

Abstract
Autophagy is an essential cellular process for bulk degradation of cytoplasmic components through the lysosome. Underlying this process is an intricate interaction between protein factors and the cell endomembrane system, leading to a gradual maturation of the autophagic membrane. This structure sequesters a portion of the cytoplasm by the formation of a double-membrane compartment called the autophagosome. The autophagosome then delivers the cargo to the lysosome to complete degradation. The molecular mechanism accounting for the generation of the autophagic membrane is a longstanding question. Here, we describe a cell-free approach we have established to understand the mechanism of early autophagic membrane generation. This system has provided insight into the membrane source of the autophagosome, the early protein-membrane associations, and the membrane remodeling that generates the autophagosomal precursors. The cell-free assay, in combination with other established approaches, e.g. cell imaging, will facilitate a deeper understanding of the mechanism of autophagy.
Keywords: Autophagy, Autophagosome, Cell-free reconstitution, LC3 lipidation
INTRODUCTION
This unit describes an in vitro approach based on cell-free reconstitution to dissect the mechanism of autophagy (see Background Information for details). The cell-free reaction here refers to the creation of an in vitro reaction to mimic a certain cellular process by combining cytosol and cellular membranes from broken cells with nucleotides in a test tube. Because they are directly derived from a living cell, the active components of the cytosol and cellular membranes are native, in contrast to the use of purified proteins and synthetic liposomes. When properly designed, the cell-free assay is able to recapitulate the key aspects of different cellular events. Indeed, cell-free approaches have been successfully employed to make groundbreaking discoveries in multiple key cellular processes including protein synthesis and translocation, vesicular transport, transcription, and cell division. Some cell-free systems are so broadly employed that they have been commercialized. A good example is the rabbit reticulocyte lysate system for in vitro translation (Jagus et al., 2003).
In the cell-free reconstitution, the cytosol and membrane are derived from broken cells. We can employ cytosol or membrane fractionation approaches to isolate the active components and subsequently identify the factors by mass spectrometry. A biochemical approach is usually a good complement to genetic approaches, e.g. the combination of a cell assay with genetic screening, to fully dissect the mechanism of a cellular process. The merit of an in vitro biochemical approach is that it allows us to directly pinpoint the role of a factor. The drawback is that not all cellular processes can be easily reconstituted in vitro. A substantial optimization is usually necessary and different controls are required to validate the physiological relevance of the established cell-free reaction.
Below we describe a set of biochemical methods to answer several key questions in autophagosome biogenesis (Burman et al., 2010; Ge et al., 2014a; Lamb et al., 2013; Mizushima et al., 2008; Rubinsztein et al., 2012) (see Background Information for details): 1) What is the membrane origin of the autophagosome? 2) How are different autophagic factors activated during each step of autophagy? 3) How is the autophagosomal membrane precursor generated from the membrane source? The biochemical methods include: 1) a cell-free LC3 lipidation assay to determine an early step of autophagosome biogenesis (Basic Protocol 1), 2) a three-step membrane fractionation method to identify the cellular membrane that triggers autophagosome biogenesis in the cell-free LC3 lipidation assay (Support Protocol 1), 3) a cell-free membrane recruitment assay to analyze the activation of the indicated autophagic factors (Basic Protocol 2), and 4) a cell-free small autophagosomal precursor formation assay to reconstitute the biogenesis of early autophagosomal precursors (Basic Protocol 3).
BASIC PROTOCOL 1: Cell-free LC3 lipidation
We have established a cell-free LC3 lipidation assay that recapitulates several major regulatory landmarks of autophagosome biogenesis (Ge et al., 2013). The key components of the cell-free assay include the cytosol, the cellular membrane, the nucleotides, and the LC3 probe. The cytosol provides the key regulatory components, such as different autophagy-related gene (ATG) proteins (see Background Information for details), for autophagosome biogenesis. The cellular membrane donates membranes to the autophagosome. The process is controlled by certain molecular determinants on the cellular membrane and the active components of the cytosol. The nucleotides provide the ATP for energy and the GTP for activating certain GTP-binding proteins which are usually required for membrane remodeling events in the cell. LC3 lipidation is the readout of autophagosome biogenesis in the cell-free reaction. Of the four steps mentioned, the preparation of nucleotide is a standard protocol (Bednarek et al., 1995) and is not described in detail. Below we describe the protocols for cytosol and membrane preparation, LC3 protein purification, and the cell-free reaction (Fig. 1).
Fig. 1. Cell-free LC3 lipidation assay (modified from (Brier et al., 2016; Ge et al., 2013)).
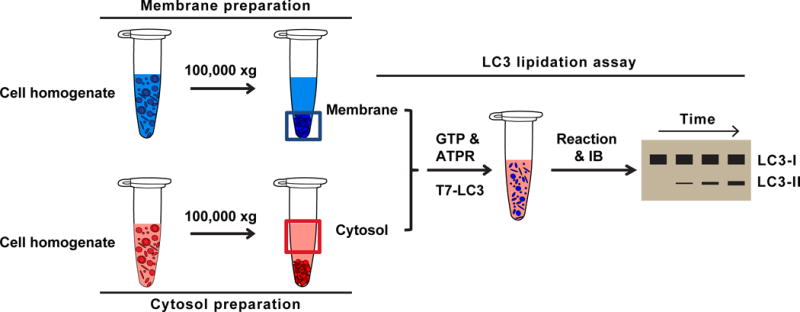
Membrane preparation: Atg5 KO MEF cells are homogenized and ultracentrifuged at 100,000×g. The membrane pellet is used as the membrane fraction.
Cytosol preparation: WT MEF or HEK293T cells are homogenized and ultracentrifuged at 100,000×g. The supernatant is used as the cytosol fraction.
LC3 lipidation reaction: cytosol, membrane, nucleotides (GTP&ATPR), and LC3-I are incubated for different times. The samples are then analyzed by immunoblotting. The lipidated LC3 appears as a faster migrating band with increased intensity according to time. The LC3-I band shows little change even when more LC3 lipidation occurs in the longer time durations because the overall lipidation efficiency is usually less than 20%.
