

Abstract
Objective:
To identify an improved measure of clinical progression in early Huntington disease (HD) using data from prospective observational cohort studies and placebo group data from randomized double-blind clinical trials.
Methods:
We studied Unified Huntington Disease Rating Scale (UHDRS) and non-UHDRS clinical measures and brain measures of progressive atrophy in 1,668 individuals with early HD followed up prospectively for up to 30 to 36 months of longitudinal clinical follow-up.
Results:
The results demonstrated that a composite measure of motor, cognitive, and global functional decline best characterized clinical progression and was most strongly associated with brain measures of progressive corticostriatal atrophy.
Conclusions:
Use of a composite motor, cognitive, and global functional clinical outcome measure in HD provides an improved measure of clinical progression more related to measures of progressive brain atrophy and provides an opportunity for enhanced clinical trial efficiency relative to currently used individual motor, cognitive, and functional outcome measures.
Affected individuals with Huntington disease (HD) exhibit a triad of neurocognitive deficits, neuropsychiatric symptoms, and movement disorder, resulting in progressive decrements in daily functioning1 that affect patients and their caregivers. Large-scale longitudinal studies of these domains can help inform the choice of clinical outcome measures for randomized trials aiming to slow clinical progression. Our study goal was to explore each of the affected individual clinical domains of HD in longitudinal observational and interventional settings and to determine which outcome measures best met the key criteria of sensitivity to longitudinal clinical change, association with measures of disease pathophysiology, and broad clinical domain coverage.
We hypothesized that a multidomain approach may better fulfill these key criteria relative to an individual clinical domain outcome approach. We focused on the early stage of manifest disease (i.e., Total Functional Capacity [TFC] stages I and II) because disability might then be more plausibly reversed or slowed, and this stage represents the cohort of participants most commonly recruited at present for trials aiming to slow clinical progression. We studied in total 1,668 individuals with up to 36 months of longitudinal clinical follow-up using all available clinical outcome measures from the Unified Huntington Disease Rating Scale (UHDRS)2 and other non-UHDRS clinical measures.
The results suggested that relative to all clinical measures investigated, a composite made up of 4 measures spanning the affected cognitive, motor, and global functional domains from the UHDRS exhibited the best signal-to-noise (S/N) ratio, the best relationship to measures of progressive brain atrophy characteristic of HD, and the best domain coverage in an early HD population. We label this outcome the composite UHDRS (cUHDRS), and here, we present the analyses to support its validity and use in clinical trials in early HD.
METHODS
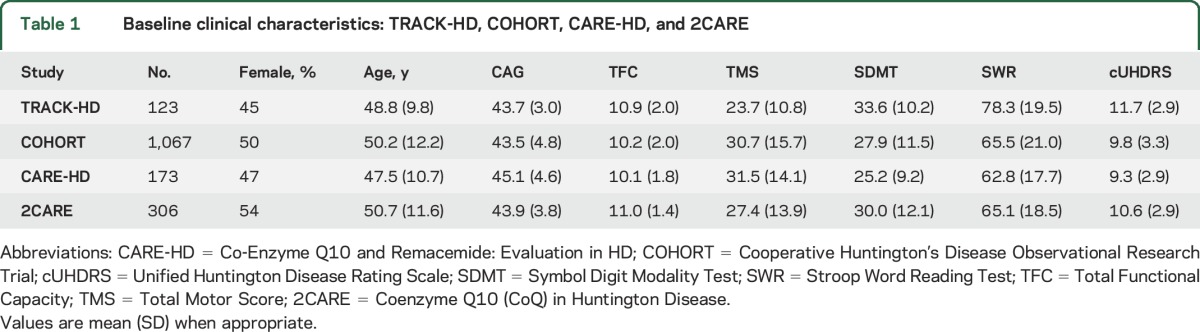
These analyses used data from 1,668 individuals with early-stage manifest HD (TFC score 7–13) and 265 controls with longitudinal clinical ratings follow-up of up to 30 months (Co-Enzyme Q10 and Remacemide: Evaluation in HD [CARE-HD]3) or up to 36 months (TRACK-HD,4 Cooperative Huntington's Disease Observational Research Trial [COHORT],5 and Coenzyme Q10 (CoQ) in Huntington Disease [2CARE]6) (see table 1 for demographics and sample size and table e-1 at Neurology.org for longitudinal loss to follow-up data). The 4 studies had multiple sites in 6 nations: TRACK-HD had 4 sites in 4 countries (Canada, France, England, and the Netherlands); COHORT had 44 sites in 3 countries (Australia, Canada, and the United States); CARE-HD had 23 sites in 2 countries (Canada and the United States); and 2CARE had 48 sites in 3 countries (Canada, Australia, and the United States).
Table 1.
Baseline clinical characteristics: TRACK-HD, COHORT, CARE-HD, and 2CARE
Standard protocol approvals, registrations, and patient consents.
All data were originally collected with appropriate preapproval of human ethics committees and written informed consent at each site in each respective study.
Statistical methods.
The 3-year TRACK-HD observational study contained the most comprehensive dataset of clinical measures, including the TFC, Total Motor Score (TMS), Symbol Digit Modality Test (SDMT), Stroop Word Reading Test (SWR), 5 quantitative motor indexes, an emotion recognition test, and the Problem Behaviors Assessment (PBA) and PBA-apathy scores. Thus, it was used as a training dataset for all analyses to generate hypotheses, which were then independently confirmed in the larger COHORT observational study. Applicability to the clinical trial setting was investigated by confirmation in the independent CARE-HD and 2CARE clinical trial studies. Additional details on study selection criteria, outcome measures, and the statistical methodology below described are available in the e-Methods.
