

The title compound, methyl 2-[5-(2-hydroxyphenyl)-2H-tetrazol-2-yl]acetate, is the major product from the reaction between 2-(2H-tetrazol-5-yl)phenol and methyl 2-bromoacetate in the presence of potassium carbonate, which gave three isomeric products.
Keywords: crystal structure, tetrazole, hydroxyphenyl tetrazole, hydrogen bonding, offset π–π interactions
Abstract
The title compound, C10H10N4O3, was synthesized by the esterification of hydroxyphenyl tetrazole. There is an intramolecular O—H⋯N hydrogen bond present involving the hydroxy group and the tetrazole ring. The tetrazole ring is inclined to the phenol ring by 2.85 (13)°, while the methyl acetate group is almost normal to the tetrazole ring, making a dihedral angle of 82.61 (14)°. In the crystal, molecules are linked by pairs of C—H⋯O hydrogen bonds, forming inversion dimers. Within the dimers, the phenol rings are linked by offset π–π interactions [intercentroid distance = 3.759 (2) Å]. There are no further significant intermolecular interactions present in the crystal. The hydroxy group is disordered about positions 2 and 6 on the benzene ring, with a refined occupancy ratio of 0.531 (5):0.469 (5).
Chemical context
Tetrazole ligands are useful building blocks for the construction of high-dimensional metal–organic frameworks by providing various binding modes toward metal centers (Karaghiosoff et al., 2009 ▸; Liu et al., 2013 ▸). Recently, we have used 5-(2-hydroxyphenyl)tetrazole as a chelating multidentate ligand and reported several interesting compounds (Park et al., 2015 ▸; 2014 ▸). It provides strong [N,O] chelation to metal centers with various additional binding modes. As part of a project on the study of the substitution effects on the tetrazole ring on the self-assembly behaviour in solution, as well as in the solid state, we have synthesized a number of substituted hydroxyphenyl tetrazole complexes. The substitution of the tetrazole group may promote supramolecular interaction by weak interactions, such as hydrogen bonding. The reaction between hydroxyphenyl tetrazole and bromo acetate methyl ester in the presence of potassium carbonate gave three isomeric products. Using column chromatography, the major product was isolated and its molecular structure was determined unambiguously by X-ray crystallography. We report herein, the synthesis and crystal structure of this compound.
Structural commentary
The molecular structure of the title compound is shown in Fig. 1 ▸. The structure analysis confirms the nature of the major product of the reaction, which yielded three isomeric compounds as described in Section 5, Synthesis and crystallization. The title molecule consists of a tetrazole ring (N1–N4/C1) and a phenol ring (C2–C7), which are connected by an intramolecular O—H⋯N hydrogen bond (Fig. 1 ▸, Table 1 ▸) and inclined to one another by 2.85 (13)°. The planar methyl acetate group [O2/O3/C8–C10; maximum deviation of 0.037 (2) Å for atom O2] is inclined to the tetrazole ring by 82.61 (14)°.
Figure 1.

A view of the molecular structure of the title compound, with the atom labelling and 30% probability displacement ellipsoids. The intramolecular O—H⋯N hydrogen bond (see Table 1 ▸) is indicated by a dashed line. Only the major component of the disordered OH group, in position 2, is shown.
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O1—H1⋯N1 | 0.84 | 1.91 | 2.659 (4) | 148 |
| C5—H5⋯O3i | 0.95 | 2.57 | 3.472 (3) | 158 |
Symmetry code: (i) 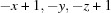 .
.
Supramolecular features
In the crystal, the molecules are linked by pairs of C—H⋯O hydrogen bonds, forming inversion dimers with an 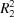 (22) loop (Table 1 ▸, Fig. 2 ▸). Within the dimers, the phenol rings are linked by offset π–π interactions [Cg⋯Cg
i = 3.759 (2) Å, interplanar distance = 3.526 (1) Å, slippage 1.305 Å; Cg is the centroid of the C2–C7 phenol ring, symmetry code: (i) −x + 1, −y, −z + 1]. There are no further significant intermolecular interactions present in the crystal.
(22) loop (Table 1 ▸, Fig. 2 ▸). Within the dimers, the phenol rings are linked by offset π–π interactions [Cg⋯Cg
i = 3.759 (2) Å, interplanar distance = 3.526 (1) Å, slippage 1.305 Å; Cg is the centroid of the C2–C7 phenol ring, symmetry code: (i) −x + 1, −y, −z + 1]. There are no further significant intermolecular interactions present in the crystal.
