

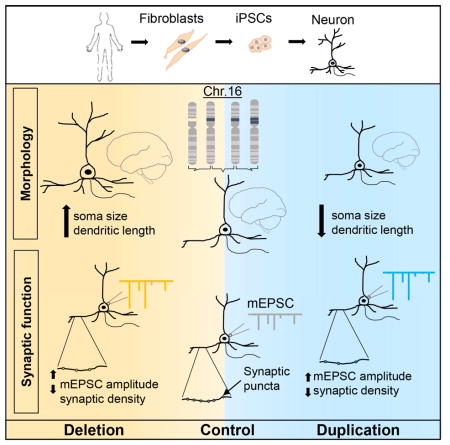
Summary
A deletion or duplication in the 16p11.2 region is associated with neurodevelopmental disorders including autism spectrum disorder and schizophrenia. In addition to clinical characteristics, carriers of the 16p11.2 copy number variant (CNV) manifest opposing neuroanatomical phenotypes, e.g. macrocephaly in deletion carriers (16pdel) and microcephaly in duplication carriers (16pdup). Using fibroblasts obtained from 16pdel and 16pdup carriers, we generated induced pluripotent stem cells (iPSCs) and differentiated them into neurons to identify causal cellular mechanisms underlying neurobiological phenotypes. Our study revealed increased soma size and dendrite length in 16pdel neurons and reduced neuronal size and dendrite length in 16pdup neurons. Functional properties of iPSC-derived neurons corroborated aspects of these contrasting morphological differences that may underlie brain size. Interestingly, both 16pdel and 16pdup neurons displayed reduced synaptic density, suggesting that distinct mechanisms may underlie brain size and neuronal connectivity at this locus.
Keywords: 16p11.2 CNV, neuron, brain size, iPSC
eTOC Blurb
Deshpande et al. show that neurons derived from individuals harboring neurodevelopmental disorders caused by the 16p11.2 deletion or duplication manifest contrasting cellular phenotypes that may underlie the macro- or microcephaly observed in carriers, respectively. Comparable functional changes in deletion- and duplication-derived neurons suggest similar mechanisms underlying common clinical features, like autism.
Introduction
The increasing prevalence of autism spectrum disorder (ASD) highlights the importance of identifying biomarkers and therapeutic targets. This complex neurodevelopmental disorder (NDD) is characterized by repetitive or stereotypic behaviors as well as communication and social deficits(American Psychiatric Association, 2013). However, neurobiological mechanisms underlying the pathology of ASD are not well understood. Of the known genetic etiologies for ASD, rare and recurrent copy number variants (CNVs) have emerged as highly penetrant risk factors(Malhotra and Sebat, 2012). The 16p11.2 CNV is a ~600kb locus harboring 29 annotated genes, most of which are highly expressed in the brain; deletion or duplication of this locus is associated with ASD(Weiss et al., 2008). Levels of mRNA expression of 16p11.2 genes correspond to copy number in human lymphoblastoid cell lines (LCLs), in induced pluripotent stem cells (iPSCs) and in the mouse brain, i.e. reduced in deletion carriers (16pdel) and increased in duplication carriers (16pdup)(Blumenthal et al., 2014; Tai et al., 2016). Studies in animal models and transcriptome analyses in LCLs have identified genes in the region involved in synaptogenesis, chromatin modulation and ciliary function (Blumenthal et al., 2014; Luo et al., 2012; Migliavacca et al., 2015). In addition to ASD, the 16p11.2 deletion is a risk factor for developmental delay (DD), language impairment, and epilepsy, while the reciprocal duplication predisposes carriers to schizophrenia, bipolar disorder, DD, and seizures(Kumar et al., 2008; McCarthy et al., 2009; Rosenfeld et al., 2010; Shinawi et al., 2010; Weiss et al., 2008). Despite common behavioral symptoms, including ASD, 16p11.2 CNV carriers manifest opposing neuroanatomical phenotypes, such as macrocephaly in 16pdel and microcephaly in 16pdup(Qureshi et al., 2014).
Interestingly, head circumference or brain size changes occur in additional genetic etiologies of ASD and NDDs. For example, loss-of-function mutations in the PTEN gene have been shown to result in extreme macrocephaly and ASD(Butler et al., 2005). Disruptive mutations in CHD8 are associated with ASD and macrocephaly(Bernier et al., 2014). Other CNVs such as the 1q21.1 duplication predispose to ASD and macrocephaly in addition to intellectual disability (ID) and schizophrenia (Mefford et al., 2008; Milone et al., 2016). In contrast, microcephaly is observed in carriers with mutations in DYRK1A along with predisposition for ID, ASD and seizures (Ji et al., 2015). Although both macrocephaly and microcephaly can be associated with ASD, studies demonstrating that the same gene (or pathway) can cause both macrocephaly and microcephaly (e.g. via gain- or loss-of-function mutations) are lacking. Thus, the 16p11.2 CNV appears to be uniquely poised to investigate whether a single cellular mechanism, dysregulated in opposite directions, could be responsible for opposing brain size phenotypes. Furthermore, the relationship between brain growth mechanisms and ASD-relevant functional characteristics could be applicable to a range of ASD risk factors.
At the cellular level, brain overgrowth or undergrowth often result from dysregulation of early developmental pathways in neural progenitor cells (NPCs). The cellular processes that can be disrupted are – i) cell proliferation & apoptosis ii) cell growth or iii) differentiation (Groszer et al., 2001; Homem et al., 2015). We hypothesize that perturbation of one or more of these cellular processes may underlie the brain growth phenotype in 16p11.2 CNV carriers, given that the CNV harbors genes known to manifest early neurobiological phenotypes when dysregulated. Noteworthy examples are: MAPK3, a key component of the Ras/MAPK pathway which regulates multiple processes including proliferation and differentiation, TAOK2, a kinase essential for dendrite and spine morphogenesis and KCTD13, a ubiquitin ligase family member shown to affect head size by altering NPC proliferation and apoptosis in a dose-dependent manner in zebrafish (Calderon de Anda et al., 2012; Golzio and Katsanis, 2013; Pucilowska et al., 2015; Yadav et al., 2017). However, the cellular mechanisms in 16p11.2 CNV-carriers underlying macro- or microcephaly remain unknown. Further, it is unclear whether behavioral symptoms common to these CNVs are a direct consequence of the same mechanism underlying differences in brain size.