Comparing longitudinal changes of individual measures using the S/N ratio.
We used the S/N ratio, defined as the mean change from baseline to a given time divided by the corresponding SD. Thus, the S/N ratio is a measure of the strength of the longitudinal change relative to the random variability of change for a given measure. A larger S/N ratio indicates greater reliable variance, which is a desirable characteristic for the general use of a clinical endpoint.7
Building a multidomain outcome variable from individual components.
To understand the relationship among the individual clinical variables in TRACK-HD that had a pattern of regular progression with high S/N, we first evaluated their association by calculating the Pearson correlation coefficient and 2-sided statistical significance (α set to 0.05) among each pair of variables using baseline and all available time points. We then performed a principal component analysis (PCA) that served as the basis to define a composite outcome variable as a linear combination of standardized individual variables. The multidomain composite generated from our PCA is the cUHDRS. In a supplemental analysis, the S/N ratio of all possible combinations of the cUHDRS relative to its individual components was investigated. We further quantified the differences in S/N between the cUHDRS and each individual component using a bootstrapping method.
Establishing the clinicometric properties of the cUHDRS.
The cUHDRS was tested against different clinical criteria, including sensitivity to disease stage; association with brain imaging changes relative to individual measures; pattern, consistency, and magnitude of longitudinal change in early HD across the datasets; and test-retest reliability with short-term data. We investigated with meta-analyses using both fixed-effect and random-effect models the pattern of longitudinal change at each available time point for the cUHDRS vs its individual component measures. The utility of the cUHDRS compared to its individual measures was evaluated in hypothetical 2- vs 3-year trials, and sample sizes were estimated.
RESULTS
Results from analyses of the TRACK-HD dataset showed that the TMS, TFC, SDMT, and SWR had relatively high longitudinal S/N ratios (≈0.6–0.9 at the 2-year follow-up) compared to all other measures (figure 1). Emotion recognition, PBA composite behavior, circle tracing, and certain quantitative motor indexes, including grip force variability and tongue force variability, had low S/N ratios (≤0.3 at the 2-year follow-up). Indexes of tapping and the PBA-apathy score had S/N in an intermediate range (≈0.5 at the 2-year follow-up). Similar analyses were performed in the healthy control group and showed that all measures had no significant progression over time (figure e-1).
Figure 1. Longitudinal S/N ratio of UHDRS and non-UHDRS clinical measures in early HD (stages I and II at baseline) across the motor, quantitative motor, cognitive, behavior, and global functional domains in the TRACK-HD sample.

HD = Huntington disease; PBA = Problem Behaviors Assessment; SDMT = Symbol Digit Modality Test; S/N = single-to-noise; SWR = Stroop Word Reading Test; TFC = Total Functional Capacity; TMS = Total Motor Score; UHDRS = Unified Huntington Disease Rating Scale.
The correlational analyses from TRACK-HD baseline data of the top-performing clinical measures revealed that the TMS, SDMT, and SWR scores were all significantly and strongly associated with each other and with the TFC score (all Pearson r = 0.41–0.59, all p < 0.001; table e-2). The PBA-apathy score was significantly although more weakly associated with the TFC, TMS, SDMT, and SWR scores (r = 0.22–0.41; table e-2). The relative strength of the associations was the same between the clinical variables when all available time points were used: the correlation coefficients ranged from 0.53 to 0.65 among the SDMT, SWR, TMS, and TFC, whereas the association between PBA-apathy and other variables was between 0.25 and 0.35.
A PCA performed on TMS, TFC, SDMT, SWR, and PBA-apathy showed that TMS, TFC, SDMT, and SWR had loadings of similar size for the primary principal component, which accounted for most of the observed variability (≈60%) (figure e-2). Principal component 2 explained only ≈18% of the variability and was associated mainly with apathy. The PCA results were consistent whether derived with data from all time points together or only baseline data (figure e-2). The overall pattern of the correlations and the PCA results indicated that TMS, TFC, SDMT, and SWR had significant overlap, but PBA-apathy did not. This provided the rationale for generating the modified UHDRS composite score among the domains of functioning, cognition, and motor with the following formula:
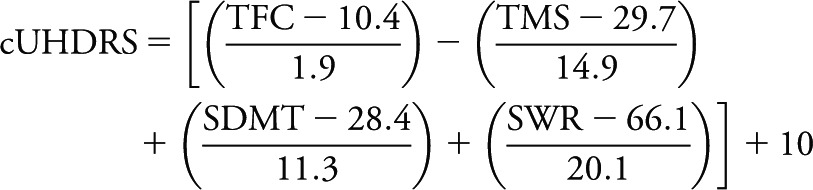 |
Note the negative sign for (scaled) TMS and the positive sign for all other variables. The centering and scaling constants for each individual variable were derived from pooled baseline data of TRACK-HD, COHORT, CARE-HD, and 2CARE. An offset of 10 was used to allow the composite measure to take on positive values in the longitudinal follow-up periods of all the studies.
The observed difference in the S/N ratio between the cUHDRS and each individual component in TRACK-HD was numerically superior in all cases (e.g., 26%–38% larger than the best individual measure, which was the TMS at 24 and 36 months, respectively) and in 10 of 12 comparisons, the S/N ratio difference between the cUHDRS and its individual components was statistically significant (p < 0.05; figure 2A and table e-3).