Materials
B88 buffer: 20 mM HEPES-KOH, pH 7.2, 250 mM sorbitol, 150 mM potassium acetate, and 5 mM magnesium acetate
B88 lysis buffer: B88 buffer with protease inhibitors (Roche), phosphatase inhibitors (Roche), and 0.3 mM DTT
PBS: 137 mM sodium chloride, 2.7 mM potassium chloride, 10 mM disodium phosphate, and 1.8 mM monopotassium phosphate; adjust pH to 7.2
EBSS – Earle’s Balanced Salt Solution (Thermo)
DMEM – Dulbecco’s Modified Eagle Medium and FBS (Fetal Bovine Serum)
10×ATP regeneration system: 400 mM creatine phosphate, 2 mg/ml creatine phosphokinase, and 10 mM ATP in B88 (provides ATP during cell-free reaction)
GTP 10 mM stock (activates GTP-binding proteins)
0.05% Trypsin-EDTA 1X (Gibco)
Phospholipids C Reagent (Wako, for measuring phosphatidylcholine (PC) level)
Trypan blue
Centrifuge tubes (1.5 ml, 15 ml, and 50 ml)
22G needles and syringes (1ml, 3ml, and 5ml)
Glass slides and coverslips
Student microscope (to check trypan blue stain)
Refrigerated benchtop swinging bucket centrifuge for 15ml and 50ml conical tubes (e.g. Sorvall ST 16R, Thermo)
Benchtop ultracentrifuge (a centrifuge capable of >150,000×g, e.g. Optima Max-XP, Beckman)
Ultracentrifuge (to remove cell debris in protein purification, e.g. Optima XE-90, Beckman)
Centrifuge for large volumes (to collect E. coli, e.g. Sorvall, RC6+)
Sonicator (to break the E. coli, e.g. Branson Sonifier 450)
Spectrophotometer (to measure OD600 with cuvettes, e.g. Spectronic Genesys 5, Thermo)
Microplate spectrophotometer (to measure protein and phospholipid concentration with 96-well plates, e.g. xMark™ Microplate Absorbance Spectrophotometer, Bio-Rad)
Protocol steps
Prepare the total cellular membrane for reaction
-
1
Culture Atg5 knockout (KO) mouse embryonic fibroblast (MEF) cells (Mizushima et al., 2001) in DMEM plus 10% FBS for 2–3 days to confluence (1–3 10cm dishes).
Use cells with passages less than 20. Membranes from cells with passages higher than 20 are less active in triggering LC3 lipidation and are prone to fragmentation, making it difficult to isolate them in the membrane fractionation described in Support Protocol 1.
-
2
Remove the medium and wash the cells once with PBS. Trypsinize the cells with 1ml/dish 1X trypsin. Then add 5ml DMEM plus 10% FBS to quench the action of trypsin.
-
3
Transfer the cell suspension to a 15ml conical tube and centrifuge at 600×g for 5 min at 4 °C to collect the cells. Remove the supernatant and suspend the cell pellet with 5 ml ice-cold PBS. Repeat the centrifugation.
-
4
Discard the supernatant, suspend the cell pellet with B88 lysis buffer (use a volume about 3 times the cell pellet volume), and pass through a 22G needle with a syringe 10 times.
Avoid bubble generation when passing through the needle.
-
5
Place 3μl of the lysate on a glass slide and stain with trypan blue. Check the fraction of cells that are trypan blue positive. If 80%–90% of cells are positive, proceed to the next step. If less than 80% of cells are positive, homogenize the cells again, with a check of cell lysis every 5 strokes until 80%–90% of the cells are lysed.
We do not recommend a complete cell lysis because some active membrane structures will be damaged with excessive homogenization.
-
6
Transfer the lysate to an ultracentrifuge tube and centrifuge at 100,000×g for 30min.
-
7
Discard the supernatant and resuspend the total membrane pellet in 50–100 μl B88 lysis buffer.
The pellet in this step is hard and difficult to resuspend. Try incubating the pellet in B88 lysis buffer on ice for 30 min–1 h, after which the pellet will soften. We do not recommend vortexing. Suspend the pellet by pipetting.
-
8
Take 1 μl of membrane suspension to measure the PC concentration with a microplate spectrophotometer. Adjust the membrane concentration to 0.6 μg/μl of PC.
PC is the most abundant lipid in the membrane. Therefore, we use PC as a standard to normalize membrane amounts in the following experiments.
Prepare the cytosol for reaction
-
9
Culture human HEK293T (human embryonic kidney) cells in DMEM plus 10% FBS for 2–3 days to confluence (usually more than 2 10cm dishes). Incubate the cells with DMEM plus 10% FBS (nutrient-rich) or EBSS (starve) for 1 h.
In addition to starvation, we could also treat the cells with drugs, such as Rapamycin and Torin-1, to activate the cytosolic components involved in autophagy. In addition to HEK293T cells, cytosol from COS7 and wild type MEF cells are also active in triggering LC3 lipidation.
-
10
Scrape the cells into the medium and transfer the cell suspension to a 15 ml conical tube. Centrifuge at 600×g for 5 min at 4 °C to collect the cells. Remove the supernatant and suspend the cell pellet with 5 ml ice-cold PBS. Repeat the centrifugation.
When suspending the cell pellets with PBS, we suggest tapping the pellets or gently pipetting. We do not recommend strong pipetting or vortexing, because this will break the cells and lead to low yield of cytosol.
-
11
Discard the supernatant, suspend the cell pellet with an equal volume of B88 lysis buffer, and pass through a 22G needle with a syringe 10 times.
-
12
Place 3 μl of the lysate on a glass slide and stain with trypan blue. Check the fraction of cells that are trypan blue positive. If 80%–90% of cells are positive, proceed to the next step. If less than 80% of cells are positive, homogenize the cells again, with a check of cell lysis every 5 strokes until 80%–90% of the cells are lysed.
We do not recommend a complete cell lysis because excessive homogenization reduces cytosol activity.
-
13
Transfer the lysate to an ultracentrifuge tube and centrifuge at 100,000×g for 30min.
-
14
Collect the supernatant and repeat step 13 if the supernatant is not clear.
Usually, if a large number of cells are harvested, a lipid layer will appear on the top after centrifugation. Try to avoid the lipid layer when collecting the supernatant. We usually collect the pure supernatant located between the cell-debris pellet and the lipid layer, sacrificing the cytosol that is near either the top or the bottom. If the lipid layer is too broad to avoid, try collecting the supernatant with some lipid contamination and repeat centrifuging the cytosol several times until you can obtain a clear supernatant of cytosol. The lipid layer usually becomes thinner after each round of centrifugation.
-
15
The clear supernatant is the cytosol ready for use in the reaction. Measure the protein concentration with the Bradford assay. Aliquot and store at −80 °C.
Avoid repeated freeze/thaws.
Purify the T7-LC3
Below is a standard His-tag protein purification protocol. T7-LC3 is easy to express and purify.
The typical yield is around 20 mg/L E.coli.
-
16
Culture 20 ml E. coli BL21 cells with pET28a-LC3 (aa1-120, human) at 37°C on a shaker overnight.