To test whether early developmental processes underlie the brain size phenotype, we generated iPSCs from fibroblasts of 16pdel, 16pdup carriers and controls. We differentiated them into forebrain cortical neurons. We assessed (i) proliferation by examining cell cycle markers, (ii) cell growth by measuring soma size, and (iii) differentiation by measuring dendritic growth and synaptic development. Finally, we assessed electrophysiological properties of iPSC-derived neurons to determine whether they reflect the observed morphological differences and assessed synaptic development to determine whether the same mechanism contributing to brain size may also influence functional properties.
Results
Generation of iPSCs from 16p11.2 CNV carriers and differentiation into NPCs
The 16p11.2 CNV carrier-derived iPSCs were comparable to control iPSCs with respect to stem cell marker expression and pluripotency (Figures 1A and 1B; Figure S1A and 1B). To confirm that iPSCs retained the CNV status after reprogramming, we assessed, by quantitative reverse-transcriptase PCR (qRT-PCR), mRNA levels of selected genes spanning the 16p11.2 CNV. We found that generally the expression of QPRT, MVP, KCTD13, TAOK2, ALDOA, TBX6, MAPK3 and CORO1A, was decreased to about 0.3–0.5 times in 16pdel lines and increased by about 1.2–1.5 times in 16pdup lines, when normalized to controls (Figure 1C; Figure S1C).
Figure 1. Characterization of 16p11.2 CNV carrier-derived cells.

(A) Diagram of the 16p11.2 CNV genes, not to scale.
(B) Images of iPSCs expressing stem cell markers nanog, OCT4 and SSEA-4.
(C) Relative mRNA expression of selected 16p11.2 genes by qRT-PCR.§
(D) Schematic of differentiation of iPSCs to forebrain neurons. Lower panel shows bright field images of stages of neural differentiation.
(E) Staining for PAX6 and EdU in NPC cultures. EdU pulse for 4h followed by a 24h chase.
(F) Average percentage of PAX6+ NPCs in 16pdel, control and 16pdup lines. No statistical difference.§
(G) Average percentage of NPCs that incorporated EdU during the 4h pulse. No statistical difference.§
Values are mean ± SE. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0005; ■p<0.1; scale bar, 100μm. §Symbols apply to all bar plots and represent all lines used in the study. See Table S4 for details about lines (3 16pdel; 3 16pdup; 4 controls), clones (1–3/line) and repeated experiments (3–5). See also Figures S1 and S2.
We then differentiated iPSCs to forebrain cortical neurons(Zhang et al., 2001) (Figure 1D). We generated NPCs from three carriers each of 16pdel and 16pdup and four controls and analyzed 2–3 clones per individual for each experiment described (Supplemental Table S4). The proportion of specific forebrain NPC markers, such as PAX6 and nestin, in 16pdel, control and 16pdup was comparable, suggesting induction of dorsal forebrain identity (Figures 1E and 1F; Figures S2C and S2D). Quantitative RT-PCR for selected 16p11.2 genes in NPCs generally showed the expected expression patterns i.e. decreased in 16pdel and increased in 16pdup (Figure S2A). We then checked NPC proliferation by EdU incorporation assay; the proportion of EdU+ NPCs was not significantly different in either 16pdel or 16pdup NPCs when compared with controls after a 24h (hour) chase following a 4h EdU pulse (p=0.56 and 1.0, respectively; Figure 1E and 1G). To assess whether the self-renewal capacity of NPCs was altered, we incubated NPCs with EdU followed 24h and 48h later with a 1h pulse of 5-bromo-2′-deoxyuridine (BrdU). We did not observe a statistically significant difference in EdU-only (cells that exited the cell cycle) in the 24h (16pdel: p=0.53; 16pdup: p=0.79) or 48h (16pdel: p=0.19; 16pdup: p=1.0) paradigm. The proportion of EdU+/BrdU+ double-labeled cells (that reentered the cell cycle) also was not significantly altered between 16pdel, control and 16pdup NPCs at 24h (16pdel: p=0.29; 16pdup: p=0.25) or 48h (16pdel: p=0.56; 16pdup: p=0.56; Figures S2B and S2E–S2J). As the proliferation rate of 16pdel and 16pdup NPCs was unchanged compared with controls, it is unlikely to account for the opposing brain size phenotype in human carriers.
16p11.2 CNV carrier-derived neurons show altered morphometric characteristics
We next investigated whether neuron morphology may contribute to brain size. We plated the 16pdel, control and 16pdup NPCs for differentiation into neurons about 4 weeks after neural induction. For visualization, we transduced the cultures with a lentivirus expressing GFP under the neuron-specific synapsin promoter (SynGFP). At 3 weeks post differentiation (wpd), the 16pdel SynGFP+ neurons showed neuronal hypertrophy compared with controls while the soma size of 16pdup neurons was reduced (16pdel: p=3.39×10−5; 16pdup: p=4.1×10−4; Figure S3A and S3B). At 6wpd, the soma size of 16pdel neurons was significantly larger (p=7.72×10−6) while that of 16pdup neurons was significantly smaller (p=6.11×10−12) compared with control neurons (Figures 2A and 2B). This prompted us to assess whether the change in neuronal size is restricted to the soma or is also reflected in other morphological features. Our analyses revealed that 16pdel neurons have increased total dendrite length (p=4.52×10−5) and more extensive dendritic arbors (p<2.2 ×10−16) compared with controls (Figures 2C and 2D). The 16pdup neurons exhibit the opposite phenotype with significantly reduced total dendrite length (p=8.54×10−3) and less complex dendritic arbors, though non-significantly (p=0.88; Figures 2C and 2D). Next, we wanted to confirm whether this difference would be observed in excitatory neurons. We transduced the cells with a lentivirus expressing GFP under the CaMKII-alpha promoter (CaMKIIaGFP) that specifically infects excitatory neurons. We observed a similar reciprocal change in the soma size (16pdel: p=2.18×10−4; 16pdup: p=9.28×10−5) and total dendrite length (16pdel: p=3.96×10−4; 16pdup: p=3.62×10−3). Dendritic complexity of CaMKIIaGFP-transduced 6wpd 16pdel neurons was significantly increased compared with controls (p=1.18×10−3) but that of 16pdup neurons was not significantly decreased (p=0.38; Figures S3C–S3F). The opposing soma size phenotype was robustly maintained through 14wpd (Figure 2E). Quantitative RT-PCR for selected 16p11.2 genes in 6wpd neurons also generally correlated with copy number i.e. decreased in 16pdel carriers and increased in 16pdup carriers (Figure S4A). Thus, neurons generated from 16pdel and 16pdup-derived NPCs, specifically excitatory neurons, manifest overt and opposing differences in soma size and total dendrite length that may contribute to head circumference phenotypes observed in carriers.