Figure 2. Longitudinal S/N ratio of individual UHDRS clinical measures (TFC, TMS, SDMT, SWR) and the cUHDRS across the 4 studies and time points.

Care HD = Co-Enzyme Q10 and Remacemide: Evaluation in HD; Cohort = Cooperative Huntington's Disease Observational Research Trial; cUHDRS = composite Unified Huntington Disease Rating Scale; SDMT = Symbol Digit Modality Test; S/N = single-to-noise; SWR = Stroop Word Reading Test; TFC = Total Functional Capacity; TMS = Total Motor Score; 2Care = Coenzyme Q10 (CoQ) in Huntington Disease; UHDRS = Unified Huntington Disease Rating Scale.
Confirming TRACK-HD findings in 3 independent studies (COHORT, CARE-HD, and 2-CARE).
The data analyses described for the TRACK-HD study were applied to the COHORT, CARE-HD, and 2CARE datasets (figure 2, B–D). The results showed an increase in the S/N ratio of the cUHDRS compared to its individual components for all comparisons and a statistically significant increase in 26 of 36 (p < 0.05) total comparisons across the datasets, with 15 of these comparisons highly significant (p < 0.001). Four of the remaining 10 comparisons had 0.05 < p < 0.10 (table e-3). Because each of the additional studies contained complete Stroop assessments (TRACK-HD lacked the color naming and interference components), a supplemental analysis compared the S/N ratio for the different components and showed that in most cases the S/N ratios for color naming and interference variables were low relative to that of the SWR variable (figures e-3 and e-4). The PCA results within individual datasets confirmed that TMS, TFC, SDMT, and SWR contributed similarly to the first principal component, accounting for 64% (COHORT), 66% (CARE-HD), and 64% (2CARE) of the total variability (figure e-2).
Sensitivity of the cUHDRS to clinical stage.
We investigated how the cUHDRS in TRACK-HD varied across time (baseline and 12, 24, and 36 months) within the 5 groups defined in that study: healthy controls, pre-HD A (≥10.8 years from HD motor diagnosis), pre-HD B (≤10.8 years from HD motor diagnosis), stage I HD (motor diagnosis and TFC score 11–13), and stage II HD (motor diagnosis and TFC score 7–10). The results showed that the cUHDRS was correlated with disease group and HD stage at baseline and showed a regular pattern of progression over time in the stage I/II HD groups, with a decline of ≈1 point per year (figure e-5). These results were replicated in COHORT (figure e-5).
Association of the cUHDRS with longitudinal brain changes.
Whereas the association between clinical measures with brain volumes was statistically significant across all comparisons at baseline and at the 12-month follow-up (all p < 0.01), the cUHDRS retained the highest magnitude, consistency, and statistically significant associations across all time points up to 36 months (figure 3). The associations between TFC and SDMT scores and brain volume loss were similar to that of the cUHDRS but weaker. The TMS showed relatively weak longitudinal associations, as did the SWR (figure 3).
Figure 3. Comparative baseline and longitudinal association of individual UHDRS clinical measures and the cUHDRS with measures of progressive corticostriatal atrophy in TRACK-HD.

Pearson correlation coefficient 2-sided p values: *p < 0.05, **p < 0.01, ***p < 0.001. cUHDRS = composite Unified Huntington Disease Rating Scale; SDMT = Symbol Digit Modality Test; TFC = Total Functional Capacity; TMS = Total Motor Score; UHDRS = Unified Huntington Disease Rating Scale.
Meta-analysis results of the cUHDRS and individual components across the datasets and test-retest reliability of short-term CARE-HD data.
Overall, the meta-analysis revealed that on average an approximate 1-point decline in the cUHDRS over the first year of follow-up corresponded to an approximate 3-point increase in TMS and an approximate 0.7-point decline in the TFC (figure e-6). SDMT and SWR showed a similar decline across datasets over time, whereas the TFC, TMS, and cUHDRS showed heterogeneity at 12 and 24 (but not 36) months. This heterogeneity was driven by a steeper pattern of decline in the CARE-HD cohort on the TMS, TFC, and cUHDRS measures. We note that participants of CARE-HD showed evidence of more progressed disease at baseline, as demonstrated by a significantly increased CAG × age product score8 (mean CAG × age product score = 516.9, SD = 139.2; F3, 1,665 = 6.49, p < 0.001), which is an index of disease burden (table 1).
We assessed the test-retest reliability of the cUHDRS using short-term baseline and 1-month data available in the CARE-HD dataset, given that over a short period of time, we would expect to see little change in disease severity. The results (ICC2,1 = 0.96)9 showed excellent test-retest reliability of the cUHDRS.
cUHDRS and sample size planning for a clinical trial.
