

Abstract
N-acylphosphatidylethanolamine phospholipase D (NAPE-PLD) is a membrane-associated zinc enzyme that catalyzes the hydrolysis of N-acylphosphatidylethanolamines (NAPEs) into fatty acid ethanolamides (FAEs). Here, we describe the identification of the first small-molecule NAPE-PLD inhibitor, the quinazoline sulfonamide derivative 2,4-dioxo-N-[4-(4-pyridyl)phenyl]-1H-quinazoline-6-sulfonamide, ARN19874.
N-acyl phosphatidylethanolamine phospholipase D (NAPE-PLD) (EC 3.1.4.4) is a membrane-associated zinc lipase that catalyzes the conversion of N-acylphosphatidylethanolamines (NAPEs) into fatty acid ethanolamides (FAEs).2–4 The FAEs are structurally and functionally diverse lipid mediators that act as agonists for G protein-coupled receptors (e. g., cannabinoid receptors),6 ligand-activated transcription factors (e. g., peroxisome proliferator-activated receptor-α)8 and ion channels (e. g., transient receptor potential subfamily V member 1, TRPV1).9 The NAPEs, on the other hand, though traditionally viewed only as precursors for the FAEs,10 are emerging as important regulators of key dynamic properties of the lipid bilayer – including stability11–13 and fusogenicity14,15 – and its interactions with intracellular signaling proteins.16 Despite the potentially important functions served by NAPE-PLD,17,18 no selective pharmacological tool for studying this enzyme is currently available.
Recent structural studies have highlighted three structural features that distinguish NAPE-PLD (Figure 1).2 First, the enzyme assembles into homodimers that form an extended lipid-binding surface, enabling a close interaction of the enzyme with cell membranes. Within this surface, a hydrophobic entry cavity accommodates the NAPEs and allows these negatively charged lipids to access the diatomic zinc center. The entry cavity also contains two recognition pockets, one in each monomer, which selectively recognize secondary bile acids such as deoxycholic acid (DCA).19 This binding event enhances dimer assembly and enables catalysis.2 The molecular architecture of NAPE-PLD offers two obvious targets for pharmacological modulation: the catalytic zinc center and the allosteric bile acid-binding site (Figure 1).
Figure 1.
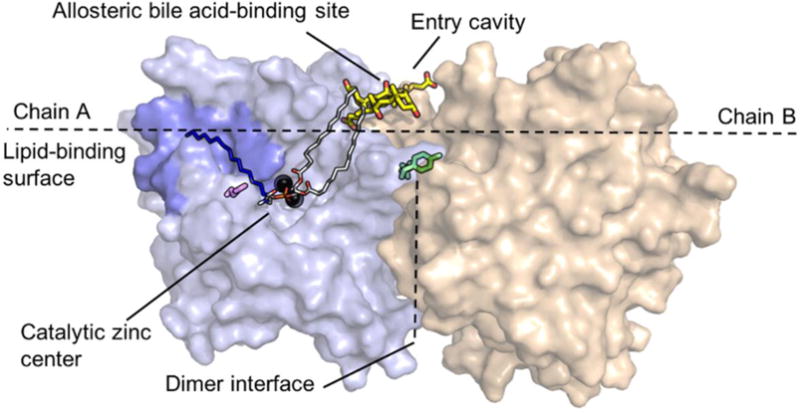
Structure of NAPE-PLD and structural details discussed in the text. Chain A and chain B are illustrated in light blue and beige, respectively. The polar head-group (red oxygens and orange phosphorus) of NAPE coordinates the diatomic zinc center (black spheres). The sn-1 and sn-2 acyl chains (grey carbons) interact with bile acid molecules (yellow carbons) that occupy a recognition pocket present at the membrane interface. The N-acyl chain of NAPE (dark blue carbons) may be accommodated within the hydrophobic pocket (blue) found in close proximity of the zinc center. Glutamine 320 (magenta) may form polar interactions with the N-acyl chain of NAPE. Also shown in figure are glutamine 158 and tyrosine 159 (green), which are important in dimer formation.