Figure 2. 16pdel- and 16pdup-derived neurons have altered neuron morphology.

(A) Representative images of SynGFP transduced neurons with their dendritic arbors, 6wpd. Nuclei counterstained with Hoechst 33342.
(B) Average soma area of SynGFP+ neurons from 16pdel, control and 16pdup lines at 6wpd.§
(C) Total dendrite length of neurons from 16pdel, control and 16pdup lines at 6wpd.§
(D) Sholl analysis of SynGFP+ 16pdel, control and 16pdup neurons at 6wpd.
(E) Representative images of SynGFP+ neurons from 16pdel, control and 16pdup lines at 14wpd. Nuclei counterstained with Hoechst 33342.
Values are mean ± SE. **p<0.01; ****p<0.0005; scale bar, 50 μm. See also Figure S3. §Symbols represent all lines used in the study. See Table S4 for details about lines (3 16pdel; 3 16pdup; 4 controls), clones (1–3/line) and repeated experiments (3–5). See also Figure S3.
Intrinsic excitability of 16pdel carrier-derived neurons corroborates the cell size phenotype
Intrinsic properties of neurons can be impacted by cell size and morphology. Our results showing altered soma size associated with 16p11.2 CNV led us to examine whether this translates to altered electrical properties, and eventually neuron function. We performed patch-clamp electrophysiology at 14wpd on SynGFP+ 16pdel, control and 16pdup neurons. We examined the ability of the 16p11.2 CNV carrier-derived neurons to fire action potentials (APs), the bona fide physiological marker of neurons. When injected with current to evoke APs, 16pdel neurons required nearly twice as much current as control neurons to fire the first AP (p=2.02×10−3; Figures 3A and 3B) and they fired far fewer APs at any particular current step than either of the other two groups (p<2.2 ×10−16; Figures 3A and 3C), demonstrating reduced excitability.
Figure 3. 16pdel neurons exhibit electrophysiological signatures of enlarged cell size.

(A) Example current-clamp recordings from 16pdel, control, and 16pdup neurons indicating action potential (AP) firing in response to current steps of increasing amplitude (20 pA (bottom traces) to 200 pA (top traces), ΔI=20 pA).
(B) Minimal current injected to obtain 1 AP (Rheobase) from a holding potential Vh=−70 mV.§
(C) Number of APs fired in response to a given positive current step from Vh.
(D) Voltage deflections in response to negative current steps from Vh.
(E) Input resistance in gigaohms (gΩ) of iPSC-derived neurons calculated from voltage-clamp recordings.§
(F) Capacitance in picofarad (pF) of GFP+ neurons from 16pdel, control and 16pdup lines.§
(G) In voltage clamp, potassium current density measured from a Vh=−70 mV.
Values are mean ± SE. **p<0.01; ****p<0.0005; ■p<0.1; ns=not significant. § Symbols represent all lines used in the study. See Table S4 for details about lines (3 16pdel; 2 16pdup; 3 controls), clones (1/line) and repeated experiments (2–3).
We wanted to determine whether the increased firing was due to altered AP properties or a decrease in resistive properties of the neurons. The voltage responses of 16pdel neurons were greatly reduced compared with control neurons (p<2.2 ×10−16; Figure 3D), implying reduced membrane resistance. This was confirmed when we calculated input resistance (Rinp), with 16pdel neurons displaying about a twofold decrease in Rinp compared with controls (p=2.33 ×10−2; Figure 3E). Capacitance (Cm), a direct correlate to cell surface area, was higher in 16pdel neurons than in controls (p=4.11×10−3; Figure 3F). Together, these results offer a potential mechanism for the observed deficit in AP firing. The 16pdup neurons, however, did not deviate significantly from controls in their AP firing properties (current at first AP: p=0.45; number of AP: p=0.72; voltage responses: p=0.96; Figures 3A–3D), or their Rin (p=0.44, Figure 3E). They showed a slight, non-significant decrease in Cm (p=0.09) compared with controls (Figure 3F).
Finally, we assayed the current required to step the neurons through a range of voltages and found that all three groups displayed highly nonlinear curves, with strong voltage dependent potassium conductances that onset at approximately the same voltage(Figure 3G). The 16pdel neurons did not differ significantly from controls in potassium leak currents (p=0.9). However, the curve for 16pdup cells was significantly steeper (p=2.2×10−4), indicating an increased potassium current density at positive voltages. Interestingly, this suggests that 16pdup neurons may compensate for their reduced somatic size by increasing outward potassium current to stabilize intrinsic excitability (Figure 3G).
Thus, the larger soma size observed in 16pdel neurons is associated with functional properties while the reciprocal phenotype observed in 16pdup neurons did not manifest in altered intrinsic properties. This may suggest that the processes establishing functional excitability, or preserving it in the face of aberrant morphological development, are more sensitive to reduction than to excess for proteins encoded by 16p11.2 genes.