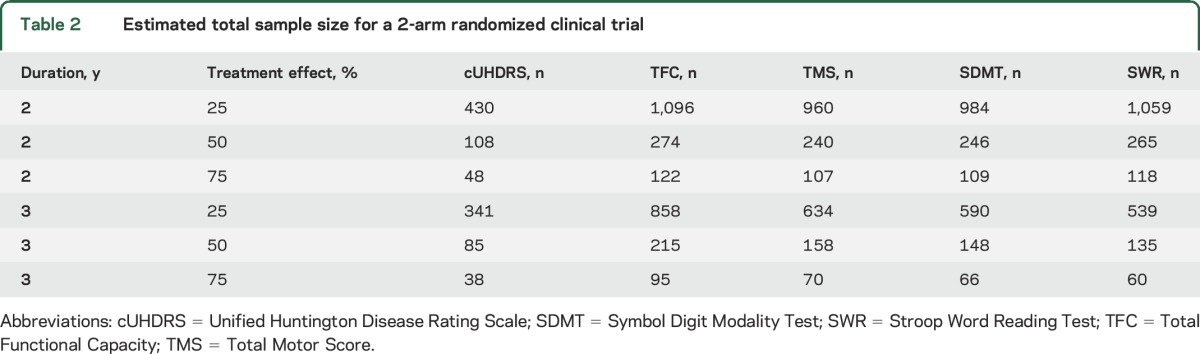
Data from TRACK-HD were used to estimate the required sample size for a significant slope difference in a randomized clinical trial (see e-Methods).10 Treatment effect was defined as the percentage improvement of the treatment group slope relative to the placebo group (25%, 50%, 75%). Trial duration of 24 and 36 months with visits every 6 months was considered. The type I error rate was 5%; the type II error rate was 20% (power = 80%); and attrition was set to 10% at 24 months and 20% at 36 months. Table 2 shows the total sample size (n/2 in each group) for the cUHDRS and for each individual measure making up the cUHDRS. Smaller sample size was associated with use of the cUHDRS relative to individual outcome measures. The required sample size using the cUHDRS could be as much as 55% smaller than the next best measure (e.g., compared to TMS at 24 months).
Table 2.
Estimated total sample size for a 2-arm randomized clinical trial
DISCUSSION
The enhanced sensitivity of the cUHDRS to clinical change in early symptomatic HD and its strong relationship to underlying brain changes relative to the TFC and the TMS suggest that the cUHDRS is an improved measure of clinical progression. The TFC3,6,11–13 and TMS14–19 provide reliable and clinically validated outcome measures of global function or motor signs characteristic of HD and thus should remain useful primary outcome measures in clinical trials. We note that the performance of the TFC was strong in the 2 clinical trial databases studied relative to the cUHDRS. However, these 2 measures either do not represent the affected underlying phenomenology of HD (i.e., the TFC does not measure underlying motor, cognition, or behavior directly) or, in the case of the TMS, provide limited domain coverage. In summary, our results suggest that the cUHDRS has greater statistical power (i.e., lower type II error rate) to detect success in clinical trials aiming to slow clinical progression relative to the TFC and TMS.
We have reached consensus that a slowing of clinical decline in the range of 20% to 30% annually on the cUHDRS (≈0.2–0.3 points per year), equivalent to ≈2.4 to 3.5 mo/y of time saved from progression, can be used to define a clinically efficacious treatment in this setting. Even a lower-magnitude slowing of decline of ≈0.1 point per year or ≈10% might be clinically meaningful, although the sample size required to demonstrate such an effect may be prohibitively large.
We have demonstrated that explicit introduction of 2 cognitive measures provides not only enhanced sensitivity to measure clinical progression in HD but also enhanced domain coverage. This was demonstrated by the consistent magnitude gain of the S/N ratio relative to all other single measures or combinations of measures studied. We note that a cognitive domain–specific composite outcome measure has been developed (i.e., the Huntington's Disease Cognitive Assessment Battery).20 This composite, although promising, has not been validated longitudinally.20 Certain components of the Huntington's Disease Cognitive Assessment Battery have been examined in a longitudinal setting (i.e., in the TRACK-HD 24-month study). It is notable that the highest sensitivity to longitudinal clinical change was indeed observed for the SDMT and SWR at 24 months, the 2 key cognitive components of the cUHDRS, along with a measure of circle tracing.21
Within the UHDRS, the PBA measure has known poor longitudinal sensitivity to clinical change,22 which we also observed. The PBA-apathy measure possessed a relatively strong pattern of progression, but it did not correlate well to either the cognitive or motor domain. Although the cUHDRS does not contain behavioral measures, our results suggest that behavioral measures either are not key clinical features of clinical progression or appear unrelated to the progressive cognitive and motor domains (i.e., the PBA-apathy score). The apathy domain merits additional investigation in clinical trials because it is clearly related to function and showed a relatively strong pattern of progression. However, other behavioral measures of emotion recognition or depressive symptoms are likely not good candidate outcome measures of clinical progression.
A critical assumption underlying the proposed use of the cUHDRS in clinical trials is that if a putative neuroprotective or mutant huntingtin-lowering therapy successfully targets disease mechanisms underlying clinical progression, then each individual measure making up the cUHDRS should move in the same direction of improvement in response to this treatment. A limitation with any composite is that such specificity is lost in the interpretation of statistical results. If the cUHDRS shows success in a clinical trial, then the conclusion is that the treatment positively affected motor, cognitive, daily functioning, or some combination thereof.
However, the association over time between the individual motor, cognitive, and global functional clinical measures as observed here, as well as the robust association between brain changes and the composite, is supportive of the assumption that the measures would tend to change together in early HD in response to a treatment designed to slow clinical progression. Another scenario might involve slowing of decline on the cUHDRS but dissociation between this and changes in behavioral measures such as apathy or depression. In such a scenario, arguably the clinical meaningfulness of a treatment effect on the cUHDRS may be limited.