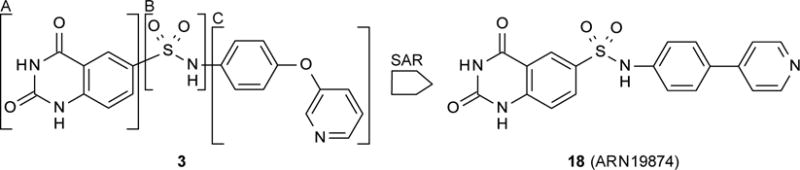
Here, we describe the discovery of a novel quinazoline sulfonamide derivative, compound 18 (2,4-dioxo-N-[4-(4-pyridyl)phenyl]-1H-quinazoline-6-sulfonamide, ARN19874) (Figure 2), as the first selective inhibitor of intracellular NAPE-PLD activity. We further show that this compound binds via a reversible mechanism to the enzyme’s zinc center and may be used in vitro to explore the biological functions of NAPE-PLD.
Figure 2.
Chemical structures of compounds reported in the present study.
Screening an in-house chemical library for NAPE-PLD inhibitors yielded compound 3 (Figure 2; Chart 1), which reduced activity of purified human recombinant NAPE-PLD by 58% at 75 μM. Despite its weak inhibitory potency, this compound attracted our attention because (i) it contains a sulfonamide group, which might form coordination bonds with the positively charged zinc atoms present in the active site of NAPE-PLD20 and, (ii) it has a modular structure that lends itself to chemical modification. As shown in Figure 2, this structure consists of a quinazolinedione ring (A) bridged to an aromatic side chain (C) by a sulfonamide linker (B). To identify determinants for NAPE-PLD inhibition by 3, we conducted a focused structure-activity relationship (SAR) study that separately targeted each of these components. We first focused on the sulfonamide group. The introduction of a methylene bridge between this group and the side chain of 3 yielded derivative 4, which was virtually inactive (Chart 1). Loss of inhibitory activity was also observed with the reverse sulfonamide 5. By contrast, activity was retained in the N-methylated sulfonamide 6. The drastic drop in inhibitory potency observed with compounds 4 and 5 suggests that the sulfonamide group plays a primary role in NAPE-PLD inhibition by this set of compounds. Next, we turned our attention to the quinazolinedione nucleus, which closely resembles scaffolds contributing to the activity of several metallo-enzyme inhibitors.21 Methylation of its endocyclic nitrogens yielded the single-methylated derivatives, 7 and 8, as well as the double-methylated derivative 9. Each of these compounds retained some inhibitory activity, although weaker than the parent molecule (Chart 1). By contrast, the unsubstituted quinazolinedione (10) had no effect (Chart 1). The results suggest that the quinazolinedione ring contributes to NAPE-PLD inhibition, but less critically than does the sulfonamide group. To investigate the role of the side chain C, we gradually rebuilt this region keeping intact both the sulfonamide linker and the quinazolinedione ring (Table 1). The phenyl (11) and the biphenyl (12) derivatives showed only limited inhibition at 75 μM, with a drastic reduction of compounds’ solubility in assay buffer (as assessed by microscopic inspection of assay wells after completion of the enzymatic reaction). To circumvent this obstacle, we prepared a series of molecules in which a nitrogen-containing heterocycle replaced the distal phenyl group of 12. While introduction of an H-bond-forming pyrazole (13) yielded a weak inhibitor, progressive increases in potency were observed with the pyridazine 14 (IC50 ≈ 124 μM) and the pyrimidine 15 (IC50 ≈ 52 μM) rings (Table 1).
Chart 1.
