

An aqueous 1:1 co-crystal salt formed from imidazole and picric acid was obtained in methanol solution. A three-dimensional hydrogen-bonded network is formed in the crystal by N—H⋯O, O—H⋯O and C—H⋯O hydrogen bonds and is further consolidated by π–π stacking interactions between pairs of imidazolium cations and picrate anions.
Keywords: crystal structure, co-crystal salt, imidazole, picric acid
Abstract
The asymmetric unit of the title co-crystal salt, 1H-imidazol-3-ium 2,4,6-trinitrophenolate monohydrate, C4H7N2 +·C6H2N3O7 −·H2O, contains one imidazolium cation, one picrate anion and one solvent water molecule of crystallization. The phenolic proton has been transferred to an imidazole N atom. In the crystal, the components are linked by N—H⋯O and O—H⋯O hydrogen bonds into a three-dimensional network which is further consolidated by weak C—H⋯O hydrogen bonds. In addition, π–π stacking interactions occur between pairs of imidazolium cations and picrate anions. If only the classical N—H⋯O and O—H⋯O hydrogen bonds are considered, the component ions are linked into a three-dimensional threefold interpenetrating network of the topological type utp [or (10,3)-d]. Hirshfeld surface analysis indicates the crystal structure is mainly stabilized by H⋯·O contacts of the hydrogen bonds.
Chemical context
Co-crystallization, which is the crystallization of more than one solid component into a new compound, is widely involved in the research fields of active pharmaceuticals (Aitipamula et al., 2015 ▸; Weyna et al., 2012 ▸; Robinson, 2010 ▸; Arenas-García et al., 2010 ▸) and crystal engineering (Manoj et al., 2014 ▸). Imidazole is an often used intermediate in pharmaceutical and chemical synthesis. Its crystallization characteristics can facilitate organic synthesis and theoretical prediction. Picric acid is a strong organic proton-donating reagent which can favor the crystallization of some basic organic complexes. By controlling one specific crystallization condition such as solvent, temperature, pressure or molar ratio of the raw materials, some polymorphs can be obtained with the same ingredients. For instance, two imidazolium picrate co-crystal salts have been reported that were crystallized from chloroform (Soriano-García et al., 1990 ▸) or dry acetonitrile (Moreno-Fuquen et al., 2011 ▸). The crystal packing in these two analogs is completely different because of their different stoichiometric compositions. In order to further research the factors affecting this crystallization process, the crystallization solvent has been adjusted to be methanol (95%). Interestingly, some yellow needle-shaped crystals were obtained after two days on the side of the vessel and when the solvent had almost evaporated, several yellow block-shaped crystals formed at the bottom of the vessel (Fig. 1 ▸
a and 1b). The results of X-ray diffraction indicates that the structure of the block-shaped crystals is the same as that reported by Moreno-Fuquen et al. (2011 ▸). Herein, the crystal structure of the needle-shaped crystals is reported.
Figure 1.

The morphologies of the two molecular salts: needle (a) of (I) and block (b) of the crystal structure reported by Moreno-Fuquen et al. (2011 ▸).
Structural commentary
The asymmetric unit of the title compound (I) contains one imidazolium cation, a picrate anion and one solvent water molecule of crystallization (Fig. 2 ▸). The phenolic proton has been transferred to a imidazole nitrogen atom, forming the solvated 1:1 co-crystal salt. In the picrate anion of (I), the C—Ophenol bond distance is 1.250 (2) Å, which is shorter by ca 0.08 Å than the value of 1.33 (2) Å in the protonated species (Bertolasi et al., 2011 ▸). Also, the two neighboring C—C bonds [1.447 (3) Å for C1—C2 and C1—C6], are significantly different from those in a benzene ring with delocalized C C bonds. The C2—C1—C6 angle [110.56 (16)°] is smaller by ca 11.3° than the averaged value of the other five ring inner angles [121.8 (1)°]. This deviation of bonds and angles can mainly be attributed to the electron-withdrawing effects of the three nitro groups, which can delocalize the negative charge on the phenolate O1 atom over the whole π-conjugated system. The nitro groups, N1/O2/O3, N2/O4/O5 and N3/O6/O7, are twisted from the central benzene ring by dihedral angles of 43.3 (3), 4.2 (3) and 48.5 (3)°, respectively. In the imidazolium cation, the C7—N4 [1.329 (3) Å] and C7—N5 [1.331 (3) Å] bond distances are the same due to the delocalizing effect and similar to those observed in other co-crystal salts (Soriano-García et al., 1990 ▸; Moreno-Fuquen et al., 2011 ▸).