The cUHDRS can assist in ensuring that trials targeting clinical progression are maximally sensitive to detect clinical change and maximally protected from failure due to measurement insensitivity. Similarly, insofar as multidomain decline is measurable in this composite measure, a more holistic clinically meaningful outcome is obtained, similar to the use of the Clinical Dementia Rating sum of boxes in Alzheimer disease, which contains multidomain cognitive and global functional elements,23 or the practice of summing parts I, II, and III of the Unified Parkinson Disease Rating Scale24 in Parkinson disease. Moreover, trials can be conducted more efficiently with smaller sample sizes with the cUHDRS. The benefits are the lessening of participant burden, using fewer resources to test hypotheses, and potentially enhancing the quality of results obtained through limiting the overall size of the trial. The observed robust gain in S/N ratio of the cUHDRS relative to the individual measures across the 4 studies suggests that our findings are generalizable to a broader early HD population. We have leveraged the knowledge accrued from these previously conducted observational and interventional studies, such that trials targeting clinical progression in early HD can now better meet the mandate of a robust primary outcome.
ACKNOWLEDGMENT
The authors acknowledge the patients and families whose cooperation was vital to generate this information. They also acknowledge Ronald Gieschke, PhD (Roche), Daniel Serafin, PhD (Roche), and Robert Strawderman, ScD (University of Rochester), for providing critical comments to data analysis contained in the manuscript; Alicia Brocht, MSc (University of Rochester), for providing data management support for the Huntington Study Group databases; and Alicia Augustine (University of Rochester) for providing critical comments on the first draft of the manuscript.
GLOSSARY
- CARE-HD
Co-Enzyme Q10 and Remacemide: Evaluation in HD
- COHORT
Cooperative Huntington’s Disease Observational Research Trial
- cUHDRS
composite Unified Huntington Disease Rating Scale
- HD
Huntington disease
- ICC
intraclass correlation
- PBA
Problem Behaviors Assessment
- PCA
principal component analysis
- SDMT
Symbol Digit Modality Test
- S/N
signal-to-noise
- SWR
Stroop Word Reading Test
- TFC
Total Functional Capacity
- TMS
Total Motor Score
- 2CARE
Coenzyme Q10 (CoQ) in Huntington Disease
- UHDRS
Unified Huntington Disease Rating Scale
Footnotes
Supplemental data at Neurology.org
Contributor Information
Collaborators: TRACK-HD, COHORT, CARE-HD, and 2CARE Huntington Study Group Investigators, John Adams, Mandar Jog, Christopher Hyson, Sarah Furtado, Andrew Duker, Melissa Armstrong, Christian Lachner, Hubert Fernandez, Michael S Okun, David Shprecher, Michael Cartwright, Clement Loy, Joohi Jiminez-Shahed, Bradley Robottom, Gregory Suter, Theresa Moore, Jane Forsyth, Andrea Hurt, Joann Belden, Katie Price, Diane Erickson, Breanna Nickels, Misty M Thompson, Linda Cole, Julie Megens, Emilija Makaji, Sara-Lynn Masse, Keith Malarick, Louisa Mook, Susan Maya, Alex Bender, Jessica Meyer, Puja Turakhia, Katherine Harwood, Kathryn Duderstadt, Sarah Lenarz, Judy Hamerlinck, Patricia Edern, Charlyne Hickey, Ashley Owens, Clare Gibbons, Carolyn Gray, Jean A Jaglin, Kimberly Janko, Holly Lawrence, Barbara Estes, Brigid Hayward, Allison Johnson, Amit Gode, Giselle Huet, Beverly Romero-Kersh, Kristine Wernette, Elizabeth Sullivan, Jamie Guyot, Julie Konkle, Christine O'Neill, Pamela King, Amanda Martin, John Bautista, Nicole Mans, Jane Griffith, Erica Surles, Sharon Halton, Alicia Palao, Nathalie Padron, Kolleen Elliott, Lynn Oelke, London Butterfield, Peggy Perry-Trice, Sarah Wyne, Carol Pantella, Lorelei Tainsh, Elaine Sperin, Sharon Evans, Maureen Gartner, Amy Duffy, Pamela Kristof, Lisa Niles, Steven Rainone, Angie Wernle, Ronda Clouse, Michelle Cines, Kelly Dustin, Maura Deeley, Stacy Merritt, Heather Ferreri, Alison McMurray, Alanna Sheinberg, Marsha Hughes-Gay, Kori Ladonna, Erin Chung, Jennifer Lee, Rachel Goldstein, Lindsay Esposito, Nicholas Scoglio, Matthew Grana, Carol Zimmerman, Lucia M Blasucci, William Thayer, Jennifer Hawkins, Julia Koch, Victoria Hunt, Jessica Bargoil, Ingrid Scott, Terry Tempkin, Linda Stewart, Emily Hayes, Donna Galea, Beatriz Belmar, Kylie Richardson, Christine Hunter, Ernesto Jimenez, Monica Quesada, Wendy Levy, Anita Blenke, Lisa Gauger, Joanna Stoner, Mary Lou Klimek, Barbara Sommerfeld, Mary Eglow, Katy Regan, Erin Neefus, Marie Malikowski, Teri Radam, Marci Zomok, Margaret Lannon, Rhonda Agramonte, Johanna Hartlein, Constance Nickerson, Samantha Gibson, Erin Hastings Monari, Camille Swartz, Anne Smith-Bova, Randi Jones, Catherine Wielinski, Shanthi Graham, David Gunn D Psych, Jillian McMillan M Clin