-
17
Expand the total culture to 1 L (1:50 expansion) and shake at 37°C for 1–2 hs until the OD600 reaches 0.5–0.8.
-
18
Induce protein expression with 100 μM IPTG at 23°C for 5 hs and collect the E. coli by centrifuging at 10,000×g for 10 min.
-
19
Suspend the E.coli pellet with 20 ml 2×PBS plus 15 mM imidazole and protease inhibitors. Add lysozyme at 0.5 mg/ml and incubate on ice for 30 min.
-
20
Add Triton X-100 to a concentration of 0.5%. Sonicate the solution with five to seven 15s bursts until the solution is not viscous.
-
21
Centrifuge the lysate at 100,000×g for 30 min, transfer the supernatant to a clean 50 ml conical tube, and incubate with 1 ml Ni Sepharose (packed beads) at 4°C for 2 hs on a rotator.
-
22
Load the mixture in an empty protein purification column and drain the flow through.
-
23
Wash the beads with 70 ml cold 2×PBS plus 25 mM imidazole and 0.2% Tween-20 followed by 50 ml cold 2×PBS plus 25 mM imidazole.
-
24
Elute the bound His-T7-LC3 with 10 ml 2×PBS plus 250 mM imidazole, concentrate the eluate to 1 ml with a centrifugal filter (10kD cut-off) and buffer exchange to PBS by running through a NAP-10 column.
-
25
Remove the His tag of His-T7-LC3 (usually ~4 mg/ml) with 1 U/ml of thrombin at room temperature for 1 h, followed by adding 1 mg/ml AEBSF to deactivate thrombin. Aliquot and store at −80 °C.
The cleaved His tag remaining in the T7-LC3 does not affect lipidation. Step 25 could be skipped.
-
26
Check protein quality and estimate concentration by running an SDS-PAGE and staining with Coomassie Blue.
Cell-free lipidation reaction
-
27
For each reaction, mix the following components on ice: cytosol (2 mg/ml final concentration), 3 μl 10×ATP regeneration system, GTP (0.15 mM final concentration), 0.2 μg T7-LC3, membrane (0.2 mg/ml PC content final concentration), and B88 to make a total of 30 μl in a low retention 1.5 ml Eppendorf tube.
-
28
Gently tap the tube to mix and incubate at 30°C for 30–90 min.
-
29
Quench the reaction by adding SDS-PAGE loading buffer to make a total of 80 μl. Load 12 μl of the sample for immunoblotting with a T7 antibody.
You should see a big band (unlipidated T7-LC3) migrating around 17kd and a faster migrating thinner band (lipidated T7-LC3) around 3–5 mm lower than the big band on the membrane. We suggest running a 12.5%, 15% or 8–16% gradient to better separate the lipidated LC3 from the unlipidated LC3.
Sometimes the unlipidated band signal is so strong that it covers the lipidated signal. In this case, we recommend adding a step of 20,000×g centrifugation after step 28. Then remove ¾ of the supernatant and add SDS-PAGE loading buffer to the pellet and remaining supernatant. The lipidated LC3 should be fully recovered in the pellet. By removing the supernatant, we only reduce the level of unlipidated LC3.
SUPPORT PROTOCOL 1: Three-step membrane fractionation and lipidation reaction
A longstanding quest in the autophagy field is to identify the membrane source of the autophagosome. The cell-free LC3 lipidation assay can be combined with membrane fractionation approaches to identify the membrane source of the autophagosome by determining the active membrane fraction that triggers LC3 lipidation.
In this section, we describe the protocol for a three-step fractionation method we have established (Ge et al., 2013), including a differential centrifugation, a sucrose gradient, and an Optiprep gradient performed sequentially (Fig. 2). The purpose is to identify a membrane fraction which triggers LC3 lipidation. In general, after each step of membrane fractionation, we perform LC3 lipidation with the obtained fractions. We select the most active fraction and further separate it with the subsequent procedures until we know the identity of the active membrane. In our case, we found that the major activity resides in the 25,000×g membrane after the differential centrifugation. We further separated the 25,000×g membrane in the sucrose gradient to obtain an L and a P fraction. The L fraction was found to be active and was further resolved in the Opti-prep gradient. In the final step, the lipidation activity co-migrates with the distribution of the ER-Golgi intermediate compartment markers ERGIC53 and SEC22B (Appenzeller-Herzog et al., 2006).
Fig. 2. Three-step membrane fractionation (modified from (Ge et al., 2013)).
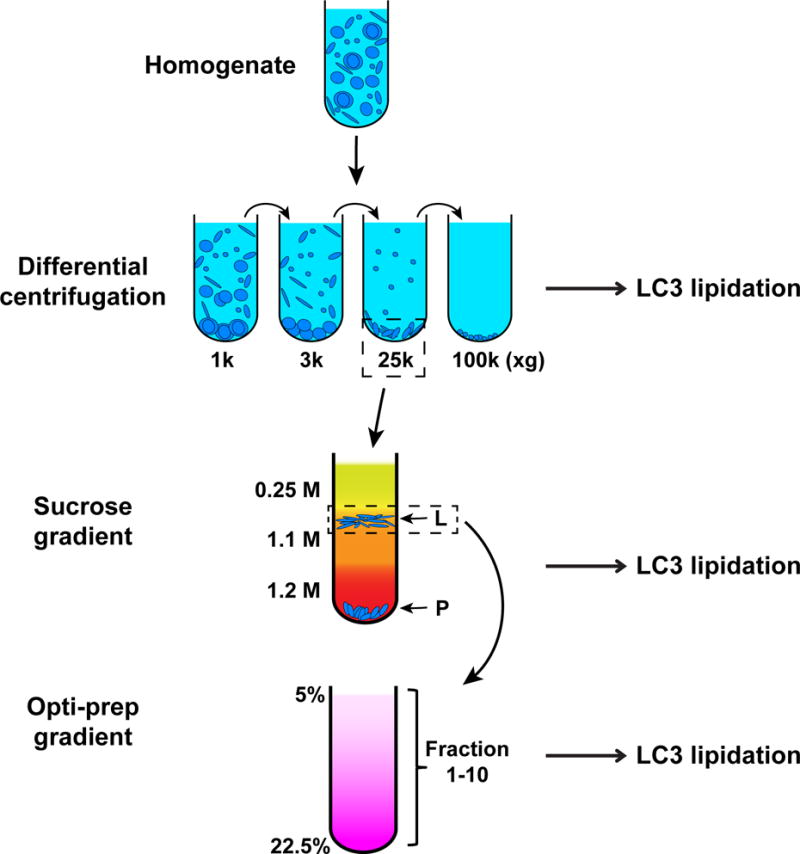
Atg5 KO MEF cells are homogenized and subjected to a differential centrifugation to separate the total membrane into 4 fractions. Membranes from these fractions are analyzed for their ability to trigger LC3 lipidation. The 25k fraction, which is most active, is selected and further separated through a sucrose gradient ultracentrifugation. An L and a P fraction are obtained and a similar LC3 lipidation assay is performed. The L fraction is selected and further resolved through a linear gradient Optiprep ultracentrifugation. Ten fractions are collected and LC3 lipidation is performed to determine the activity of each membrane fraction.