Figure 2.

A view along the a axis of the crystal packing of the title compound. The intra- and intermolecular hydrogen bonds (see Table 1 ▸) are indicated by dashed lines. The offset π–π interactions are shown as dashed double arrows. Only H atoms H1 and H5, and the major component of the disordered OH group in position 2, have been included.
Database survey
A search of the Cambridge Structural Database (Version 5.38, update May 2017; Groom et al., 2016 ▸) for the methyl 2-(5-phenyl-2H-tetrazol-2-yl)acetate skeleton revealed only two hits, viz. ethyl (Z)-3-phenyl-2-(5-phenyl-2H-tetrazol-2-yl)-2-propenoate (SAKVIM; Ramazani et al., 2017 ▸) and methyl (5-phenyl-2H-tetrazol-2-yl)acetate (WUKNUN; Saeed et al., 2015 ▸). In WUKNUN, the 5-phenyl substituent is inclined to the tetrazole ring by 3.89 (7)°, compared to 2.85 (13)° in the title compound. In contrast, the corresponding dihedral angle in SAKVIM is 19.97 (16)°. The methyl/ethyl acetate groups are inclined to the plane of the tetrazole ring by 84.99 (7)° in WUKNUN and 84.57 (7)° in SAKVIM, similar to the value observed in the title compound, viz. 82.61 (14)°.
Synthesis and crystallization
The synthesis of the title compound is illustrated in Fig. 3 ▸. 2-(2H-Tetrazol-5-yl)phenol (100 mg, 0.62 mmol) and potassium carbonate (85.0 mg, 0.62 mmol) were dissolved in acetonitrile at 273 K while stirring for 30 min. To the resulting solution methyl 2-bromoacetate (207 µl, 2.18 mmol) was added and stirring was continued for 24 h. The white solid that was obtained was filtered and the solvent removed under reduced pressure. The residue was purified by column chromatography on silica gel using ether:hexane (2:3) as eluent. Three isomeric compounds were obtained, as shown in Fig. 3 ▸. The major product (I) (yield = 59%), was recrystallized in dichloromethane and yielded needle-like colourless crystals of the title compound. Spectroscopic data: 1H NMR (CDCl3, 400MHz): δ = 9.59 (s, 1H, OH), 8.06 (d, 1H, Ph), 7.41 (t, 1H, Ph), 7.11 (d, 1H, Ph), 6.99 (t, 1H, Ph), 5.51 (s, 2H), 3.85 (s, 3H). 13C NMR (125 MHz, CDCl3): 165.06, 164.68, 156.42, 132.44, 127.50, 120.06, 117.62, 53.41, 53.38 ppm.
Figure 3.

Reaction scheme for the synthesis of the title compound, (I).
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 2 ▸. The hydroxy group is disordered about positions 2 and 6 on the phenol ring, with a refined occupancy ratio of 0.531 (5):0.469 (5). All the H atoms were included in calculated positions using a riding model: O—H = 0.84 Å, C-H = 0.95–1.00 Å with U iso(H) = 1.5 U eq(O-hydroxyl, C-methyl) and 1.2U eq(C) for other H atoms.
Table 2. Experimental details.