Neuropsych, W. Koroshetz, M. McDermott, M.F. Beal, J.T. Greenamyre, C.A. Ross, I. Shoulson, M.E. Cudkowicz, P. Sexton, C. Skeuse, D. Rosas, B. Jenkins, A. Rosenblatt, M. Sherr, S. Hersch, J. Cellar, M. Guttman, C. Brown, P.G. Como, F. Marshall, J.A. DeMarcaida, C. Zimmerman, K. Marder, C. Moskowitz, C. Polanco, J. Beltre, K. Shannon, J.A. Jaglin, F. Niederman, T. Ashizawa, C. Hunter, C. Hunter, R. Hauser, A. Walker, L. Gauger, M. A. Hahn, H. Delgado, T. Dawson, E. Siemers, L. Seeberger, C. Dingmann, R. Dubinsky, C. Gray, W.J. Weiner, D. Rodriguez–Batemen, R.L. Albin, K. Wernette, M. Harrison, E. Rost–Ruffner, J. Paulsen, R.L. Rodnitzky, J. Dobson, L. Vining, O. Suchowersky, C. Pantella, M.H. Saint–Hilaire, J. Furtado, W. Martin, M. Wieler, F. Walker, V. Hunt, L. Raymond, E. Almqvist, A. Rubin, B. Blair, K. Marek, K. Caplan, D. Baker, A. Brocht, C. Casaceli, C. Orme, A. Rudolph, L. Rumfola, G. Schifitto, A. Shinaman, K. Sulimowicz, A. Watts, S. Eapen, L. Zhang, P. Tariot, S. Fahn, W. Hall, W. Koller, J. Gorell, R. Woolson, I. Shoulson, James Gusella, Tatiana Foroud, Daniel P. Van Kammen, Frederick Marshall, Charlyne Hickey, Karen Marder, Steven Frucht, Carol Moskowitz, Ronda Clouse PaulaWasserman, Lisa Muratori, Elan Louis, Kathleen Shannon, Jeana Jaglin, Joseph Jankovic, Alicia Palao, Madaline Harrison, Robert Davis, Susan Dietrich, Carlos Singer, Monica Quesada, Steven Hersch, Diana Rosas, Kaloyan Tanev, Keith Malarick, Robert McInnis, Juan Sanchez-Ramos, Sandra Kostyk, Nick Doucette, Jane Paulsen, Ergun Uc, Robert Rodnitzky, Anne Leserman, Stacie Vik, Joel Perlmutter, Samer Tabbal, Amy Schmidt, Stacey Barton, Christopher Ross, Ray Dorsey, Frederick Nucifora, Richard Dubinsky, Hilary Dubinsky, Janice Broyles, Oksana Suchowersky, Mary Lou Klimek, Randi Jones, Claudia Testa, Elaine Sperin, Claudia Testa, Dana Jennings, Donald Higgins, Eric Molho, John Adams, Samuel Frank, Marie Saint-Hilaire, Melissa Diggin, Sarah Furtado, FrancisWalker Christine O'Neill, Kimberly Quaid, Melissa Wesson, S. Elizabeth Zauber, Mark LeDoux, Lynn Raymond, Blair Leavitt, Joji Decolongon, Susan Perlman, Jody Corey-Bloom, Guerry Peavy, Rajeev Kumar, Vicki Segro, Diane Erickson, Elizabeth McCusker, Jane Griffith, Clement Loy, Vicki Wheelock, Terry Tempkin, Amanda Martin, Martha Nance, Un Kang, William Mallonee, Greg Suter, Fred Revilla, Carolyn Drazinic, Mary Jane Fitzpatrick, Kevin Duff, Burton Scott, Bradley Robottom, Edmond Chiu, Olga Yastrubetskaya, Andrew Churchyard, Timothy John Greenamyre, Nancy Lucarelli, Pinky Agarwal, Irina Antonijevic, Jeanen DeLaRosa, Peter Panegyres, Allison Coleman, David Oakes, Christopher Beck, Suzanne Robertson, Dustina Holt, Ken Eaton, Patricia Lindsay, Lisa Deuel, Marcy MacDonald, Tatiana Foroud, Ray Dorsey, Joseph Giuliano, Louise Vetter, Oksana Suchowersky, Christopher Beck, David Oakes, Sarah J Tabrizi, Alexandra Durr, Blair Leavitt, RA Roos, Beth Borowsky, Bernhard Landwehrmeyer, C Frost, H Johnson, Douglas Langbehn, David Craufurd, Ralf Feilmann, Julie C Stout, AustraliC Campbell, M Campbell, I Labuschagne, C Milchman, Canada A Coleman, R Dar Santos, J Decolongon, A Sturrock, France C Jauff ret, D Justo, S Lehericy, C Marelli, K Nigaud, R Valabregue, Germany N Bechtel, S Bohlen, Netherlands S J A van den Bogaard, E M Dumas, J van der Grond, E P ‘t Hart, UK N Arran, J Callaghan, C Stopford, D M Cash, H Crawford, N C Fox, N Z Hobbs, N Lahiri, I Malone, J Read, M J Say, D Whitehead, E Wild, R Jones, USA E Axelson, S Queller, and C Campbell
AUTHOR CONTRIBUTIONS
Scott Schobel designed the study, reviewed and interpreted data analyses, and wrote the first draft of the manuscript. Giuseppe Palermo codesigned the study and generated all analysis on the TRACK-HD dataset, cowrote the first-draft of the manuscript, interpreted all data analyses, and generated figures and tables. Douglas Langbehn codesigned the study, contributed analysis from TRACK-HD and brain imaging data for the article, reviewed and interpreted all data analyses, and cowrote the first draft of the manuscript. Jeff Long codesigned the study, interpreted all data analyses, contributed further analysis from the TRACK-HD dataset regarding hypothetical clinical trials and underlying assumptions, and cowrote the first draft of the manuscript. Peggy Auinger and Shiyang Ma contributed all analysis from the COHORT, CARE-HD, and 2-CARE studies and provided edits to the first draft of the manuscript. Merit Cudkowicz contributed data from the 2CARE study and provided edits to the first draft of the manuscript. Ray Dorsey, Blair Leavitt, and Karl Kieburtz shared data from the CARE-HD, COHORT, and 2CARE studies, provided input to the design of the study, and provided edits to the first draft of the manuscript. Omar Khwaja, Jeff Sevigny, Dylan Trundell, Steven Hersch, and Cristina Sampaio provided input into the design of the study and provided critical comments and edits to the first draft of the manuscript. Sarah Tabrizi shared the 3-year TRACK-HD dataset, designed the study, interpreted all data analyses, and cowrote the first draft of the manuscript.