Materials
In addition to those described above:
HB1 buffer: 20 mM HEPES-KOH, pH 7.2, 400 mM sucrose and 1 mM EDTA
HB1 lysis buffer: HB1 buffer with protease inhibitors (Roche), phosphatase inhibitors (Roche), and 0.3 mM DTT
1.25 M, 1.1 M, and 0.25M sucrose buffers (Golgi isolation kit, Sigma)
50% Optiprep solution: 50% Optiprep (Sigma), 20 mM Tricine-KOH, pH 7.4, 42 mM sucrose and 1 mM EDTA
Optiprep dilution buffer: 20 mM Tricine-KOH, pH 7.4, 250 mM sucrose and 1 mM EDTA
Ultracentrifuge tubes (11×34 mm Ultra-clear and polycarbonate, 13×51 mm Ultra-clear and polycarbonate)
Cell lysis
-
1
Culture Atg5 KO MEF (ten 15 cm dishes) in DMEM plus 10% FBS for 2–3 days to confluence.
Use cells with passages less than 20.
-
2
Remove the medium and wash the cells once with PBS. Trypsinize the cells with 1 ml/dish 1× trypsin. Then add 5ml DMEM plus 10% FBS to quench the action of trypsin.
-
3
Transfer the cell suspension to 15 ml conical tubes and centrifuge at 600×g for 5 min at 4 °C to collect the cells. Remove the supernatant and suspend the cell pellet with 5 ml ice-cold PBS. Repeat the centrifugation.
-
4
Discard the supernatant, suspend the cell pellet with HB1 lysis buffer (use a volume about 3 times the cell pellet volume), and pass through a 22G needle with a syringe 10 times.
Again, avoid any bubble generation.
-
5
Pipet 3 μl of the lysate on a glass slide and stain with trypan blue. Check the fraction of cells that are trypan blue positive. If 80%–90% of cells are positive, proceed to the next step. If less than 80% of cells are positive, homogenize the cells again, with a check of cell lysis every 5 strokes until 80%–90% of the cells are lysed.
Avoid more than 90% lysis.
Differential centrifugation
-
6
Centrifuge the cell lysate at 1000×g in a swinging bucket rotor for 10 min. Transfer the supernatant to a new 15 ml centrifuge tube. Suspend the pellet with HB1 lysis buffer (use a volume about 3 times the pellet volume) and centrifuge for 10 min at 1000×g. Keep the pellet (1k membrane) and combine the supernatant from the two centrifugations.
The pellet after the 1000×g spin is difficult to see because of turbidity due to the remaining membranes in the supernatant. Try to look at the pellet against a monochromatic background. Dark blue is usually a good choice. Carefully transfer the supernatant. If the pellet is not visible, try increasing the speed to 1200–1500×g. To increase recovery, we suggest washing the pellet with lysis buffer and repeating the centrifugation.
-
7
Centrifuge the supernatant at 3000×g in a swinging bucket rotor for 10 min. Keep the pellet (3k membrane) and transfer the supernatant to a 13×51 mm polycarbonate ultracentrifuge tube.
-
8
Ultracentrifuge the supernatant at 25,000×g in a TLA100.3 rotor for 20 min. Keep the pellet (25k membrane) and transfer the supernatant to 13×51 mm polycarbonate ultracentrifuge tubes.
-
9
Ultracentrifuge the supernatant at 100,000×g in a TLA100.3 rotor for 30 min. Keep the pellet (100k membrane) and discard the supernatant.
We performed the cell-free lipidation assay with the four membrane fractions. The 25k membrane is the active membrane. Therefore, we further separated the 25k with the sucrose gradient indicated below.
Sucrose gradient
-
10
Suspend the 25k membrane with 0.75 ml 1.25 M sucrose buffer.
-
11
Pass the membrane suspension through a 22G needle 10 times to fully disperse the membrane pellet.
-
12
Transfer the suspension into an 11×34 mm clear ultracentrifuge tube. Layer on top of the membrane suspension with 0.5 ml 1.1 M sucrose buffer and 0.5 ml 0.25 M sucrose.
-
13
Ultracentrifuge at 120,000×g for 2 hs in a TLS-55 rotor.
-
14
Collect the membrane layer located at the boundary of 0.25/1.1 M sucrose (L fraction) as well as the pellet (P fraction).
Sometimes the L fraction layer is broad. In that case, collect all floating membranes.
-
15
Dilute the L fraction 3 times with HB1 buffer in a 13×51 mm polycarbonate ultracentrifuge tubes and ultracentrifuge at 100,000×g in a TLA100.3 rotor for 40 min.
We have performed the cell-free lipidation assay with both of these membrane fractions. The L fraction is the active membrane. Therefore, we further separated the L fraction with the following Optiprep gradient
Optiprep gradient ultracentrifugation
-
16
Make 22.5%, 19%, 16%, 12%, 8% and 5% Optiprep solutions by diluting the 50% Optiprep with Optiprep dilution buffer.
-
17
Suspend the L fraction membrane pellet with 1 ml 19% Optiprep gradient by passing through a 22G needle 10 times.
-
18
In a 13×51 mm clear ultracentrifuge tube, layer Optiprep gradients of 0.5 ml 22.5%, 1 ml 19% (with the membrane), 0.9 ml 16%, 0.9 ml 12%, 1 ml 8%, 0.5 ml 5% and 0.2 ml HB1 buffer.
Carefully load the gradient and minimize disturbance.
-
19
Ultracentrifuge at 150,000×g in a SW-55 rotor for 3 hs.
Multiple membrane layers should show up after this step.
-
20
Collect 10 fractions, 0.5 ml each, from the top to the bottom. Dilute with 2 ml B88 buffer in a 13×51 mm polycarbonate centrifuge tube and ultracentrifuge for 40 min at 100,000×g.
-
21
Keep the membrane pellets (#1–10).
Lipidation reaction
-
22
Suspend all membrane pellets with B88 buffer and centrifuge to collect the membrane with the same speed used before.
The purpose is to buffer exchange to B88 because all reactions are performed in B88 buffer.