| Crystal data | |
| Chemical formula | C10H10N4O3 |
| M r | 234.22 |
| Crystal system, space group | Monoclinic, P21/c |
| Temperature (K) | 100 |
| a, b, c (Å) | 10.060 (2), 8.2538 (17), 13.536 (3) |
| β (°) | 104.479 (10) |
| V (Å3) | 1088.2 (4) |
| Z | 4 |
| Radiation type | Mo Kα |
| μ (mm−1) | 0.11 |
| Crystal size (mm) | 0.15 × 0.10 × 0.10 |
| Data collection | |
| Diffractometer | Bruker APEXII CCD |
| Absorption correction | Multi-scan (SADABS; Krause et al., 2015 ▸) |
| T min, T max | 0.987, 0.989 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 14003, 2372, 1252 |
| R int | 0.044 |
| (sin θ/λ)max (Å−1) | 0.642 |
| Refinement | |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.057, 0.137, 1.02 |
| No. of reflections | 2372 |
| No. of parameters | 167 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.14, −0.17 |
Supplementary Material
Crystal structure: contains datablock(s) I, Global. DOI: 10.1107/S205698901701698X/su5409sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S205698901701698X/su5409Isup2.hkl
Supporting information file. DOI: 10.1107/S205698901701698X/su5409Isup3.cml
CCDC reference: 1587621
Additional supporting information: crystallographic information; 3D view; checkCIF report
supplementary crystallographic information
Crystal data
| C10H10N4O3 | F(000) = 488 |
| Mr = 234.22 | Dx = 1.430 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| a = 10.060 (2) Å | Cell parameters from 3134 reflections |
| b = 8.2538 (17) Å | θ = 2.9–24.3° |
| c = 13.536 (3) Å | µ = 0.11 mm−1 |
| β = 104.479 (10)° | T = 100 K |
| V = 1088.2 (4) Å3 | Needle, colourless |
| Z = 4 | 0.15 × 0.10 × 0.10 mm |
Data collection
| Bruker APEXII CCD diffractometer | 1252 reflections with I > 2σ(I) |
| φ and ω scans | Rint = 0.044 |
| Absorption correction: multi-scan (SADABS; Krause et al., 2015) | θmax = 27.2°, θmin = 2.1° |
| Tmin = 0.987, Tmax = 0.989 | h = −12→12 |
| 14003 measured reflections | k = −10→10 |
| 2372 independent reflections | l = −17→17 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.057 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.137 | H-atom parameters constrained |
| S = 1.01 | w = 1/[σ2(Fo2) + (0.0479P)2 + 0.3171P] where P = (Fo2 + 2Fc2)/3 |
| 2372 reflections | (Δ/σ)max < 0.001 |
| 167 parameters | Δρmax = 0.14 e Å−3 |
| 0 restraints | Δρmin = −0.17 e Å−3 |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | Occ. (<1) | |
| O1 | 0.3781 (3) | 0.2337 (4) | 0.3644 (3) | 0.0745 (14) | 0.531 (5) |
| H1 | 0.4534 | 0.2755 | 0.3623 | 0.112* | 0.531 (5) |
| O2 | 0.92819 (16) | 0.46762 (19) | 0.26859 (14) | 0.0718 (5) | |
| O3 | 0.92563 (18) | 0.2756 (2) | 0.38419 (16) | 0.0879 (6) | |
| O1A | 0.5939 (5) | 0.1877 (6) | 0.7094 (3) | 0.1031 (19) | 0.469 (5) |
| H1A | 0.6557 | 0.2377 | 0.6900 | 0.155* | 0.469 (5) |
| N1 | 0.61929 (19) | 0.3786 (2) | 0.43476 (16) | 0.0618 (5) | |
| N2 | 0.7415 (2) | 0.4500 (2) | 0.46016 (19) | 0.0679 (6) | |