STUDY FUNDING
Funding was received from the CHDI foundation (TRACK-HD and COHORT studies); the National Institute of Neurological Disorders and Stroke (NS R01-35284), General Clinical Research Centers (grants RR00645, RR00042, RR00044, RR01066, RR07122), AstraZeneca, and Vitaline (the CARE-HD study); the National Institute of Neurological Disorders and Stroke (grants NS052592 and NS052619 for the 2CARE study); and Roche (support of Roche-led and non–Roche-led data analyses contained in the manuscript as a part of a University College London, University of Iowa, Harvard/Massachusetts General Hospital, Huntington Study Group, University of Rochester, CHDI, and Roche collaboration on endpoint development in HD).
DISCLOSURE
S. Schobel and G. Palermo are full-time employees of F. Hoffman-La Roche, Ltd. P. Auinger reports no disclosures relevant to the manuscript. J. Long consults with NeuroPhage, is a paid consultant for Roche and Azevan Pharmaceuticals, and receives funding from CHDI Inc, Michael J. Fox Foundation, and the NIH. S. Ma reports no disclosures relevant to the manuscript. O. Khwaja and D. Trundell are full-time employees of F. Hoffman-La Roche, Ltd. M. Cudkowicz consults with Denali, Biohaven, Karyopharm, Lilly, MT Pharma, and Biogen. S. Hersch reports research support from the NIH, Food and Drug Administration Orphan Products Development Program, Huntington's Disease Society of America, Hereditary Disease Foundation, PRANA, Novartis Institutes for Biomedical Research, California Institute for Regenerative Medicine, and Dake Foundation and consults with Azevan, Link Medicine, Mitochon Pharmaceuticals, Pfizer, PRANA, Roche, Wave Life Sciences, and Voyager Therapeutics. C. Sampaio reports no disclosures relevant to the manuscript. E. Ray Dorsey has received honoraria for speaking at American Academy of Neurology courses; received compensation for consulting activities from 23andMe, Clintrex, GlaxoSmithKline, Grand Rounds, Lundbeck, MC10, MedAvante, Medico Legal services, the National Institute of Neurological Disorders and Stroke, Shire, Teva, and UCB; research support from AMC Health, Burroughs Wellcome Fund, Davis Phinney Foundation, Duke University, GlaxoSmithKline Great Lakes Neurotechnologies, Greater Rochester Health Foundation, Huntington Study Group, Michael J. Fox Foundation, National Science Foundation, Patient-Centered Outcomes Research Institute, Prana Biotechnology, Raptor Pharmaceuticals, Roche, Safra Foundation, and University of California Irvine; and stock options from Grand Rounds. B. Leavitt is the co–editor-in-chief of The Journal of Huntington's Disease and reports personal fees for consulting and nonfinancial research support from Ionis Pharmaceuticals, research grants and personal fees for consulting from LifeMax Pharmaceuticals and Teva Pharmaceuticals, personal fees for consulting from Raptor Pharmaceuticals, personal fees for consulting from Roche, and personal fees as co-chair of the Huntington Study Group. K. Kieburtz consults for the NIH, Acorda, Astellas Pharma, AstraZeneca, BioMarin, Biotie, Britannia, CHDI, Chondrial, Clearpoint Strategy Group, Clintrex, Corium International, Cynapsus, EMD Serono, Dart Neuroscience, Forward Pharma, Genzyme, INC Research, Intec, Jazz, Knopp, Kyowa Kirin, Lundbeck, Medivation, Melior Discovery, Monosol, Neurocrine, Neuroderm, Neuropore, Neurmedix, Novartis, Orion Pharma, Otsuka, Pfizer, Pharma2B, Photopharmics, Prana Biotechnology, Prothena/Neotope/Elan Pharmaceutical, PTC, Raptor Pharmaceuticals, Remedy Pharmaceuticals, Roche/Genentech, Sage Bionetworks, Sanofi, Serina, Sunovion, Synagile, Teva, UCB Pharma, Ultragenyx, Uniqure, US WorldMeds, Vaccinex, vTv Therapeutics, Voyager, and Weston Brain Institute. J. Sevigny is a full-time employee of F. Hoffman-La Roche, Ltd. D. Langbehn reports consulting fees and research support from CHDI and is a consultant for Roche, Voyager, and Teva. S. Tabrizi has undertaken consultancy services, including advisory boards, with F. Hoffmann-La Roche Ltd, Ixico Technologies, Shire Human Genetic Therapies, Takeda Pharmaceuticals International, TEVA Pharmaceuticals, GSK, Heptares Therapeutics, and University College Irvine, and receives research support from Medical Research Council UK, the Wellcome Trust, the EU FP7 Health Call, the Rosetrees Trust, Takeda Pharmaceuticals, the National Institute for Health Research (NIHR) University College London Biomedical Research Centre, the NIHR North Thames Local Clinical Research Network, the Wolfson Foundation for Neurodegeneration, and the CHDI Foundation. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Ross CA, Tabrizi SJ. Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 2011;10:83–98. [DOI] [PubMed] [Google Scholar]