-
23
Suspend the membrane pellets with minimum amount of B88 lysis buffer, measure PC concentration, and adjust to 0.6 μg/μl of PC.
-
24
Perform the LC3 lipidation reaction with the membranes using the protocol indicated above.
BASIC PROTOCOL 2: Membrane recruitment assay
Autophagosome biogenesis requires the assembly of cytosolic ATG proteins on special sites of the cellular membrane. These ATG proteins are recruited to the membrane in a hierarchical manner. Membrane targeting of these ATG proteins is key for their activation. In this section, we describe a cell-free membrane recruitment assay to determine the membrane targeting of certain ATG proteins to reflect the activation of different stages of autophagosome biogenesis (Ge et al., 2013).
The basic steps include a cell-free reaction that allows the targeting of cytosolic ATG proteins to the membrane, and a membrane flotation assay that separates the membrane from the reaction to determine the membrane-associated ATG proteins after the reaction (Fig. 3).
Fig. 3. Membrane recruitment assay.
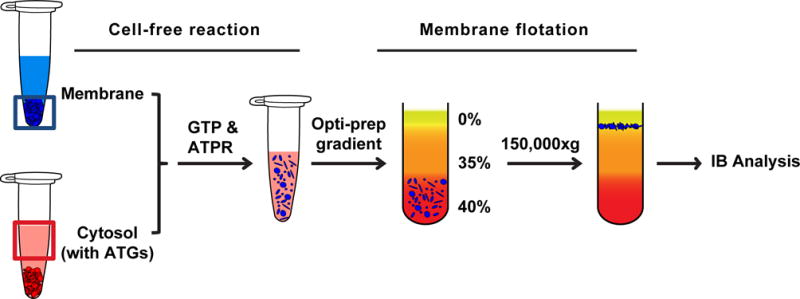
The post-nuclear supernatant of Atg5 KO MEF membrane is collected. Cytosol from HEK293T cells with exogenously expressed ATG proteins is prepared. The membrane, cytosol, and nucleotides (GTP&ATPR) are incubated together, during which the ATG proteins in the cytosol are recruited to the membrane. After incubation, the reaction mixture is loaded in the bottom of an Optiprep gradient and subjected to ultracentrifugation. The membrane fraction together with the bound ATG proteins will float to the top after centrifugation. The amount of the ATG proteins in the top fraction corresponds to their membrane recruitment.
Materials
In addition to what are described above:
X-tremeGene HP cell transfection reagent (Roche)
50% Optiprep in B88: 50% Optiprep, 20 mM HEPES-KOH, pH 7.2, 250 mM sorbitol, 150 mM potassium acetate, and 5 mM magnesium acetate
Cytosol preparation
-
1
Transfect HEK293T cells with plasmids encoding the ATG proteins that are going to be tested.
-
2
Prepare the cytosol according to basic protocol 1.
Membrane preparation
-
3
Collect and lyse Atg5 KO MEF according to basic protocol 1.
-
4
Centrifuge the lysate at 1000×g and collect supernatant.
-
5
Ultracentrifuge the supernatant (post-nuclear supernatant) at 100,000×g in a TLA100.3 rotor for 40 min and collect the pellet.
-
6
Suspend the pellet in B88 lysis buffer and dilute to 0.6 μg/μl of PC.
Cell-free reaction
-
7
In a low retention 1.5 ml Eppendorf tube, make a similar cell-free reaction containing the cytosol (2 μg/μl), 1× ATP regeneration system, GTP (0.15 mM), and membrane (0.2 μg/μl PC). Add B88 buffer to bring the total volume to 50 μl.
-
8
Gently tap to mix and incubate the mixture at 30°C for 1 h.
Membrane flotation
-
9
After the reaction, add 200 μl 50% Optiprep in B88 to adjust the final concentration of Optiprep to 40%.
-
10
Transfer to an 11×34 mm polycarbonate ultracentrifuge tube and layer with 200 μl 35% Optiprep in B88 followed by 50 μl B88.
-
11
Ultracentrifuge at 150,000×g in a TLS-55 rotor for 1.5 hs.
There will be a membrane band between the B88 and 35% Optiprep layers.
-
12
Collect 7 fractions from the top to the bottom (70 μl) each.
-
13
Determine membrane recruitment of the relevant ATG proteins by immunoblot.
The 1st fraction contains ~70–80% of the membrane and the 2nd contains ~20%. The soluble components are in the last 4 fractions.
BASIC PROTOCOL 3: Small autophagosomal precursor formation assay
Autophagic membranes are derived from intracellular organelles. The membrane conversion process involves multiple membrane remodeling processes. We found that small membrane precursors of the autophagosome are generated during starvation. These precursors are active for LC3 lipidation. Therefore, we established the following cell-free assay to determine the generation of small autophagosomal precursors under different conditions (Ge et al., 2014b). The protocol includes two major steps: the cell-free small membrane formation assay and the cell-free lipidation assay. The former one is adapted from the established COPII vesicle formation assay (Kim et al., 2005; Schindler et al., 2009) to generate different kinds of small membranes (including the small autophagosomal precursors) in a test tube. A differential centrifugation is used to isolate the small vesicles generated. The latter one is to determine the generation of the autophagosomal precursors in the small membrane fraction based on the fact that these autophagosomal precursors are active for LC3 lipidation (Fig. 4).
Fig. 4. Cell-free autophagosomal precursor formation assay (modified from (Ge et al., 2014b)).
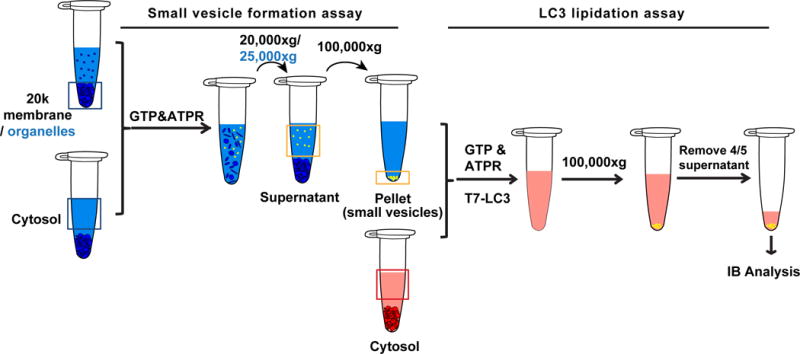
Small vesicle formation assay: The donor membrane consists of the membrane fraction from the 20,000×g-centrifugation of the Atg5 KO MEF membrane after cell lysis, or organelle fractions obtained from the three-step membrane fractionation. The cytosol is from Atg5 KO MEF cells. The donor membrane, cytosol, and nucleotides (GTP&ATPR) are incubated together, during which the small vesicles are formed in the cell-free reaction. The mixture is centrifuged at 20,000×g (25,000×g for organelles) to remove the donor membrane. The supernatant is ultracentrifuged at 100,000×g to enrich the small vesicles which may contain the small autophagosomal precursors.