Figure 2.

Molecular structure of (I), showing the atom-numbering scheme. Displacement ellipsoids are drawn at the 50% probability level.
Supramolecular features
In the crystal of (I), the three components are linked into a three-dimensional network by N—H⋯O and O—H⋯O hydrogen bonds (Table 1 ▸, Fig. 3 ▸). In order to understand the structure simply, we can analyze it in the terms below. Firstly, the imidazolium cations, picrate anions and water molecules are linked by each three N—H⋯O and three O—H⋯O hydrogen bonds, forming a three-dimensional framework structure (Fig. 3 ▸; Spek, 2003 ▸, 2009 ▸). It is worthy mentioning that if both the water molecule and the picrate anion are regarded as 3-connected nodes by hydrogen-bonding and the imidazole cation as a 2-connected node, then the three-dimensional framework can be viewed topologically as a 3-connected utp network with a short Schläfli symbol of (103)-d (Blatov et al., 2014 ▸; Baburin & Blatov, 2007 ▸) (Fig. 3 ▸). Secondly, the three-dimensional hydrogen-bonded framework is consolidated by π–π interactions between pairs of imidazolium cations and picrate anions, both with centroid-to-centroid distances of 3.553 (4) Å, and weak intermolecular C—H⋯O interactions (Table 1 ▸). It should be mentioned that the short O2⋯O7( − x, y −
− x, y −  , + z) contact of 2.837 (4) Å may be the result of an inclined NO2⋯π(NO2) interaction (Daszkiewicz, 2013 ▸)
, + z) contact of 2.837 (4) Å may be the result of an inclined NO2⋯π(NO2) interaction (Daszkiewicz, 2013 ▸)
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N4—H4⋯O8i | 0.93 (3) | 1.83 (3) | 2.763 (3) | 172 (3) |
| N5—H5A⋯O1ii | 0.89 (3) | 1.94 (3) | 2.812 (2) | 165 (3) |
| N5—H5A⋯O3ii | 0.89 (3) | 2.46 (3) | 2.956 (3) | 116 (2) |
| O8—H8A⋯O4iii | 0.82 (4) | 2.29 (4) | 3.034 (2) | 151 (3) |
| O8—H8B⋯O1ii | 0.88 (4) | 2.04 (4) | 2.899 (2) | 165 (3) |
| O8—H8B⋯O6ii | 0.88 (4) | 2.44 (4) | 2.943 (3) | 117 (3) |
| C5—H5⋯O2iv | 0.95 | 2.49 | 3.383 (3) | 157 |
| C7—H7⋯O5v | 0.95 | 2.37 | 3.187 (3) | 144 |
| C8—H8⋯O4vi | 0.95 | 2.44 | 3.188 (3) | 135 |
| C8—H8⋯O8 | 0.95 | 2.53 | 3.255 (3) | 133 |
| C9—H9⋯O7vii | 0.95 | 2.48 | 3.349 (3) | 152 |
Symmetry codes: (i)  ; (ii)
; (ii)  ; (iii)
; (iii)  ; (iv)
; (iv)  ; (v)
; (v) 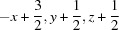 ; (vi)
; (vi)  ; (vii)
; (vii)  .
.
Figure 3.