Altered synaptic development in 16p11.2 CNV carrier-derived neurons
Previous studies using iPSC-derived neurons have indicated altered synaptogenesis as a likely cause for cognitive deficits in NDDs(Habela et al., 2016). The 16pdel, control and 16pdup neurons did not show significant differences in their maturation in our differentiation paradigm (Figures S4B and 4C). To assess whether the morphological changes in the 16pdel and 16pdup neurons affect synapse development, we analyzed the expression of the presynaptic protein synapsin1 (SYN1) and postsynaptic marker homer1 (HOM1) at 6wpd (Figures 4A–A′). Interestingly, we found lower density of SYN1+HOM1+ punctae, representing synapses, in both SynGFP transduced 16pdel- and 16pdup-derived 6wpd neurons compared with controls (16pdel: p=6.17×10−12; 16pdup: p=4.17×10−12; Figure 4C). Further, co-localization of punctae containing the excitatory presynaptic marker VGLUT2 and the postsynaptic scaffolding protein PSD95 revealed a lower density of excitatory synapses on the SynGFP transduced 16pdel and 16pdup neurons compared with controls (16pdel: p=7.31×10−6; 16pdup: p=3.53×10−4; Figures 4B–B′ and 4D). When we transduced the cells with CaMKIIaGFP to specifically measure synaptic density of excitatory neurons, the density of VGLUT2+PSD95+ punctae was also reduced in both 16pdel and 16pdup neurons compared with controls (16pdel: p=1.24×10−10; 16pdup: p=1.99×10−11; Figure S4D–D′ and S4E). The lower synaptic density may ultimately result in altered network activity and behavioral symptoms found in both 16pdel and 16pdup carriers.
Figure 4. 16pdel neurons display reduction in synaptic density.

(A) High magnification images of SynGFP+ dendrites of 16pdel, control and 16pdup neurons, 6wpd stained for GFP (green), presynaptic marker SYN1 (red) and postsynaptic marker HOM1 (white). A′ depicts SYN1+HOM1+ co-localized punctae (yellow arrows).
(B) High magnification images of SynGFP+ dendrites of 16pdel, control and 16pdup neurons, 6wpd stained for GFP (green), presynaptic marker VGLUT2 (red) and postsynaptic marker PSD95 (white). B′ depicts VGLUT2+PSD95+ co-localized punctae (yellow arrows).
(C) Synaptic density quantified as number of SYN1+HOM1+ punctae/dendrite divided by total dendrite length.§
(D) Density of excitatory synapses quantified as number of VGLUT2+PSD95+ punctae/dendrite divided by total dendrite length§
Values are mean ± SE. ****p<0.0005; scale bar, 10μm. §Symbols represent all lines used in the study. See Table S4 for details about lines (3 16pdel; 3 16pdup; 4 controls), clones (1–2/line) and repeated experiments (2–3). See also Figure S4.
To test whether the reduced excitatory synaptic density is accompanied by a decrease in synaptic function, we recorded miniature excitatory postsynaptic currents (mEPSCs) in 8–9wpd CaMKIIaGFP+ neurons. We did not observe a significant difference in the kinetics (16pdel: p0.29; 16pdup: p=0.88) or frequency (16pdel: p=0.46; 16pdup: p=0.33) of the mEPSCs in 16pdel and 16pdup neurons compared with controls (Figures S4F and S4H). But, the amplitude of mEPSCs was significantly increased in both 16pdel and 16dup neurons (16pdel: p<2.2×10−16; 16pdup: p<2.2×10−16; Figure S4G and S4H). Thus, it appears that although there are fewer excitatory synapses in 16pdel and 16pdup neurons, they have increased synaptic strength.
Discussion
The 16p11.2 CNV is one of the common genetic etiologies of NDDs. Yet, molecular and cellular mechanisms underlying carrier phenotypes remain largely unclear. Traditionally, mouse models are used to study human NDDs and delineate disease mechanisms. However, 16pdel and 16dup mice do not faithfully recapitulate human phenotypes: 16pdel mice exhibit reduced brain volume while 16pdel carriers have macrocephaly, opposite of the human conditions(Pucilowska et al., 2015; Qureshi et al., 2014). Hence, we thought using carrier-derived iPSCs might prove more informative in recapitulating the human disorder. Indeed, using 16pdel and 16pdup iPSC-derived neurons, we identified reciprocal cellular phenotypes that may underlie opposing brain size differences in 16pdel and 16pdup carriers as well as other observations. We have uncovered cell growth as a potential undescribed mechanism for microcephaly. Contrary to morphological phenotypes, we identified similar reduction in synapse density that may lead to similarities in human clinical outcomes.
With respect to opposing brain size differences, neuroimaging studies in 16p11.2 CNV carriers have shown significant effects on gray matter volume, specifically increase in the cortical surface area in 16pdel and reciprocal decrease in 16pdup carriers(Qureshi et al., 2014). Early developmental processes such as i) progenitor proliferation, ii) cell growth, or iii) differentiation can influence cortical surface area by altering cell number, soma size or morphology(Bizzotto and Francis, 2015; Gilmore and Walsh, 2013). In our studies, proliferation appeared not to be altered in 16pdel and 16pdup NPCs. However, contrasting effects on soma size and dendrite length found in 16p11.2 CNV carrier-derived neurons could be a major contributor to overall brain size. Hence, our data suggest that cell growth-related pathways are influenced by the 16p11.2 CNV.