LC3 lipidation assay: To determine the amount of autophagosomal precursors in the small vesicle fraction, the LC3 lipidation assay is performed with these vesicles. After reaction, the mixture is ultracentrifuged at 100,000×g and 4/5 of the supernatant is carefully removed. Immunoblot is performed with the remaining supernatant and pellet. The level of LC3 lipidation is a reflection of the amount of autophagosomal precursors generated in the above reaction.
Materials
In addition to those described above
Refrigerated bench top microfuge (for centrifuging 1.5 ml Eppendorf tubes to collect total donor membrane, e.g. Eppendorf Centrifuge 5430R)
Total donor membrane preparation
We usually use total donor membrane in most of the experiments
-
1
Collect and lyse Atg5 KO MEF in B88 lysis buffer according to Basic Protocol 1.
-
2
Centrifuge the lysate at 20,000×g for 20 min in a refrigerated microfuge.
In this case, we do not need the 100,000×g small vesicle fraction.
-
3
Discard the supernatant and suspend the pellet with B88.
-
4
Repeat the centrifugation and suspend the donor membrane pellet with B88 lysis buffer.
-
5
Adjust concentration to 1 μg/μl PC.
Donor membrane preparation from membrane fractionation
The purpose is to find the membrane fraction active for generating autophagosomal precursors.
-
6
Collect and lyse Atg5 KO MEF in HB1 lysis buffer according to Support Protocol 1.
-
7
3k fraction: Collect 3k membrane fraction according to Support Protocol 1.
-
8
P fraction: Collect P membrane fraction according to Support Protocol 1, except that after suspending the pellet with B88 buffer, centrifuge the membrane at 25,000×g for 40 min. Keep the pellet and suspend it in B88 lysis buffer.
-
9
Four fractions from Optiprep gradient: Perform the Optiprep gradient and collect ten fractions according to Support Protocol 1. Combine Fractions to four fractions F1 (1&2), F2 (3&4), F3 (5&6), and F4 (7–10). Dilute the combined fractions with B88 buffer (at least 3 times the volume of the Optiprep fraction) and collect the membrane at 25,000×g for 40 min. Suspend in B88 lysis buffer.
-
10
Adjust all membrane fractions to the concentration of 1 μg/μl PC.
Cytosol preparation
In Atg5 KO MEF, we observed accumulation of small autophagosomal precursors. So we used cytosol from Atg5 KO MEF to reconstitute the formation of autophagosomal precursor in the cell-free assay.
-
11
Prepare Atg5 KO MEF cytosol and HEK293T cytosol according to Basic Protocol 1.
Be careful not to contaminate the cytosol with any membrane.
Cell-free small vesicle formation assay
-
12
In a low retention 1.5 ml Eppendorf tube, make a similar cell-free reaction containing the Atg5 KO MEF cytosol (2 μg/μl), 1×ATP regeneration system, GTP (0.15 mM), and membrane (0.2 μg/μl PC). Add B88 buffer to bring the total volume to 50 μl.
-
13
Gently tap to mix and incubate the mixture at 30°C for 1 hour.
-
14
Centrifuge the reaction mixture at 20,000×g (total donor membrane) or 25,000×g (membrane fractions) for 20 min.
The purpose of this step is to remove the donor membrane.
-
15
Transfer 35 μl supernatant to a new 1.5 ml Eppendorf tube and ultracentrifuge at 100,000×g for 30 min in a TLA100.3 rotor with adapters or a TLA-55 rotor.
-
16
Discard the supernatant and keep the pellet.
The pellet consists of the small membranes generated in the test tube. Sometimes the pellet is hard to see, but it is there.
Analyzing the generation of small autophagosomal precursors via LC3 lipidation
-
17
Make a master mix of HEK293T cytosol (2 μg/μl), 1× ATP regeneration system, GTP (0.15 mM), and T7-LC3 (10 ng/ml) in B88.
-
18
Suspend the pellet from step 16 with 15 μl master mix by pipetting.
-
19
Incubate the mixture at 30°C for 1 h.
-
20
Ultracentrifuge the mixture at 100,000×g for 30 min. Remove 12 μl supernatant carefully.
Because there is not much membrane in the small membrane fraction, the lipidated LC3 level is low. By removing the supernatant, the amount of unlipidated LC3 is reduced, which prevents the strong signal of the unlipidated LC3 from obscuring the lipidated LC3 signal.
-
21
Add 60ul 1× SDS-PAGE loading buffer to the pellet plus the 3 μl supernatant. Determine the LC3 lipidation level using a 12 μl sample by immunoblot.
REAGENTS AND SOLUTIONS
Cytosol (usually 5–10 μg/μl, aliquot to 100–200 μl/tube, snap freeze with liquid nitrogen; can be stored at −80°C for at least two years)
Membrane (prepare fresh; can be kept on ice overnight)
B88 Buffer: 20 mM HEPES-KOH, pH 7.2, 250 mM sorbitol, 150 mM potassium acetate, and 5 mM magnesium acetate (can be stored at 4°C for at least two years)
The pH of the B88 buffer is crucial. Lower pH leads to low lipidation efficiency and higher pH leads to non-specific lipidation.
B88 lysis buffer: B88 buffer with protease inhibitors (Roche), phosphatase inhibitors (Roche), and 0.3 mM DTT (can be stored at −20°C for at least two years)
10× ATP regeneration system: 400 mM creatine phosphate, 2 mg/ml creatine phosphokinase, and 10 mM ATP in B88 (aliquot to 100 μl/tube and can be stored at −80°C for at least two years)
GTP 10 mM stock solution (aliquot to 10 μl/tube and can be stored at −80°C for at least two years)
HB1 buffer: 20 mM HEPES-KOH, pH 7.2, 400 mM sucrose and 1 mM EDTA (can be stored at 4°C for at least two years)
HB1 lysis buffer: HB1 buffer with protease inhibitors (Roche), phosphatase inhibitors (Roche), and 0.3 mM DTT (can be stored at −20°C for at least two years)
50% Optiprep solution: 50% Optiprep (sigma), 20 mM Tricine-KOH, pH 7.4, 42 mM sucrose and 1 mM EDTA (can be stored at 4°C for at least two years)
Optiprep dilution buffer: 20 mM Tricine-KOH, pH 7.4, 250 mM sucrose and 1 mM EDTA (can be stored at 4°C for at least two years)
All other buffers and media are stored at 4°C.