Part of the crystal structure of (I), showing (a) the formation of the three-dimensional hydrogen-bonded network by N—H⋯O and O—H⋯O hydrogen bonds as dashed lines, (b) the simplified network when picrate, water and imidazolium ions are considered as 3-, 3- and 2-connected nodes, respectively, and (c) the simplified utp network.
Hirshfeld surface analysis
An alternative way to asses the intermolecular interactions quantitatively around one specific molecule is through Hirshfeld surface analysis (Wolff et al., 2012 ▸; McKinnon et al., 2004 ▸). The Hirshfeld surface can define the environment of each crystallographically independent molecule within a crystal. Fingerprint plots (Fig. 4 ▸) and show that for the picrate anion 55.2% and 8.8% of the area is concerned with the O⋯H (hydrogen-bonding) and C⋯C (π–π interaction) contacts, respectively. In the imidazole cation, 51.5% and 9.7% of the area is concerned with the the O⋯H (hydrogen-bonding) and C⋯C/N (π–π interaction) contacts, respectively. This quantitative analysis of the intermolecular interactions again shows that the three-dimensional network is defined mainly by hydrogen bonds.
Figure 4.

Fingerprint plots of co-crystal salt (I) showing the percentage of the O⋯H and C⋯C contacts around the environment of picrate and imidazolium ions.
Database survey
A search of the Cambridge Structural Database (CSD version 5.37 plus one update, Groom et al., 2016 ▸) indicates some analogs have been reported, viz. BEZGEU (Dhanabal et al., 2013 ▸), QAKYOS (Dutkiewicz et al., 2011 ▸), QAKGUG (Moreno-Fuquen et al., 2011 ▸) and SEZREU (Soriano-García et al., 1990 ▸). A structural comparison between these compounds indicates that the two nitrogen atoms in the imidazolium cations are preferably hydrogen-bonded to the picrate anions, in which they can be in a bifurcated or a linear mode. For instance, in the 1:1 organic salt imidazolium picrate (QAKGUG; Moreno-Fuquen et al., 2011 ▸), the imidazole N7 atom is linearly hydrogen-bonded to the phenolate oxygen atom O1. However, in the 1:2 salt (SEZREU; Soriano-García et al., 1990 ▸), the imidazole N1 atom is involved in bifuracted hydrogen bonding to the phenolate O1 and nitro O2 atoms. Both of these compounds crystallize in orthorhombic space groups (Pbca or P212121). Further research about polymorphism in this system is being carried out in our lab.
Synthesis and crystallization
All the reagents and solvents were used as obtained without further purification. Equivalent molar amounts of imidazole (1.0 mmol, 68.0 mg) and picric acid (1.0 mmol, 229.0 mg) were dissolved in 95% methanol (40.0 ml). The mixture was stirred for half an hour at ambient temperature and then filtered. The resulting yellow solution was kept in air for two weeks. Yellow needle-shaped crystals of (I) suitable for single-crystal X-ray diffraction analysis formed on the side of the vessel after two days. The crystals were separated manually (yield: 75%, ca 0.24 g).
Refinement
Crystal data, data collection and structure refinement details of compound (I) are summarized in Table 2 ▸. H atoms bonded to C atoms were positioned geometrically with C—H = 0.93 Å (aromatic) and refined in riding mode [U iso(H) = 1.2U eq(C)]. H atoms bonded to N and O atoms were found in difference-Fourier maps and refined freely with constraints of U iso(H)= 1.2U eq(N) or 1.5U eq(N).
