doi: 10.1073/pnas.1313192110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Itoh N, Ohta H, Nakayama Y, Konishi M. Roles of FGF Signals in Heart Development, Health, and Disease. Front Cell Dev Biol. 2016;4:110. doi: 10.3389/fcell.2016.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korf-Klingebiel M, et al. Conditional transgenic expression of fibroblast growth factor 9 in the adult mouse heart reduces heart failure mortality after myocardial infarction. Circulation. 2011;123:504–514. doi: 10.1161/CIRCULATIONAHA.110.989665. [DOI] [PubMed] [Google Scholar]
- 28.Singla DK, Singla RD, Abdelli LS, Glass C. Fibroblast growth factor-9 enhances M2 macrophage differentiation and attenuates adverse cardiac remodeling in the infarcted diabetic heart. PLoS One. 2015;10:e0120739. doi: 10.1371/journal.pone.0120739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gong G, et al. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science. 2015;350:aad2459. doi: 10.1126/science.aad2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patterson M, et al. Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat Genet. 2017 doi: 10.1038/ng.3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bersell K, et al. Moderate and high amounts of tamoxifen in alphaMHC-MerCreMer mice induce a DNA damage response, leading to heart failure and death. Disease models & mechanisms. 2013;6:1459–1469. doi: 10.1242/dmm.010447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koitabashi N, et al. Avoidance of transient cardiomyopathy in cardiomyocyte-targeted tamoxifen-induced MerCreMer gene deletion models. Circ Res. 2009;105:12–15. doi: 10.1161/circresaha.109.198416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pugach EK, Richmond PA, Azofeifa JG, Dowell RD, Leinwand LA. Prolonged Cre expression driven by the alpha-myosin heavy chain promoter can be cardiotoxic. Journal of molecular and cellular cardiology. 2015;86:54–61. doi: 10.1016/j.yjmcc.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sam F, et al. Progressive left ventricular remodeling and apoptosis late after myocardial infarction in mouse heart. American journal of physiology. Heart and circulatory physiology. 2000;279:8. doi: 10.1152/ajpheart.2000.279.1.H422. [DOI] [PubMed] [Google Scholar]
- 35.Wang J, et al. A simple and fast experimental model of myocardial infarction in the mouse. Texas Heart Institute journal/from the Texas Heart Institute of St Luke’s Episcopal Hospital, Texas Children’s Hospital. 2006;33:290–293. [PMC free article] [PubMed] [Google Scholar]
- 36.Nascimento DS, et al. MIQuant–semi-automation of infarct size assessment in models of cardiac ischemic injury. PLoS One. 2011;6:e25045. doi: 10.1371/journal.pone.0025045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bergmann O, et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell. 2015;161:1566–1575. doi: 10.1016/j.cell.2015.05.026. [DOI] [PubMed] [Google Scholar]
- 38.Sanz E, et al. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13939–13944. doi: 10.1073/pnas.0907143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giudice J, et al. Alternative splicing regulates vesicular trafficking genes in cardiomyocytes during postnatal heart development. Nat Commun. 2014;5:3603. doi: 10.1038/ncomms4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lukas ED, et al. A pipeline for the generation of shRNA transgenic mice. Nature Protocols. 2012;7:374–393. doi: 10.1038/nprot.2011.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morikawa Y, Heallen T, Leach J, Xiao Y, Martin JF. Dystrophin glycoprotein complex sequesters Yap to inhibit cardiomyocyte proliferation. Nature. 2017 doi: 10.1038/nature22979. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data in this manuscript is available through the accession number on GEO: GSE100532.