COMMENTARY
Background Information
Autophagy is a catabolic cellular process broadly related to many physiological processes and pathological conditions, such as cancer, neurodegeneration, and metabolic syndromes (Levine et al., 2008; Mizushima et al., 2008). Autophagy is usually activated by stress conditions, such as starvation. During starvation-induced autophagy, a cascade of cytosolic signals induces the activation of the ATG proteins to act sequentially at special sites in the cell leading to the formation of a double-membrane vesicle called the autophagosome. The autophagosome encapsulates part of the cytoplasm and transports the trapped components to the lysosome for degradation (Burman et al., 2010; Feng et al., 2014; Ge et al., 2014a).
Biogenesis of the autophagosome is a central step of autophagy. Since its discovery in the 1950s, a long-standing question has been how the autophagosome is generated in the cell. In the 1990s, a breakthrough was made by genetic studies which identified the key regulatory factors, the ATG genes, which are essential for autophagy. Subsequently, cell imaging-based approaches characterized the phagophore, a cup-shaped membrane precursor of the autophagosome, and indicated hot spots of autophagic membrane assembly, called phagophore assembly sites (PAS). However, it is still unclear how these ATG proteins act together with the intracellular membrane system (which generates the autophagic membrane) to build the autophagosome (Levine et al., 2017; Ohsumi, 2014).
There have been two puzzles in the field. 1) What is the membrane origin of the autophagosome? 2) How is that source membrane converted into the autophagic membrane? In the early stages of autophagosome biogenesis, the membrane origin of the autophagosome is buried in the endomembrane system and the nascent autophagic membrane precursors lack a distinct morphology, such as a cup-shaped phagophore or a double-membrane autophagosome. These problems made it difficult to answer the two questions through traditional cell imaging-based approaches.
To answer the first question, we sought to establish a biochemical approach based on cell-free reconstitution to functionally determine the early stage of autophagosome biogenesis (Ge et al., 2013). The rationale is: through a successful cell-free reconstitution, we are able to directly determine which cellular membrane triggers autophagosome biogenesis by combining this assay with a membrane fractionation approach.
It has been well recognized that LC3 lipidation is a key step of autophagy. The process involves a covalent linkage of a cytosolic protein LC3 (LC3-I) to phosphatidylethanolamine (PE) located on the autophagic membrane precursors (Ichimura et al., 2000; Kabeya et al., 2000). Under physiological conditions, the lipidated LC3 (LC3-II) decorates different stages of the autophagic membrane. Therefore, the LC3 lipidation is a good marker for autophagosome biogenesis. Cytosolic catalysts of the lipidation reaction include these ATG proteins: ATG12-5/16, ATG3, and ATG7. The process is carried out through a ubiquitin-like conjugation process (Feng et al., 2014). In vitro studies using purified lipidation catalysts, LC3, and synthetic liposomes with PE have reconstituted the pure enzymatic reaction of LC3 lipidation (Hanada et al., 2007; Oh-oka et al., 2008). Inspired by the in vitro assay and the importance of LC3 lipidation as a marker, we established a cell-free reaction based on LC3 lipidation to reconstitute an early step of autophagosome biogenesis (Basic Protocol 1). LC3 lipidation in this system recapitulates many key regulatory landmarks of cellular autophagy regulation including starvation induction, MTORC1 regulation, and dependence on a series of key ATG proteins (Ge et al., 2013).
We combined this cell-free assay with a three-step membrane fractionation approach (Support Protocol 1) to trace the active membrane fraction and identified the ERGIC as a key membrane origin of the autophagosome. In search of the interaction between the ERGIC and ATG proteins, we extended the cell-free reaction to a membrane flotation assay (Basic Protocol 2) based on the fact that the majority of the ATG proteins need to associate with the membrane to perform their function in autophagic membrane assembly. With this approach, we found that an autophagic phosphatidylinositol 3-kinase (PI3K) associates with the ERGIC and therefore determines the ERGIC as a membrane origin of the autophagosome (Ge et al., 2013).
To answer the second question, we established a small autophagosomal precursor generation assay (Basic Protocol 3) because we observed the accumulation of small autophagosomal precursors in Atg5 KO MEF. With this assay, we found a double requirement of PI3K and COPII machinery for generating small autophagosomal precursors from the ERGIC. Through the cooperative action, the ERGIC membrane is converted into the autophagic membrane as an essential step for supplying membranes for the autophagosome (Ge et al., 2014b).
This unit describes reconstituting autophagy during a cell-free system. In addition, another form of reconstitution based on purified protein and synthetic liposomes has also been developed in the field. Together, these in vitro approaches have set the basis for in vitro reconstitution to deepen the understanding of the mechanistic actions of autophagic factors and the membrane underlying biogenesis of the autophagosome.
Critical Parameters and Troubleshooting
Detailed critical parameters and troubleshooting have been indicated in each protocol. Please pay special attention to the following points:
Cell-free assay (Basic Protocol 1)
- Preserving the activity of cytosol and membrane.
- the cell passages should not be higher than 20;
- strictly follow the 80–90% lysis efficiency when collecting cytosol and membrane;
- make sure protease inhibitors, phosphatase inhibitors, and DTT are in the lysis buffer;
- for membrane preparation, the membrane buffer must be exchanged to B88 by washing with B88 buffer once;
- for cytosol preparation, the amount of lysis buffer added should not be more than 2 times that of the cell pellet volume in order to keep protein concentration high.
The pH of B88 buffer must be 7.2.
The incubation temperature must be 30 °C.
The consistency of the total composition of the buffer. For example, in comparing LC3 lipidation activity with different drugs, be sure to add equal amounts of solvents (e.g., DMSO) used to dissolve the drugs in all samples, including controls.
Membrane fractionation (Support Protocol 1)
- Maintaining membrane integrity.
- the cell passages should not be higher than 20;
- strictly follow the 80–90% lysis efficiency when collecting cytosol and membrane;
- the size of the needle should be 22G;
- Avoid excessive bubble formation when homogenizing the cells;
- Avoid vortexing.
Avoid fraction contamination and carefully collect each fraction.
Gradient consistency; pre-make sucrose and Optiprep gradients for 10–15 experiments and refrigerate.
Membrane recruitment assay (Basic Protocol 2)
Preserve cytosol and membrane activity and membrane integrity (see above)
Small autophagosome precursor generation assay (Basic Protocol 3)
Preserve cytosol and membrane activity and membrane integrity (see above)
Avoid donor membrane contamination. After the reaction and the medium speed centrifugation, carefully transfer the supernatant (2/3 of supernatant) and avoid even a tiny amount of the pellet.