Table 2. Experimental details.
| Crystal data | |
| Chemical formula | C3H5N2 +·C6H2N3O7 −·H2O |
| M r | 315.21 |
| Crystal system, space group | Orthorhombic, P n a21 |
| Temperature (K) | 100 |
| a, b, c (Å) | 21.577 (11), 3.5533 (18), 16.096 (8) |
| V (Å3) | 1234.0 (11) |
| Z | 4 |
| Radiation type | Mo Kα |
| μ (mm−1) | 0.15 |
| Crystal size (mm) | 0.40 × 0.04 × 0.02 |
| Data collection | |
| Diffractometer | Bruker APEXII CCD |
| Absorption correction | Multi-scan (SADABS; Bruker, 2001 ▸) |
| T min, T max | 0.937, 0.997 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 11664, 3837, 3367 |
| R int | 0.038 |
| (sin θ/λ)max (Å−1) | 0.724 |
| Refinement | |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.035, 0.086, 1.04 |
| No. of reflections | 3837 |
| No. of parameters | 211 |
| No. of restraints | 1 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.35, −0.22 |
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2056989017016401/lh5861sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989017016401/lh5861Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989017016401/lh5861Isup3.cml
CCDC reference: 1585713
Additional supporting information: crystallographic information; 3D view; checkCIF report
supplementary crystallographic information
Crystal data
| C3H5N2+·C6H2N3O7−·H2O | Dx = 1.697 Mg m−3 |
| Mr = 315.21 | Mo Kα radiation, λ = 0.71073 Å |
| Orthorhombic, Pna21 | Cell parameters from 4015 reflections |
| a = 21.577 (11) Å | θ = 2.3–31.2° |
| b = 3.5533 (18) Å | µ = 0.15 mm−1 |
| c = 16.096 (8) Å | T = 100 K |
| V = 1234.0 (11) Å3 | Needle, yellow |
| Z = 4 | 0.40 × 0.04 × 0.02 mm |
| F(000) = 648 |
Data collection
| Bruker APEXII CCD diffractometer | 3367 reflections with I > 2σ(I) |
| φ and ω scans | Rint = 0.038 |
| Absorption correction: multi-scan (SADABS; Bruker, 2001) | θmax = 31.0°, θmin = 1.9° |
| Tmin = 0.937, Tmax = 0.997 | h = −28→30 |
| 11664 measured reflections | k = −4→5 |
| 3837 independent reflections | l = −23→22 |
Refinement
| Refinement on F2 | Hydrogen site location: mixed |
| Least-squares matrix: full | H atoms treated by a mixture of independent and constrained refinement |
| R[F2 > 2σ(F2)] = 0.035 | w = 1/[σ2(Fo2) + (0.0494P)2 + 0.0144P] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.086 | (Δ/σ)max < 0.001 |
| S = 1.04 | Δρmax = 0.35 e Å−3 |
| 3837 reflections | Δρmin = −0.22 e Å−3 |
| 211 parameters | Absolute structure: Flack x determined using 1456 quotients [(I+)-(I-)]/[(I+)+(I-)] (Parsons et al., 2013) |