- 2.Unified Huntington's Disease Rating Scale: reliability and consistency: Huntington Study Group. Mov Disord 1996;11:136–142. [DOI] [PubMed] [Google Scholar]
- 3.Huntington Study Group. A randomized, placebo-controlled trial of coenzyme Q10 and remacemide in Huntington's disease. Neurology 2001;57:397–404. [DOI] [PubMed] [Google Scholar]
- 4.Tabrizi SJ, Scahill RI, Owen G, et al. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington's disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol 2013;12:637–649. [DOI] [PubMed] [Google Scholar]
- 5.Dorsey ER, Beck CA, Darwin K, et al. Natural history of Huntington disease. JAMA Neurol 2013;70:1520–1530. [DOI] [PubMed] [Google Scholar]
- 6.McGarry A, McDermott M, Kieburtz K, et al. A randomized, double-blind, placebo-controlled trial of coenzyme Q10 in Huntington disease. Neurology 2017;88:152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elkum N, Shoukri MM. Signal-to-noise ratio (SNR) as a measure of reproducibility: design, estimation, and application. Health Serv Outcomes Res Methodol 2008;8:119–133. [Google Scholar]
- 8.Zhang Y, Long JD, Mills JA, et al. Indexing disease progression at study entry with individuals at-risk for Huntington disease. Am J Med Genet B Neuropsychiatr Genet 2011;156B:751–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shrout PE, Fleiss JL. Intraclass correlations: uses in assessing rater reliability. Psychol Bull 1979;86:420–428. [DOI] [PubMed] [Google Scholar]
- 10.Yi Q, Panzarella T. Estimating sample size for tests on trends across repeated measurements with missing data based on the interaction term in a mixed model. Control Clin Trials 2002;23:481–496. [DOI] [PubMed] [Google Scholar]
- 11.Kremer B, Clark CM, Almqvist EW, et al. Influence of lamotrigine on progression of early Huntington disease: a randomized clinical trial. Neurology 1999;53:1000–1011. [DOI] [PubMed] [Google Scholar]
- 12.Landwehrmeyer GB, Dubois B, de Yebenes JG, et al. Riluzole in Huntington's disease: a 3-year, randomized controlled study. Ann Neurol 2007;62:262–272. [DOI] [PubMed] [Google Scholar]
- 13.Shoulson I, Odoroff C, Oakes D, et al. A controlled clinical trial of baclofen as protective therapy in early Huntington's disease. Ann Neurol 1989;25:252–259. [DOI] [PubMed] [Google Scholar]
- 14.NCT02006472. PRIDE HD clinical trial (Pridopidine, Teva Pharmaceuticals). Available at: ClinicalTrials.gov. Accessed August 21, 2017.
- 15.NCT02197130. Amaryllis clinical trial (PDE10A inhibitor; Pfizer). Available at: ClinicalTrials.gov. Accessed August 21, 2017.
- 16.NCT02101957. Cysteamine clinical trial (Raptor Pharmaceuticals). Available at: ClinicalTrials.gov. Accessed August 21, 2017.
- 17.NCT02215616. LEGATO HD trial (Laquinimod, Teva Pharmaceuticals). Available at: ClinicalTrials.gov. Accessed August 21, 2017.
- 18.Huntington Study Group, Frank S, Testa CM, et al. Effect of deutetrabenazine on chorea among patients with Huntington disease: a randomized clinical trial. JAMA 2016;316:40–50. [DOI] [PubMed] [Google Scholar]
- 19.Huntington Study Group. A randomized, double-blind, placebo-controlled trial of pridopidine in Huntington's disease. Mov Disord 2013;28:1407–1415. [DOI] [PubMed] [Google Scholar]
- 20.Stout JC, Queller S, Baker KN, et al. HD-CAB: a cognitive assessment battery for clinical trials in Huntington's disease 1,2,3. Mov Disord 2014;29:1281–1288. [DOI] [PubMed] [Google Scholar]
- 21.Stout JC, Jones R, Labuschagne I, et al. Evaluation of longitudinal 12 and 24 month cognitive outcomes in premanifest and early Huntington's disease. J Neurol Neurosurg Psychiatry 2012;83:687–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salem L, Saleh N, Youssov K, et al. The most appropriate primary outcomes to design clinical trials on Huntington's disease: meta-analyses of cohort studies and randomized placebo-controlled trials. Fundam Clin Pharmacol 2014;28:700–710. [DOI] [PubMed] [Google Scholar]
- 23.Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry 1982;140:566–572. [DOI] [PubMed] [Google Scholar]
- 24.Fahn S, Elton R; Members of the UPDRS Development Committee. Recent developments in Parkinson's disease. Macmillan Health Care Inf 1987;2:153–163, 293–304. [Google Scholar]