The two important pathways known to affect cell growth are the PI3K/AKT and the Ras/MAPK pathways. They are interconnected and can co-regulate one another. Neuron size typically positively correlates with dendrite growth. Activation of PI3K or its downstream effector, AKT, is sufficient to increase soma size and dendritic complexity in mouse hippocampal neurons(Kumar et al., 2005). In humans, dysregulation of the PI3K/AKT pathway due to mutation of PTEN, an inhibitor of the pathway, is known to cause macrocephaly, at least in part mediated by neuronal hypertrophy(Kwon et al., 2001; Zhou et al., 2009). PTEN-associated macrocephaly is strongly associated with development of ASD(Varga et al., 2009). Similarly, loss of TSC2, which inhibits mTOR, an effector of the PI3K/AKT pathway, often results in macrocephaly associated with ASD, ID and seizures. Loss of TSC2 in human iPSC-derived neurons results in increased soma size, dendritic arborization and total dendrite length (similar to our observations in 16pdel) mediated by a hyperactive PI3K/AKT pathway(Costa et al., 2016). The 16p11.2 region harbors genes encoding proteins that are known to interact with the PI3K/AKT or Ras/MAPK pathway, hence it may affect cellular phenotypes in a dosage-dependent manner via this interaction. For example, MVP binds PTEN and functions as a scaffold protein for ERK, encoded by MAPK3, and hence may regulate the PI3K/AKT and Ras/MAPK pathways(Kolli et al., 2004; Yu et al., 2002). Another candidate, TAOK2, activates the Ras/MAPK pathway and has been shown to regulate basal dendrite formation in cortical neurons(Calderon de Anda et al., 2012). Mutations in the Ras/MAPK pathway are known to cause NDDs (RASopathies) and have macrocephaly as one of the clinical symptoms(Rauen, 2013). Neurons generated from RASopathy (Costello syndrome) subject-derived iPSCs show a significant increase in soma size, consistent with over-expression of Ras. However, the study reported a reduction in neurite length, which is contradictory to Ras hyperactivation(Rooney et al., 2016). Thus, it will be important to study the role and regulation of ERK and AKT signaling in neuronal growth parameters in human models. Interestingly, KCTD13, a 16p11.2 gene reported to cause dose-dependent macrocephaly in zebrafish, was shown to do so via proliferation(Golzio et al., 2012), not soma size or dendrite morphology as we observe in our human iPSC model.
In contrast to 16pdel, 16pdup neurons showed a significant reduction in soma size and total dendrite length, with no decrease in dendritic complexity. Causes of primary microcephaly have been reported to involve aberrant cellular processes like reduced neuron number, premature neurogenesis and increased apoptosis(Bizzotto and Francis, 2015). Known causative genes belong to centrosome-associated, cell division or DNA damage repair pathways. Interestingly, the targeted deletion of one allele of the kinase Dyrk1a in mice resulted in decreased brain volume, and the authors observed reduced neuronal size and dendritic complexity, similar to our observations in 16pdup neurons(Benavides-Piccione et al., 2005; Fotaki et al., 2002). This kinase is involved in regulation of the PI3K/AKT and Ras/MAPK pathways and hence it is likely that its disruption may result in aberrant neuronal differentiation via dysregulation of these pathways(Ji et al., 2015). However, in humans, microcephaly has not previously been linked with somatic growth defects(Gilmore and Walsh, 2013). In this light, our results are interesting because – i) they present a mechanism for microcephaly, ii) although known single-gene causes for macro- and microcephaly are not currently overlapping, it appears plausible that the opposing brain size phenotype in 16pdel and 16pdup may be driven by same gene(s) in the 16p11.2 CNV in a dosage-dependent manner.
Altered neuron morphology is likely to affect neuron function. In line with previous studies, patch clamp recordings from 16pdel neurons showed intrinsic electrical properties consistent with their larger size, i.e., increased Cm, decreased Rinp and reduced excitability(Costa et al., 2016; Williams et al., 2015). Based on increased dendrite length of 16pdel neurons, we would predict increased number of synapses on the dendrites. Our study, however, revealed a significant decrease in excitatory synaptic density in 16pdel neurons, suggesting that distinct mechanisms underlie cell growth and synaptogenesis. Reduced synaptic density, predominantly excitatory, has been demonstrated in several iPSC studies modeling NDDs(Russo et al., 2015) such as Rett syndrome and tuberous sclerosis. In fact, the TSC2 deficient iPSC-derived neurons show neuronal and synaptic alterations(Costa et al., 2016) that closely resemble those we found in 16pdel neurons. This suggests that 16p11.2 genes may regulate (or dysregulate) the same synaptic pathways that are impaired in other NDDs, implying shared pathophysiological mechanisms. The increased mEPSC amplitude in 16pdel indicates that these neurons may compensate for the reduced number of synapses by increasing the strength of excitatory synapses using homeostatic plasticity mechanisms(Turrigiano and Nelson, 2004). However, further careful analyses of synaptic function and maturation are warranted.
Although we observed that reduced soma size was corroborated by a trend of reduced Cm in 16pdup neurons, other intrinsic excitability parameters such as AP firing or Rinp, did not deviate from controls. This suggests either a compensation for morphological changes or a subtle phenotype that would not be readily observed in our model system. For example the upregulation in potassium conductance may serve to preserve a normal amount of potassium flux, which helps preserve excitability, indicative of compensation by 16pdup neurons for their reduced size. But consistent with the synaptic defects in 16pdel neurons, 16pdup neurons showed a reduced synaptic density and increased mEPSC amplitude. Thus, although 16pdup neurons may retain the capacity to rebalance membrane conductance to preserve excitability, they exhibit substantial deficits in synaptic connectivity and function.
The more evident cellular phenotypes observed in 16pdel neurons compared to 16pdup neurons may result from the more dramatic fold-change in gene expression for deletion (0.5X) compared with duplication (1.5X) CNVs. Alternatively, it may result from under-expression generally being more impactful than over-expression, as having less of a critical protein could be more difficult to overcome than excess protein (e.g. if other limiting factors exist). This pattern may underlie the failure of the hypertrophic 16pdel neurons to functionally compensate for increased size (e.g. AP firing), resulting in striking physiological defects that affect formation and function of neural networks. The hypotrophic 16pdup neurons, on the other hand, are capable of homeostatically regulating their excitability. These findings at the cellular level are consistent with clinical features, given that neurological symptoms, although incompletely penetrant, are more common among 16pdel carriers than among 16pdup carriers, who often show milder or no clinical features(Golzio and Katsanis, 2013; Steinman et al., 2016). Moreover, the cellular phenotypes were extremely consistent in each group (across individuals and clones), supporting the robustness of our conclusions despite inter-individual variation. In summary, our iPSC-derived model of 16p11.2 CNVs provides evidence of causal neuronal and synaptic phenotypes that might contribute to neuropathology associated with 16p11.2 CNVs, are present in a system amenable to manipulation and, in future studies, will be important in establishing a gene/pathway-phenotype correlation.