| 1 restraint | Absolute structure parameter: 0.4 (5) |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| C1 | 0.67480 (9) | −0.0182 (6) | 0.64727 (12) | 0.0122 (3) | |
| C2 | 0.72241 (9) | 0.0454 (6) | 0.70899 (11) | 0.0125 (4) | |
| C3 | 0.78154 (9) | 0.1698 (6) | 0.69303 (11) | 0.0131 (4) | |
| H3 | 0.8106 | 0.2010 | 0.7368 | 0.016* | |
| C4 | 0.79782 (9) | 0.2488 (6) | 0.61128 (12) | 0.0128 (3) | |
| C5 | 0.75625 (8) | 0.1917 (6) | 0.54609 (12) | 0.0133 (4) | |
| H5 | 0.7681 | 0.2377 | 0.4902 | 0.016* | |
| C6 | 0.69788 (9) | 0.0674 (6) | 0.56500 (12) | 0.0125 (3) | |
| C8 | 0.48877 (9) | 0.4718 (7) | 0.77357 (13) | 0.0172 (4) | |
| H8 | 0.4715 | 0.4535 | 0.7194 | 0.021* | |
| C9 | 0.46050 (9) | 0.3770 (7) | 0.84591 (13) | 0.0181 (4) | |
| H9 | 0.4197 | 0.2809 | 0.8522 | 0.022* | |
| N1 | 0.70790 (8) | −0.0342 (5) | 0.79547 (11) | 0.0143 (3) | |
| N2 | 0.85824 (8) | 0.4007 (5) | 0.59405 (11) | 0.0150 (3) | |
| N3 | 0.65576 (8) | 0.0095 (5) | 0.49511 (11) | 0.0149 (3) | |
| C7 | 0.55452 (10) | 0.5801 (7) | 0.87530 (13) | 0.0172 (4) | |
| H7 | 0.5908 | 0.6495 | 0.9049 | 0.021* | |
| N4 | 0.50257 (9) | 0.4472 (6) | 0.90866 (12) | 0.0170 (3) | |
| H4 | 0.4957 (14) | 0.398 (9) | 0.9649 (19) | 0.020* | |
| N5 | 0.54696 (8) | 0.5991 (5) | 0.79335 (11) | 0.0157 (3) | |
| H5A | 0.5763 (13) | 0.682 (8) | 0.7586 (18) | 0.019* | |
| O1 | 0.62270 (6) | −0.1576 (5) | 0.66176 (9) | 0.0156 (3) | |
| O4 | 0.89440 (7) | 0.4486 (5) | 0.65267 (10) | 0.0226 (3) | |
| O5 | 0.87159 (7) | 0.4829 (5) | 0.52189 (10) | 0.0226 (4) | |
| O8 | 0.50725 (7) | 0.6717 (6) | 0.57776 (10) | 0.0214 (3) | |
| H8A | 0.4849 (16) | 0.841 (10) | 0.594 (2) | 0.032* | |
| H8B | 0.5441 (17) | 0.745 (10) | 0.595 (2) | 0.032* | |
| O2 | 0.74665 (8) | −0.1992 (5) | 0.83731 (10) | 0.0227 (4) | |
| O3 | 0.65759 (7) | 0.0718 (5) | 0.82223 (10) | 0.0219 (3) | |
| O6 | 0.60348 (7) | 0.1417 (5) | 0.50023 (10) | 0.0222 (4) | |
| O7 | 0.67538 (7) | −0.1656 (5) | 0.43500 (9) | 0.0211 (3) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| C1 | 0.0097 (7) | 0.0125 (9) | 0.0143 (8) | 0.0002 (6) | 0.0006 (6) | 0.0003 (7) |
| C2 | 0.0109 (8) | 0.0138 (10) | 0.0127 (8) | 0.0000 (7) | 0.0006 (6) | 0.0008 (7) |
| C3 | 0.0099 (8) | 0.0138 (10) | 0.0157 (8) | 0.0014 (7) | −0.0005 (6) | −0.0006 (7) |
| C4 | 0.0085 (7) | 0.0128 (9) | 0.0171 (8) | −0.0001 (6) | 0.0008 (6) | −0.0002 (6) |
| C5 | 0.0115 (8) | 0.0135 (10) | 0.0148 (8) | −0.0003 (7) | 0.0010 (6) | 0.0006 (7) |