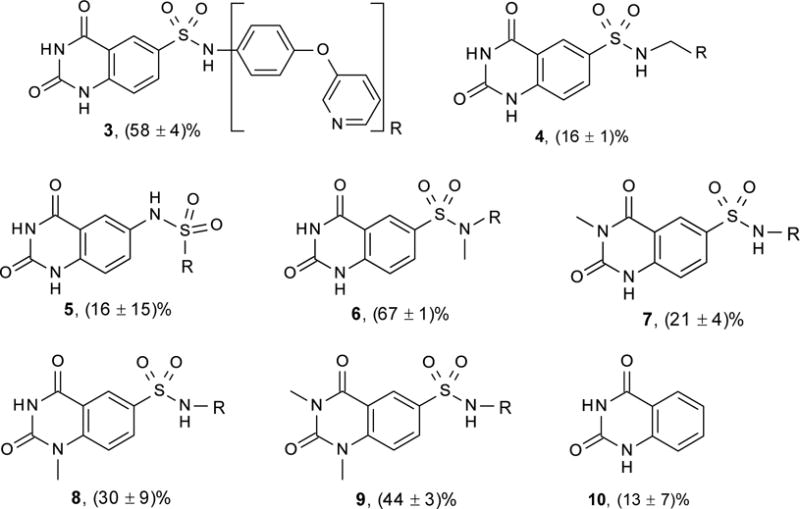
Chemical structure and NAPE-PLD inhibition by compounds 3 – 10.
Compounds tested at 75 μM on purified human recombinant NAPE-PLD. Values are expressed as the mean ± SD of percent inhibition relative to vehicle (n=3).
Table 1.
Structures of compounds 11 – 24 and NAPE-PLD inhibition.
| ||
|---|---|---|
| Compound | R | Inhibition |
| 11 |
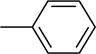
|
26.2 ± 0.7a |
| 12 |
|
23.4 ± 7.3a |
| 13 |
|
254.0 ± 1.3b |
| 14 |
|
124.0 ± 1.1b |
| 15 |
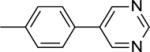
|
52.4 ± 1.2b |
| 16 |
|
120.9 ± 1.2b |
| 17 |
|
25.9 ± 1.2b |
| 18 |
|
33.7 ± 9.8b |
| 19 |
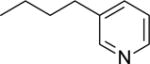
|
NAc |
| 20 |
|
NAc |
| 21 |
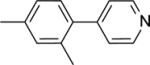
|
39.9 ± 1.2b |
| 22 |
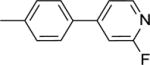
|
36.3 ± 1.2b |
| 23 |
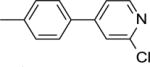
|
36.9 ± 2.3b |
| 24 |
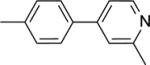
|
50.3 ± 1.3b |
Compounds tested on purified human recombinant NAPE-PLD.
Percent inhibition at 75 μM.
IC50 value (μM) in the interval 0.3 – 300 μM.
Not active. Values are the mean ± SD of three independent experiments, except for 18, where n=15.
This finding directed our attention toward the possible existence of polar interactions between the nitrogen atoms of 14 and 15 and specific residues in NAPE-PLD. To explore this idea, we prepared a small set of pyridyl-phenyl derivatives that differed in the position of the pyridyl nitrogen. While the 2-pyridyl derivative 16 was as active as 14 (IC50 ≈ 120 μM), a significant increase in potency was observed with the 3-pyridyl derivative 17 (IC50 ≈ 26 μM) and the 4-pyridyl derivative 18 (IC50 ≈ 34 μM) (Table 1). The greater inhibitory potency demonstrated by 17 and 18 supports the possibility that the pyridyl nitrogen interacts with a recognition site in NAPE-PLD. Confirming this, we found that replacement of the phenyl ring with a flexible n-propyl chain (19) or of the pyridyl ring with an n-butyl chain (20) resulted in complete loss of activity (Table 1). By contrast, addition of a methyl group at the meta position of the phenyl ring of 18 afforded derivative 21 which retained inhibitory activity (IC50 ≈ 40 μM). The scaffold of 18 also tolerated the introduction of a limited steric bulk in position alpha to the pyridyl nitrogen, such as that of a small halogen atom in derivatives 22 (IC50 ≈ 36 μM) and 23 (IC50 ≈ 37 μM), or, to a lesser extent, a methyl group in derivative 24 (IC50 ≈ 50 μM). In sum, these focused analyses suggest that determinants of NAPE-PLD inhibition include: (i) an appropriately positioned sulfonamide group, and (ii) an arylic side chain where rigidity is combined with the distal H-bonding function afforded by the 4-pyrydyl nitrogen. Subsequent studies were centered on compound 18, which integrates these pharmacophoric elements with adequate solubility in aqueous buffers.