Experimental Procedures
16p11.2 CNV carriers
Skin fibroblasts of three 16p11.2 deletion and three duplication carriers were obtained from Simons Variation in Individuals Project (VIP) and used for reprogramming to iPSCs(The Simons VIP Consortium, 2012). Fibroblasts of control individuals were obtained by punch biopsies using published protocols(Rooney et al., 2016). The control line BJ4 was a kind gift from the Hsiao lab(Matsumoto et al., 2013). Carrier and control lines are described in Supplemental tables. All human research was performed in accordance with Institutional Review Board and Committee on Human Research codes of practice(IRB protocol#: 10-03304, 10-01426, 10-02794).
Generation of iPSCs using episomal vectors
Fibroblasts were cultured in DMEM with 10% FBS(Life Technologies) and reprogramed to iPSCs with episomal plasmids pCXLE-hOct3/4-shp53-F, pCXLE-hSox2-Klf4, pCXLE-hcmyc-Lin28 (Addgene) using published protocols (Okita et al., 2011). Briefly, cells were electroporated with 3μg plasmid mixture using Neon transfection device (Invitrogen) according to the manufacturer protocol (1650V, 10ms width, and 3 pulses). Transfected cells were plated on mitomycin C-treated mouse embryonic fibroblasts (Millipore; MEFs) in human embryonic stem (hES) media: Knockout DMEM/F-12, 1x Knockout Serum Replacement, 1% Non-essential amino acids, 1% Penicillin/Streptomycin (Life Technologies) with bFGF. The iPSC colonies were picked manually after 2 weeks, transferred to MEF feeders in hES media. After a few passages on MEFs, iPSCs were transferred to Matrigel (BD Biosciences)-coated plates and maintained in mTeSR-1 media (Stem Cell Technologies). All experiments were performed with 2–3 independent iPSC clones per line.
Characterization of iPSCs from 16p11.2 CNV carriers
Induced PSCs were stained for nanog, OCT4, SOX2 and SSEA-4. For pluripotency analysis, iPSCs were allowed to differentiate spontaneously in mTeSR-1 media or randomly differentiated using the Human Pluripotent Stem Cell Functional Identification Kit (#SC027; R&D Systems) and stained for markers of the three germ layers. All iPSCs generated cells of all three germ layers, assessed by positive immunoreactivity for β-III-tubulin for ectoderm, smooth muscle actin for mesoderm and SOX17 for endoderm. For the array CGH, DNA was extracted using the QIAamp DNA Blood Mini kit (Qiagen) according to manufacturer’s protocol and eluted in nuclease-free water. Eluted DNA was processed for analysis by Illumina CytoSNP 850K array per manufacturer protocol. Data was processed and CNV calls generated with BlueFuse Multi Software v4.2 (Illumina). CNV calls by SNP array were reported using standard clinical thresholds in the UCSF Clinical Cytogenetics Laboratory, including annotation of any CNVs >200kb. Interpretation of clinical significance was done per standard American College of Medical Genetics guidelines (Kearney et al., 2011). See Table S2 for array CGH results.
Statistical Analyses
All results are represented as bar or line plots using ggplot2 library in R 3.2.0 environment(r-project.org). Bar plots show means ± standard error (SE) for 16pdel, control and 16pdup groups. Symbols represent all lines used in the study. Statistical significance between the groups were assessed with the Mann-Whitney Wilcoxon Signed-Rank test with wilcox.test() function in R. Line plots show means ± SE for each group for repeated measurements. Statistical significance was calculated using repeated measures ANOVA with post hoc Tukey’s HSD correction using aov() and TukeyHSD() function in R.
Supplementary Material
Document S1. Figures S1–S4, supplemental experimental procedures and Tables S1–3. Supplemental Table S4. Summary of cell lines used per experiment and summary of number of cells of each genotype analyzed.
Highlights.
Cellular model of NDDs established using hiPSCs from 16pdel and 16pdup carriers.
Opposing neuronal size phenotype may model macro- and microcephaly.
Synaptic impairment in 16pdel and 16pdup neurons may reflect behavioral features.
Functional compensation observed in 16pdup compared with 16pdel neurons.
Acknowledgments
We thank Curtis Tom, Federico Mendoza-Camacho, Cheuk Tam, John Paul Kwok, Tracy Hyunh, and Shriya Perati for their technical assistance. We also thank Dr. Edward Hsaio for kindly sharing the BJ4 iPSC line and Dr. Michela Traglia for her expertise in using the R software. We thank Michael Ho of the UCSF Clinical Cytogenetics Laboratory for processing samples for array-CGH. We are grateful to all families participating in the Simons VIP. We appreciate access to phenotypic data on SFARI Base. This work was supported by a grant from the Simons Foundation (SFARI 345471, L.A.W.). This work was funded by NIH DP2 OD007449, NIH R21 MH105745, CIRM Bridges grant to CCSF and IMHRO(L.A.W.). A.D. was supported by the Autism Speaks Meixner Postdoctoral Fellowship in Translational Research. S.Y. was supported by the NIH ‘Pathway to Independence’ grant 5K99MH108648-02. D.Q.D. and K.C.H. were funded by the National Institutes of Health T32 Training Grant EY007120. E.M.U. was funded by Research to Prevent Blindness Walt and Lilly Disney Award for Amblyopia Research.