| C6 | 0.0101 (8) | 0.0142 (10) | 0.0134 (8) | −0.0012 (7) | −0.0020 (6) | 0.0002 (7) |
| C8 | 0.0135 (9) | 0.0198 (11) | 0.0182 (9) | −0.0029 (7) | 0.0000 (7) | −0.0001 (8) |
| C9 | 0.0125 (8) | 0.0203 (11) | 0.0215 (9) | −0.0020 (8) | 0.0028 (7) | 0.0004 (8) |
| N1 | 0.0133 (8) | 0.0159 (9) | 0.0138 (7) | −0.0034 (6) | −0.0006 (6) | −0.0001 (6) |
| N2 | 0.0097 (7) | 0.0151 (9) | 0.0203 (8) | 0.0000 (6) | 0.0013 (6) | 0.0000 (7) |
| N3 | 0.0129 (8) | 0.0181 (9) | 0.0136 (7) | −0.0029 (6) | −0.0025 (5) | 0.0031 (6) |
| C7 | 0.0132 (9) | 0.0198 (11) | 0.0185 (9) | 0.0008 (7) | 0.0013 (7) | −0.0004 (8) |
| N4 | 0.0143 (7) | 0.0193 (9) | 0.0176 (8) | 0.0004 (7) | 0.0034 (6) | 0.0004 (7) |
| N5 | 0.0122 (8) | 0.0184 (9) | 0.0163 (7) | −0.0012 (6) | 0.0021 (6) | 0.0006 (7) |
| O1 | 0.0095 (6) | 0.0210 (8) | 0.0163 (6) | −0.0035 (5) | −0.0002 (5) | 0.0011 (6) |
| O4 | 0.0112 (6) | 0.0316 (10) | 0.0248 (7) | −0.0045 (6) | −0.0036 (6) | 0.0025 (7) |
| O5 | 0.0166 (7) | 0.0318 (10) | 0.0195 (7) | −0.0063 (6) | 0.0057 (6) | −0.0003 (6) |
| O8 | 0.0122 (7) | 0.0340 (10) | 0.0181 (7) | 0.0016 (7) | −0.0005 (5) | −0.0032 (7) |
| O2 | 0.0228 (8) | 0.0294 (10) | 0.0158 (7) | 0.0039 (7) | −0.0032 (6) | 0.0040 (7) |
| O3 | 0.0141 (7) | 0.0333 (10) | 0.0182 (7) | −0.0005 (6) | 0.0056 (6) | −0.0026 (6) |
| O6 | 0.0124 (7) | 0.0318 (10) | 0.0224 (7) | 0.0042 (6) | −0.0030 (5) | 0.0031 (7) |
| O7 | 0.0187 (7) | 0.0287 (10) | 0.0159 (6) | −0.0031 (6) | −0.0002 (6) | −0.0056 (6) |
Geometric parameters (Å, º)
| C1—O1 | 1.250 (2) | C9—N4 | 1.381 (3) |
| C1—C2 | 1.447 (3) | C9—H9 | 0.9500 |
| C1—C6 | 1.447 (3) | N1—O2 | 1.223 (2) |
| C2—C3 | 1.374 (3) | N1—O3 | 1.227 (2) |
| C2—N1 | 1.455 (3) | N2—O5 | 1.232 (2) |
| C3—C4 | 1.391 (3) | N2—O4 | 1.236 (2) |
| C3—H3 | 0.9500 | N3—O6 | 1.225 (2) |
| C4—C5 | 1.395 (3) | N3—O7 | 1.226 (3) |
| C4—N2 | 1.438 (3) | C7—N4 | 1.329 (3) |
| C5—C6 | 1.369 (3) | C7—N5 | 1.331 (3) |
| C5—H5 | 0.9500 | C7—H7 | 0.9500 |
| C6—N3 | 1.461 (3) | N4—H4 | 0.93 (3) |
| C8—C9 | 1.357 (3) | N5—H5A | 0.89 (3) |
| C8—N5 | 1.372 (3) | O8—H8A | 0.82 (4) |
| C8—H8 | 0.9500 | O8—H8B | 0.88 (4) |
| O1—C1—C2 | 124.91 (18) | C8—C9—H9 | 126.7 |
| O1—C1—C6 | 124.28 (17) | N4—C9—H9 | 126.7 |
| C2—C1—C6 | 110.56 (16) | O2—N1—O3 | 123.96 (18) |
| C3—C2—C1 | 125.53 (17) | O2—N1—C2 | 118.23 (17) |
| C3—C2—N1 | 116.18 (16) | O3—N1—C2 | 117.81 (17) |
| C1—C2—N1 | 118.28 (17) | O5—N2—O4 | 122.65 (17) |
| C2—C3—C4 | 118.43 (17) | O5—N2—C4 | 118.87 (17) |