| N3 | 0.8043 (2) | 0.4384 (3) | 0.5571 (2) | 0.0890 (7) | |
| N4 | 0.7203 (2) | 0.3567 (3) | 0.59958 (17) | 0.0826 (7) | |
| C1 | 0.6085 (2) | 0.3209 (3) | 0.5240 (2) | 0.0574 (6) | |
| C2 | 0.4922 (2) | 0.2276 (2) | 0.53816 (19) | 0.0555 (6) | |
| C3 | 0.4899 (3) | 0.1677 (3) | 0.6336 (3) | 0.0749 (7) | |
| H3 | 0.5638 | 0.1911 | 0.6908 | 0.090* | 0.531 (5) |
| C4 | 0.3811 (4) | 0.0744 (3) | 0.6462 (3) | 0.0916 (10) | |
| H4 | 0.3810 | 0.0334 | 0.7117 | 0.110* | |
| C5 | 0.2743 (3) | 0.0412 (3) | 0.5647 (3) | 0.0913 (10) | |
| H5 | 0.2001 | −0.0237 | 0.5734 | 0.110* | |
| C6 | 0.2733 (3) | 0.1010 (3) | 0.4701 (3) | 0.0831 (8) | |
| H6 | 0.1978 | 0.0790 | 0.4137 | 0.100* | |
| C7 | 0.3818 (3) | 0.1929 (3) | 0.4565 (2) | 0.0652 (7) | |
| H7 | 0.3808 | 0.2330 | 0.3905 | 0.078* | 0.469 (5) |
| C8 | 0.8045 (3) | 0.5246 (3) | 0.3866 (2) | 0.0778 (8) | |
| H8A | 0.8619 | 0.6170 | 0.4189 | 0.093* | |
| H8B | 0.7322 | 0.5670 | 0.3288 | 0.093* | |
| C9 | 0.8923 (2) | 0.4048 (3) | 0.3474 (2) | 0.0635 (7) | |
| C10 | 1.0223 (3) | 0.3749 (3) | 0.2257 (2) | 0.0834 (8) | |
| H10A | 1.0402 | 0.4334 | 0.1674 | 0.125* | |
| H10B | 0.9817 | 0.2691 | 0.2031 | 0.125* | |
| H10C | 1.1087 | 0.3594 | 0.2776 | 0.125* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.063 (2) | 0.086 (3) | 0.071 (3) | −0.0122 (18) | 0.0098 (17) | 0.0043 (19) |
| O2 | 0.0666 (11) | 0.0501 (10) | 0.1035 (13) | 0.0043 (8) | 0.0303 (10) | 0.0086 (10) |
| O3 | 0.0893 (13) | 0.0446 (10) | 0.1425 (17) | 0.0147 (9) | 0.0529 (12) | 0.0194 (11) |
| O1A | 0.127 (4) | 0.118 (4) | 0.062 (3) | 0.006 (3) | 0.018 (3) | 0.011 (3) |
| N1 | 0.0533 (12) | 0.0445 (11) | 0.0897 (15) | 0.0028 (10) | 0.0215 (11) | −0.0002 (11) |
| N2 | 0.0570 (13) | 0.0463 (12) | 0.1035 (18) | 0.0007 (10) | 0.0260 (13) | −0.0040 (12) |
| N3 | 0.0684 (15) | 0.0838 (18) | 0.111 (2) | −0.0131 (14) | 0.0151 (15) | −0.0115 (16) |
| N4 | 0.0699 (15) | 0.0835 (16) | 0.0897 (17) | −0.0084 (13) | 0.0113 (13) | −0.0090 (13) |
| C1 | 0.0534 (15) | 0.0414 (12) | 0.0779 (17) | 0.0082 (11) | 0.0172 (13) | −0.0068 (13) |
| C2 | 0.0568 (14) | 0.0374 (12) | 0.0754 (17) | 0.0095 (11) | 0.0225 (13) | −0.0017 (12) |
| C3 | 0.085 (2) | 0.0605 (17) | 0.082 (2) | 0.0139 (15) | 0.0271 (18) | 0.0054 (16) |
| C4 | 0.118 (3) | 0.0586 (18) | 0.119 (3) | 0.0109 (19) | 0.069 (2) | 0.0116 (18) |
| C5 | 0.096 (2) | 0.0504 (17) | 0.151 (3) | −0.0090 (16) | 0.074 (2) | −0.015 (2) |
| C6 | 0.0686 (18) | 0.0668 (18) | 0.121 (3) | −0.0077 (15) | 0.0376 (17) | −0.0194 (18) |
| C7 | 0.0604 (16) | 0.0517 (15) | 0.088 (2) | 0.0008 (12) | 0.0273 (15) | −0.0047 (14) |
| C8 | 0.0703 (16) | 0.0433 (14) | 0.130 (2) | 0.0011 (12) | 0.0439 (17) | 0.0049 (15) |
| C9 | 0.0474 (13) | 0.0384 (13) | 0.106 (2) | −0.0044 (11) | 0.0219 (13) | −0.0004 (14) |
| C10 | 0.0779 (18) | 0.0708 (18) | 0.110 (2) | 0.0042 (15) | 0.0398 (16) | −0.0052 (16) |
Geometric parameters (Å, º)
| O1—C7 | 1.282 (4) | N4—C1 | 1.351 (3) |
| O2—C9 | 1.315 (3) | C1—C2 | 1.452 (3) |