Figure 3a illustrates the effects of 18 on purified human recombinant NAPE-PLD. Under identical conditions, the cephalosporin antibiotic nitrocefin, which was reported to inhibit NAPE-PLD activity in cell homogenates with an IC50 of ≈ 1 mM,1 was inactive (Figure 3a). Kinetic analyses revealed that 18 acts through an uncompetitive mechanism characterized by an increase in Km and a decrease in Vmax (Figure 3b; Table S1). Moreover, NAPE-PLD inhibition by 18 was rapid (Figure 3c) and readily reversed upon dilution of the enzyme-inhibitor complex (Figure 3d).7 Collectively, these findings suggest that 18 is a reversible uncompetitive inhibitor of NAPE-PLD activity.
Figure 3.
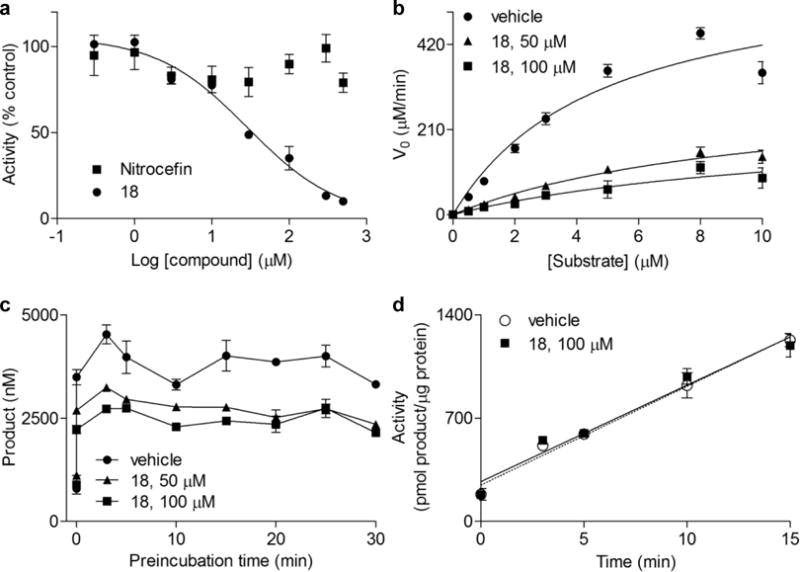
a) Inhibition of NAPE-PLD by 18 (filled circles) or nitrocefin1 (filled squares). b) Michaelis-Menten kinetics5 and (c) time-course of NAPE-PLD in the presence of vehicle (1% DMSO, filled circles) or 18 at 50 μM (filled triangles) or 100 μM (filled squares). d) Rapid dilution assay7 of NAPE-PLD preincubated with vehicle (1% DMSO, empty circles) or 18 (100 μM, filled squares).
Next, we conducted a series of site-directed mutagenesis and computational studies aimed at probing the molecular interactions between 18 and NAPE-PLD. We first hypothesized that 18 might interact with the bile acid-binding pockets of NAPE-PLD (Figure 1), which have been implicated in regulating dimer formation and catalysis.2 To test this idea, we assessed the effects of 18 on the double NAPE-PLD mutant Q158S/Y159S, which is unable to bind bile acids.2 As previously shown, the mutant does not form dimers and displays reduced activity compared to the wild-type enzyme (Figure S1; Table S2).2 The mutant was, however, as susceptible to inhibition by 18 as wild-type NAPE-PLD (Figure S2; Table S3), suggesting that compound 18 does not interact with the bile acid-binding pocket of NAPE-PLD.