| C2—C3—H3 | 120.8 | O4—N2—C4 | 118.47 (16) |
| C4—C3—H3 | 120.8 | O6—N3—O7 | 124.46 (18) |
| C3—C4—C5 | 121.33 (17) | O6—N3—C6 | 117.85 (18) |
| C3—C4—N2 | 119.15 (17) | O7—N3—C6 | 117.69 (17) |
| C5—C4—N2 | 119.50 (17) | N4—C7—N5 | 108.37 (19) |
| C6—C5—C4 | 118.12 (17) | N4—C7—H7 | 125.8 |
| C6—C5—H5 | 120.9 | N5—C7—H7 | 125.8 |
| C4—C5—H5 | 120.9 | C7—N4—C9 | 108.85 (19) |
| C5—C6—C1 | 126.00 (17) | C7—N4—H4 | 126.4 (19) |
| C5—C6—N3 | 116.53 (17) | C9—N4—H4 | 124.7 (19) |
| C1—C6—N3 | 117.46 (16) | C7—N5—C8 | 108.99 (18) |
| C9—C8—N5 | 107.10 (19) | C7—N5—H5A | 123.3 (18) |
| C9—C8—H8 | 126.4 | C8—N5—H5A | 127.7 (18) |
| N5—C8—H8 | 126.4 | H8A—O8—H8B | 102 (3) |
| C8—C9—N4 | 106.69 (18) | ||
| O1—C1—C2—C3 | 174.4 (2) | C3—C2—N1—O2 | −42.2 (3) |
| C6—C1—C2—C3 | 0.0 (3) | C1—C2—N1—O2 | 136.7 (2) |
| O1—C1—C2—N1 | −4.3 (3) | C3—C2—N1—O3 | 137.2 (2) |
| C6—C1—C2—N1 | −178.67 (19) | C1—C2—N1—O3 | −44.0 (3) |
| C1—C2—C3—C4 | 1.1 (3) | C3—C4—N2—O5 | −177.4 (2) |
| N1—C2—C3—C4 | 179.82 (18) | C5—C4—N2—O5 | 1.2 (3) |
| C2—C3—C4—C5 | −2.3 (3) | C3—C4—N2—O4 | 1.5 (3) |
| C2—C3—C4—N2 | 176.28 (19) | C5—C4—N2—O4 | −179.89 (19) |
| C3—C4—C5—C6 | 2.3 (3) | C5—C6—N3—O6 | −132.1 (2) |
| N2—C4—C5—C6 | −176.28 (19) | C1—C6—N3—O6 | 49.1 (3) |
| C4—C5—C6—C1 | −1.1 (3) | C5—C6—N3—O7 | 47.3 (3) |
| C4—C5—C6—N3 | −179.77 (19) | C1—C6—N3—O7 | −131.5 (2) |
| O1—C1—C6—C5 | −174.5 (2) | N5—C7—N4—C9 | 0.3 (3) |
| C2—C1—C6—C5 | 0.0 (3) | C8—C9—N4—C7 | 0.0 (3) |
| O1—C1—C6—N3 | 4.2 (3) | N4—C7—N5—C8 | −0.6 (3) |
| C2—C1—C6—N3 | 178.65 (18) | C9—C8—N5—C7 | 0.6 (3) |
| N5—C8—C9—N4 | −0.3 (3) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N4—H4···O8i | 0.93 (3) | 1.83 (3) | 2.763 (3) | 172 (3) |
| N5—H5A···O1ii | 0.89 (3) | 1.94 (3) | 2.812 (2) | 165 (3) |
| N5—H5A···O3ii | 0.89 (3) | 2.46 (3) | 2.956 (3) | 116 (2) |
| O8—H8A···O4iii | 0.82 (4) | 2.29 (4) | 3.034 (2) | 151 (3) |
| O8—H8B···O1ii | 0.88 (4) | 2.04 (4) | 2.899 (2) | 165 (3) |
| O8—H8B···O6ii | 0.88 (4) | 2.44 (4) | 2.943 (3) | 117 (3) |
| C5—H5···O2iv | 0.95 | 2.49 | 3.383 (3) | 157 |
| C7—H7···O5v | 0.95 | 2.37 | 3.187 (3) | 144 |
| C7—H7···O3ii | 0.95 | 2.47 | 2.955 (3) | 112 |
| C8—H8···O4vi | 0.95 | 2.44 | 3.188 (3) | 135 |
| C8—H8···O8 | 0.95 | 2.53 | 3.255 (3) | 133 |
| C9—H9···O7vii | 0.95 | 2.48 | 3.349 (3) | 152 |
Symmetry codes: (i) −x+1, −y+1, z+1/2; (ii) x, y+1, z; (iii) x−1/2, −y+3/2, z; (iv) −x+3/2, y+1/2, z−1/2; (v) −x+3/2, y+1/2, z+1/2; (vi) x−1/2, −y+1/2, z; (vii) −x+1, −y, z+1/2.