Anticipated Results
Cell-free assay (Basic Protocol 1)
A time dependent increase of LC3 lipidation starting from 30 min of incubation and peaking around 90 min of incubation;
2–3 fold increase of LC3 lipidation with membrane and cytosol from starved cells versus non-starved cells;
LC3 lipidation inhibited by PI3K inhibitors in a dose-dependent manner;
LC3 lipidation compromised in Ulk1 KO cells and totally abolished in Atg5, Atg3, or Atg7 KO cells;
LC3 lipidation blocked using membranes from H89 treated cells (ERGIC-depleted).
Membrane fractionation (Support Protocol 1)
1k membrane: enriched in nucleus, mitochondria and ER;
3k membrane: enriched in mitochondria, ER and lysosomes;
25k membrane: enriched in plasma membrane, endosomes, Golgi, ERGIC, ER, and peroxisomes;
100 k membrane: enriched in Golgi, endosomes, and vesicles; not ER?
L membrane: enriched in plasma membrane, endosomes, Golgi, ERGIC, some light membranes, and a small amount of ER and mitochondria;
P membrane: enriched in ER and light mitochondria;
Optiprep gradient #1–2 is enriched in light membrane fractions;
Optiprep gradient #3–4 is enriched in ERGIC;
Optiprep gradient #5–6 is enriched in plasma membrane and endosomes;
Optiprep gradient #7–8 is enriched in some ER and mitochondria;
Optiprep gradient #9–10 is enriched in Golgi.
Membrane recruitment assay (Basic Protocol 2)
Starvation-enhanced fractionation of ATG14 and DFCP1 in the top membrane fraction;
Depletion of the ERGIC membrane abolishes the fractionation of ATG14 and DFCP1 in the top membrane fraction.
Small autophagosome precursor generation assay (Basic Protocol 3)
Starvation-enhanced and PI3K dependent generation of LC3 lipidation-active small membranes;
The ERGIC is the donor membrane that generates the LC3 lipidation-active small membranes;
COPII inhibitors blocks the generation of the small LC3 lipidation-active small membrane.
Time Considerations
See table 1
Table 1.
Time consideration of the key procedures
| Procedure | Time (h) |
|---|---|
| Cytosol preparation | 6 |
| Total membrane preparation | 2 |
| Lipidation reaction | 1.5 |
| LC3 protein purification | 24 |
| Three-step membrane fractionation | 24 |
| Membrane recruitment | 1.5 + 1.5 (reaction + centrifugation) |
| Small autophagosome precursor formation | 1.5 + 1.5 lipidation |
Acknowledgments
L.G. is supported by the NIH Pathway to Independence Award (Parent K99/R00) National Institute of General Medical Sciences (Grant Number: 1K99GM114397-02). L.G. is deeply grateful for the training provided by Dr. Randy Schekman at the University of California Berkeley. We thank Bob Lesch at University of California Berkeley for critical reading and helpful editing of the manuscript.
LITERATURE CITED
- Appenzeller-Herzog C, Hauri HP. The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. J Cell Sci. 2006;119:2173–83. doi: 10.1242/jcs.03019. [DOI] [PubMed] [Google Scholar]
- Bednarek SY, Ravazzola M, Hosobuchi M, Amherdt M, Perrelet A, Schekman R, Orci L. COPI- and COPII-coated vesicles bud directly from the endoplasmic reticulum in yeast. Cell. 1995;83:1183–96. doi: 10.1016/0092-8674(95)90144-2. [DOI] [PubMed] [Google Scholar]
- Brier LW, Zhang M, Ge L. Mechanistically Dissecting Autophagy: Insights from In Vitro Reconstitution. J Mol Biol. 2016;428:1700–13. doi: 10.1016/j.jmb.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burman C, Ktistakis NT. Autophagosome formation in mammalian cells. Semin Immunopathol. 2010;32:397–413. doi: 10.1007/s00281-010-0222-z. [DOI] [PubMed] [Google Scholar]
- Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res. 2014;24:24–41. doi: 10.1038/cr.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge L, Baskaran S, Schekman R, Hurley JH. The protein-vesicle network of autophagy. Curr Opin Cell Biol. 2014a;29C:18–24. doi: 10.1016/j.ceb.2014.02.005. [DOI] [PubMed] [Google Scholar]
- Ge L, Melville D, Zhang M, Schekman R. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Elife. 2013;2:e00947. doi: 10.7554/eLife.00947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge L, Zhang M, Schekman R. Phosphatidylinositol 3-kinase and COPII generate LC3 lipidation vesicles from the ER-Golgi intermediate compartment. Elife. 2014b;3:e04135. doi: 10.7554/eLife.04135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282:37298–302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, Noda T, Ohsumi Y. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408:488–92. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- Jagus R, Beckler GS. Overview of eukaryotic in vitro translation and expression systems. Curr Protoc Cell Biol. 2003:1. doi: 10.1002/0471143030.cb1101s00. Chapter 11: Unit 11. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Hamamoto S, Ravazzola M, Orci L, Schekman R. Uncoupled packaging of amyloid precursor protein and presenilin 1 into coat protein complex II vesicles. J Biol Chem. 2005;280:7758–68. doi: 10.1074/jbc.M411091200. [DOI] [PubMed] [Google Scholar]
- Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14:759–74. doi: 10.1038/nrm3696. [DOI] [PubMed] [Google Scholar]
- Levine B, Klionsky DJ. Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: Breakthroughs in baker’s yeast fuel advances in biomedical research. Proc Natl Acad Sci U S A. 2017;114:201–5. doi: 10.1073/pnas.1619876114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–68. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh-oka K, Nakatogawa H, Ohsumi Y. Physiological pH and acidic phospholipids contribute to substrate specificity in lipidation of Atg8. J Biol Chem. 2008;283:21847–52. doi: 10.1074/jbc.M801836200. [DOI] [PubMed] [Google Scholar]
- Ohsumi Y. Historical landmarks of autophagy research. Cell Res. 2014;24:9–23. doi: 10.1038/cr.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Shpilka T, Elazar Z. Mechanisms of autophagosome biogenesis. Curr Biol. 2012;22:R29–34. doi: 10.1016/j.cub.2011.11.034. [DOI] [PubMed] [Google Scholar]
- Schindler AJ, Schekman R. In vitro reconstitution of ER-stress induced ATF6 transport in COPII vesicles. Proc Natl Acad Sci U S A. 2009;106:17775–80. doi: 10.1073/pnas.0910342106. [DOI] [PMC free article] [PubMed] [Google Scholar]