Footnotes
Author Contributions
Conceptualization, A.D. and L.A.W; Methodology, A.D., L.A.W., S.Y. D.Q.D. and E.M.U.; Investigation, A.D., S.Y. D.Q.D., Z-Y.W., K.C.H., M.K.C. and A.P.W.; Writing – Original Draft, L.A.W., A.D.; Writing –Review & Editing, L.A.W., A.D., S.Y. D.Q.D., Z-Y.W., K.C.H., E.M.U, Y-N.J.; Funding Acquisition, L.A.W., E.M.U, Y-N.J.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. Arlington, VA: Americal Psychiatric Publishing; 2013. [Google Scholar]
- Benavides-Piccione R, Dierssen M, Ballesteros-Yáñez I, Martínez de Lagrán M, Arbonés ML, Fotaki V, DeFelipe J, Elston GN. Alterations in the phenotype of neocortical pyramidal cells in the Dyrk1A+/− mouse. Neurobiol Dis. 2005;20:115–122. doi: 10.1016/j.nbd.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Bernier R, Golzio C, Xiong B, Stessman HA, Coe BP, Penn O, Witherspoon K, Gerdts J, Baker C, Vulto-van Silfhout AT, et al. Disruptive CHD8 Mutations Define a Subtype of Autism Early in Development. Cell. 2014;158:263–276. doi: 10.1016/j.cell.2014.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bizzotto S, Francis F. Morphological and functional aspects of progenitors perturbed in cortical malformations. Front Cell Neurosci. 2015;9:30. doi: 10.3389/fncel.2015.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal I, Ragavendran A, Erdin S, Klei L, Sugathan A, Guide JR, Manavalan P, Zhou JQ, Wheeler VC, Levin JZ, et al. Transcriptional consequences of 16p11.2 deletion and duplication in mouse cortex and multiplex autism families. Am J Hum Genet. 2014;94:870–883. doi: 10.1016/j.ajhg.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, Miles JH, Wang CH, Stratton R, Pilarski R, et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42:318–321. doi: 10.1136/jmg.2004.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon de Anda F, Rosario AL, Durak O, Tran T, Gräff J, Meletis K, Rei D, Soda T, Madabhushi R, Ginty DD, et al. Autism spectrum disorder susceptibility gene TAOK2 affects basal dendrite formation in the neocortex. Nat Neurosci. 2012;15:1022–1031. doi: 10.1038/nn.3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa V, Aigner S, Vukcevic M, Sauter E, Behr K, Ebeling M, Dunkley T, Friedlein A, Zoffmann S, Meyer CA, et al. mTORC1 Inhibition Corrects Neurodevelopmental and Synaptic Alterations in a Human Stem Cell Model of Tuberous Sclerosis. Cell Rep. 2016;15:86–95. doi: 10.1016/j.celrep.2016.02.090. [DOI] [PubMed] [Google Scholar]
- Fotaki V, Dierssen M, Alcántara S, Martínez S, Martí E, Casas C, Visa J, Soriano E, Estivill X, Arbonés ML. Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol Cell Biol. 2002;22:6636–6647. doi: 10.1128/MCB.22.18.6636-6647.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore EC, Walsh CA. Genetic causes of microcephaly and lessons for neuronal development. Wiley Interdiscip Rev Dev Biol. 2013;2:461–478. doi: 10.1002/wdev.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golzio C, Katsanis N. Genetic architecture of reciprocal CNVs. Curr Opin Genet Dev. 2013;23:240–248. doi: 10.1016/j.gde.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golzio C, Willer J, Talkowski ME, Oh EC, Taniguchi Y, Jacquemont S, Reymond A, Sun M, Sawa A, Gusella JF, et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 2012;485:363–367. doi: 10.1038/nature11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groszer M, Erickson R, Scripture-Adams DD, Lesche R, Trumpp A, Zack JA, Kornblum HI, Liu X, Wu H, Gimm O, et al. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science. 2001;294:2186–2189. doi: 10.1126/science.1065518. [DOI] [PubMed] [Google Scholar]
- Habela CW, Song H, Ming GL. Modeling synaptogenesis in schizophrenia and autism using human iPSC derived neurons. Mol Cell Neurosci. 2016;73:52–62. doi: 10.1016/j.mcn.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homem CCF, Repic M, Knoblich JA. Proliferation control in neural stem and progenitor cells. Nat Rev Neurosci. 2015;16:647–659. doi: 10.1038/nrn4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji J, Lee H, Argiropoulos B, Dorrani N, Mann J, Martinez-Agosto JA, Gomez-Ospina N, Gallant N, Bernstein JA, Hudgins L, et al. DYRK1A haploinsufficiency causes a new recognizable syndrome with microcephaly, intellectual disability, speech impairment, and distinct facies. Eur J Hum Genet. 2015;23:1473–1481. doi: 10.1038/ejhg.2015.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med. 2011;13:680–685. doi: 10.1097/GIM.0b013e3182217a3a. [DOI] [PubMed] [Google Scholar]
- Kolli S, Zito CI, Mossink MH, Wiemer EAC, Bennett AM. The major vault protein is a novel substrate for the tyrosine phosphatase SHP-2 and scaffold protein in epidermal growth factor signaling. J Biol Chem. 2004;279:29374–29385. doi: 10.1074/jbc.M313955200. [DOI] [PubMed] [Google Scholar]
- Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA, Gilliam TC, Nowak NJ, Cook EH, Dobyns WB, et al. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 2008;17:628–638. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- Kumar V, Zhang MX, Swank MW, Kunz J, Wu GY. Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J Neurosci. 2005;25:11288–11299. doi: 10.1523/JNEUROSCI.2284-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, Eberhart CG, Burger PC, Baker SJ. Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet. 2001;29:404–411. doi: 10.1038/ng781. [DOI] [PubMed] [Google Scholar]