Funding Statement
This work was funded by Wuhan University grant .
References
- Aitipamula, S., Mapp, L. K., Wong, A. B. H., Chow, P. S. & Tan, R. B. H. (2015). CrystEngComm, 17, 9323–9335.
- Arenas-García, J. I., Herrera-Ruiz, D., Mondragón-Vásquez, K., Morales-Rojas, H. & Höpfl, H. (2010). Cryst. Growth Des. 10, 3732–3742.
- Baburin, I. A. & Blatov, V. A. (2007). Acta Cryst. B63, 791–802. [DOI] [PubMed]
- Bertolasi, V., Gilli, P. & Gilli, G. (2011). Cryst. Growth Des. 11, 2724–2735.
- Blatov, V. A., Shevchenko, A. P. & Proserpio, D. M. (2014). Cryst. Growth Des. 14, 3576–3586.
- Brandenburg, K. (2006). DIAMOND. Crystal Impact GbR, Bonn, Germany.
- Bruker (2001). APEX2, SAINT & SADABS . Bruker AXS, Inc., Madison, Wisconsin, USA.
- Daszkiewicz, M. (2013). CrystEngComm, 15, 10427–10430.
- Dhanabal, T., Sethuram, M., Amirthaganesan, G. & Das, S. K. (2013). J. Mol. Struct. 1045, 112–123.
- Dutkiewicz, G., Samshuddin, S., Narayana, B., Yathirajan, H. S. & Kubicki, M. (2011). Acta Cryst. E67, o235. [DOI] [PMC free article] [PubMed]
- Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. (2016). Acta Cryst. B72, 171–179. [DOI] [PMC free article] [PubMed]
- Manoj, K., Tamura, R., Takahashi, H. & Tsue, H. (2014). CrystEngComm, 16, 5811–5819.
- McKinnon, J. J., Spackman, M. A. & Mitchell, A. S. (2004). Acta Cryst. B60, 627–668. [DOI] [PubMed]
- Moreno-Fuquen, R., De Almeida Santos, R. & Aguirre, L. (2011). Acta Cryst. E67, o139. [DOI] [PMC free article] [PubMed]
- Robinson, D. I. (2010). Org. Process Res. Dev. 14, 946–959.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sheldrick, G. M. (2015). Acta Cryst. C71, 3–8.
- Soriano-García, M., Schatz-Levine, M., Toscano, R. A. & Villena Iribe, R. (1990). Acta Cryst. C46, 1556–1558.
- Spek, A. L. (2003). J. Appl. Cryst. 36, 7–13.
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Weyna, D. R., Cheney, M. L., Shan, N., Hanna, M., Wojtas, Ł. & Zaworotko, M. J. (2012). CrystEngComm, 14, 2377–2380.
- Wolff, S. K., Grimwood, D. J., McKinnon, J. J., Turner, M. J., Jayatilaka, D. & Spackman, M. A. (2012). Crystal Explorer. University of Western Australia.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2056989017016401/lh5861sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989017016401/lh5861Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989017016401/lh5861Isup3.cml
CCDC reference: 1585713
Additional supporting information: crystallographic information; 3D view; checkCIF report