| O2—C10 | 1.447 (3) | C2—C7 | 1.387 (3) |
| O3—C9 | 1.190 (3) | C2—C3 | 1.388 (3) |
| O1A—C3 | 1.280 (5) | C3—C4 | 1.383 (4) |
| N1—C1 | 1.328 (3) | C4—C5 | 1.362 (4) |
| N1—N2 | 1.329 (3) | C5—C6 | 1.370 (4) |
| N2—N3 | 1.310 (3) | C6—C7 | 1.378 (3) |
| N2—C8 | 1.445 (3) | C8—C9 | 1.508 (3) |
| N3—N4 | 1.319 (3) | ||
| C9—O2—C10 | 117.19 (18) | O1A—C3—C4 | 119.1 (4) |
| C1—N1—N2 | 101.8 (2) | O1A—C3—C2 | 119.9 (3) |
| N3—N2—N1 | 114.2 (2) | C4—C3—C2 | 120.7 (3) |
| N3—N2—C8 | 122.4 (2) | C5—C4—C3 | 120.0 (3) |
| N1—N2—C8 | 123.2 (2) | C4—C5—C6 | 120.3 (3) |
| N2—N3—N4 | 105.9 (2) | C5—C6—C7 | 120.1 (3) |
| N3—N4—C1 | 106.5 (2) | O1—C7—C6 | 116.2 (3) |
| N1—C1—N4 | 111.6 (2) | O1—C7—C2 | 122.9 (3) |
| N1—C1—C2 | 124.2 (2) | C6—C7—C2 | 120.7 (3) |
| N4—C1—C2 | 124.2 (2) | N2—C8—C9 | 111.09 (19) |
| C7—C2—C3 | 118.1 (2) | O3—C9—O2 | 126.0 (2) |
| C7—C2—C1 | 121.0 (2) | O3—C9—C8 | 124.7 (2) |
| C3—C2—C1 | 120.8 (2) | O2—C9—C8 | 109.3 (2) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1···N1 | 0.84 | 1.91 | 2.659 (4) | 148 |
| C5—H5···O3i | 0.95 | 2.57 | 3.472 (3) | 158 |
Symmetry code: (i) −x+1, −y, −z+1.
Funding Statement
This work was funded by National Research Foundation of Korea grants 2015R1A4A1041036 and 2016R1D1A1B03930507.
References
- Bruker (2014). APEX2 and SAINT. Bruker AXS Inc., Madison, Wisconsin, USA.
- Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. (2016). Acta Cryst. B72, 171–179. [DOI] [PMC free article] [PubMed]
- Karaghiosoff, K., Klapötke, T. M. & Miró Sabaté, C. (2009). Chem. Eur. J. 15, 1164–1176. [DOI] [PubMed]
- Krause, L., Herbst-Irmer, R., Sheldrick, G. M. & Stalke, D. (2015). J. Appl. Cryst. 48, 3–10. [DOI] [PMC free article] [PubMed]
- Liu, Z.-Y., Zou, H.-A., Hou, Z.-J., Yang, E.-C. & Zhao, X.-J. (2013). Dalton Trans. 42, 15716–15725. [DOI] [PubMed]
- Macrae, C. F., Bruno, I. J., Chisholm, J. A., Edgington, P. R., McCabe, P., Pidcock, E., Rodriguez-Monge, L., Taylor, R., van de Streek, J. & Wood, P. A. (2008). J. Appl. Cryst. 41, 466–470.
- Park, K. H., Lee, K. M., Go, M. J., Choi, S. H., Park, H. R., Kim, Y. & Lee, J. (2014). Inorg. Chem. 53, 8213–8220. [DOI] [PubMed]
- Park, Y. J., Ryu, J. Y., Begum, H., Lee, M. H., Stang, P. J. & Lee, J. (2015). J. Am. Chem. Soc. 137, 5863–5866. [DOI] [PubMed]
- Ramazani, A., Nasrabadi, F. Z., Ślepokura, K., Lis, T. & Joo, S. W. (2017). J. Heterocycl. Chem. 54, 55–64.
- Saeed, A., Qasim, M., Hussain, M., Flörke, U. & Erben, M. F. (2015). Spectrochim. Acta Part A, 150, 1–8. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sheldrick, G. M. (2015). Acta Cryst. C71, 3–8.
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, Global. DOI: 10.1107/S205698901701698X/su5409sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S205698901701698X/su5409Isup2.hkl
Supporting information file. DOI: 10.1107/S205698901701698X/su5409Isup3.cml
CCDC reference: 1587621
Additional supporting information: crystallographic information; 3D view; checkCIF report