- Luo R, Sanders SJ, Tian Y, Voineagu I, Huang N, Chu SH, Klei L, Cai C, Ou J, Lowe JK, et al. Genome-wide transcriptome profiling reveals the functional impact of rare de novo and recurrent CNVs in autism spectrum disorders. Am J Hum Genet. 2012;91:38–55. doi: 10.1016/j.ajhg.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra D, Sebat J. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell. 2012;148:1223–1241. doi: 10.1016/j.cell.2012.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto Y, Hayashi Y, Schlieve CR, Ikeya M, Kim H, Nguyen TD, Sami S, Baba S, Barruet E, Nasu A, et al. Induced pluripotent stem cells from patients with human fibrodysplasia ossificans progressiva show increased mineralization and cartilage formation. Orphanet J Rare Dis. 2013;8:190. doi: 10.1186/1750-1172-8-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy SE, Makarov V, Kirov G, Addington AM, McClellan J, Yoon S, Perkins DO, Dickel DE, Kusenda M, Krastoshevsky O, et al. Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet. 2009;41:1223–1227. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, Sharp AJ, Baker C, Itsara A, Jiang Z, Buysse K, Huang S, Maloney VK, Crolla JA, Baralle D, et al. Recurrent Rearrangements of Chromosome 1q21.1 and Variable Pediatric Phenotypes. N Engl J Med. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliavacca E, Golzio C, Männik K, Blumenthal I, Oh EC, Harewood L, Kosmicki JA, Loviglio MN, Giannuzzi G, Hippolyte L, et al. A Potential Contributory Role for Ciliary Dysfunction in the 16p11.2 600 kb BP4-BP5 Pathology. Am J Hum Genet. 2015;96:784–796. doi: 10.1016/j.ajhg.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milone R, Valetto A, Battini R, Bertini V, Valvo G, Cioni G, Sicca F. Focal cortical dysplasia, microcephaly and epilepsy in a boy with 1q21.1–q21.3 duplication. Eur J Med Genet. 2016;59:278–282. doi: 10.1016/j.ejmg.2016.03.003. [DOI] [PubMed] [Google Scholar]
- Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, Hong H, Nakagawa M, Tanabe K, Tezuka K, et al. A more efficient method to generate integration-free human iPS cells. Nat Methods. 2011;8:409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- Pucilowska J, Vithayathil J, Tavares EJ, Kelly C, Karlo JC, Landreth GE. The 16p11.2 deletion mouse model of autism exhibits altered cortical progenitor proliferation and brain cytoarchitecture linked to the ERK MAPK pathway. J Neurosci. 2015;35:3190–3200. doi: 10.1523/JNEUROSCI.4864-13.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi AY, Mueller S, Snyder AZ, Mukherjee P, Berman JI, Roberts TPL, Nagarajan SS, Spiro JE, Chung WK, Sherr EH, et al. Opposing brain differences in 16p11.2 deletion and duplication carriers. J Neurosci. 2014;34:11199–11211. doi: 10.1523/JNEUROSCI.1366-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauen KA. The RASopathies. Annu Rev Genomics Hum Genet. 2013;14:355–369. doi: 10.1146/annurev-genom-091212-153523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney GE, Goodwin AF, Depeille P, Sharir A, Schofield CM, Yeh E, Roose JP, Klein OD, Rauen KA, Weiss LA, et al. Human iPS Cell-Derived Neurons Uncover the Impact of Increased Ras Signaling in Costello Syndrome. J Neurosci. 2016;36:142–152. doi: 10.1523/JNEUROSCI.1547-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld JA, Ballif BC, Torchia BS, Sahoo T, Ravnan JB, Schultz R, Lamb A, Bejjani BA, Shaffer LG. Copy number variations associated with autism spectrum disorders contribute to a spectrum of neurodevelopmental disorders. Genet Med. 2010;12:694–702. doi: 10.1097/GIM.0b013e3181f0c5f3. [DOI] [PubMed] [Google Scholar]
- Russo FB, Cugola FR, Fernandes IR, Pignatari GC, Beltrão-Braga PCB. Induced pluripotent stem cells for modeling neurological disorders. World J Transplant. 2015;5:209–221. doi: 10.5500/wjt.v5.i4.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinawi M, Liu P, Kang SHL, Shen J, Belmont JW, Scott DA, Probst FJ, Craigen WJ, Graham BH, Pursley A, et al. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J Med Genet. 2010;47:332–341. doi: 10.1136/jmg.2009.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman KJ, Spence SJ, Ramocki MB, Proud MB, Kessler SK, Marco EJ, Green Snyder L, D’Angelo D, Chen Q, Chung WK, et al. 16p11.2 deletion and duplication: Characterizing neurologic phenotypes in a large clinically ascertained cohort. Am J Med Genet Part A. 2016 doi: 10.1002/ajmg.a.37820. [DOI] [PubMed] [Google Scholar]
- Tai DJC, Ragavendran A, Manavalan P, Stortchevoi A, Seabra CM, Erdin S, Collins RL, Blumenthal I, Chen X, Shen Y, et al. Engineering microdeletions and microduplications by targeting segmental duplications with CRISPR. Nat Neurosci. 2016;19:517–522. doi: 10.1038/nn.4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Simons VIP Consortium. Simons Variation in Individuals Project (Simons VIP): A Genetics-First Approach to Studying Autism Spectrum and Related Neurodevelopmental Disorders. Neuron. 2012;73:1063–1067. doi: 10.1016/j.neuron.2012.02.014. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Varga EA, Pastore M, Prior T, Herman GE, McBride KL. The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genet Med. 2009;11:111–117. doi: 10.1097/GIM.0b013e31818fd762. [DOI] [PubMed] [Google Scholar]
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- Williams MR, DeSpenza T, Li M, Gulledge AT, Luikart BW, Luikart BW. Hyperactivity of newborn Pten knock-out neurons results from increased excitatory synaptic drive. J Neurosci. 2015;35:943–959. doi: 10.1523/JNEUROSCI.3144-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav S, Oses-Prieto JA, Peters CJ, Zhou J, Pleasure SJ, Burlingame AL, Jan LY, Jan YN. TAOK2 Kinase Mediates PSD95 Stability and Dendritic Spine Maturation through Septin7 Phosphorylation. Neuron. 2017;93:379–393. doi: 10.1016/j.neuron.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Fotouhi-Ardakani N, Wu L, Maoui M, Wang S, Banville D, Shen SH. PTEN associates with the vault particles in HeLa cells. J Biol Chem. 2002;277:40247–40252. doi: 10.1074/jbc.M207608200. [DOI] [PubMed] [Google Scholar]
- Zhang SC, Wernig M, Duncan ID, Brüstle O, Thomson JA. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat Biotechnol. 2001;19:1129–1133. doi: 10.1038/nbt1201-1129. [DOI] [PubMed] [Google Scholar]
- Zhou J, Blundell J, Ogawa S, Kwon CH, Zhang W, Sinton C, Powell CM, Parada LF. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci. 2009;29:1773–1783. doi: 10.1523/JNEUROSCI.5685-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Figures S1–S4, supplemental experimental procedures and Tables S1–3. Supplemental Table S4. Summary of cell lines used per experiment and summary of number of cells of each genotype analyzed.