Because the sulfonamide linker is an important determinant of NAPE-PLD inhibition, we speculated that the effects of this group might be mediated via coordination of the enzyme’s zinc metal center.20,22 As an initial test of this idea, we performed a series of docking studies using chain A of the enzyme’s crystal structure.2 In the best docking pose obtained, 18 is positioned within the catalytic site, with the sulfonamide group interacting with both zinc atoms (Figure 4a) and mimicking the phosphate group of the co-crystallized phosphatidylethanolamine (PE) molecule, which presumably occupies the same space taken by NAPE.2 Additional stabilization is afforded by a polar interaction between the sulfonamide group of 18 and the carboxyl group of aspartate 189. Importantly, the pose is stabilized by an H-bond between one oxygen of the quinazolinedione ring and the side chain of glutamine 320, which is thought to interact with the carboxyamide group of NAPE.2 The 4-pyridyl-phenyl moiety is located within the hydrophobic cavity occupied by the fatty acyl chains of PE, with the pyridine ring making a face-to-edge aromatic interaction with tyrosine 188. The pyridyl nitrogen of 18 is at moderate distance from the flexible chain of lysine 256, which may heighten the binding of the compound to NAPE-PLD (Figure 4a).
Figure 4.
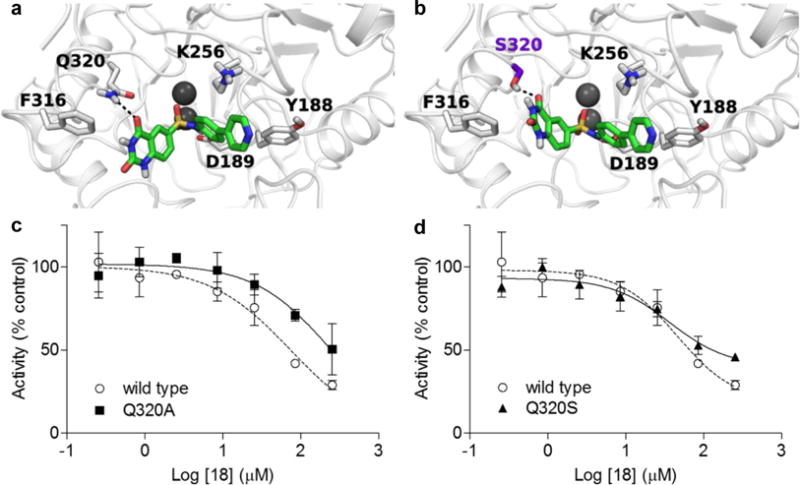
a) Docking solution for 18 in NAPE-PLD active site. b) 18 assumed a similar pose with Q320S NAPE-PLD. c) IC50 of 18 with Q320A or d) Q320S NAPE-PLD mutants.
To test the validity of this model, we generated two NAPE-PLD mutants that targeted glutamine 320: Q320A, in which the H-bonding capacity of glutamine is lost; and Q320S, in which H-bonding capacity is partially retained (purification is reported in Figure S3). Time-course (Figure S4) and kinetic analyses (Table S4) showed that both Q320A and Q320S mutants have reduced enzyme activity, compared to wild-type NAPE-PLD, confirming that glutamine 320 plays a significant role in supporting catalysis.2 The residual activity was sufficient, however, to investigate the effects of compound 18. The compound was significantly less potent on the Q320A mutant (IC50 = 89.2 ± 2.3 μM, n=3) than on wild-type NAPE-PLD (IC50 = 46.2 ± 1.6 μM, n=3) (Figure 4c), which is suggestive of a role for glutamine 320 in the inhibitory effects of 18. Interestingly, mutating glutamine 320 to serine (Figure 4b) did not significantly affect inhibition (36.7 ± 1.6 μM in Q320S NAPE-PLD, n=3) (Figure 4d), suggesting that serine may substitute for glutamine in the interaction with 18. We interpret the results of these studies as indicating that 18 inhibits NAPE-PLD activity by binding to the diatomic zinc center of the enzyme, via its sulfonamide moiety, and to glutamine 320, via its quinazolinedione ring.
Because our studies implicated an interaction of 18 with the diatomic zinc center of NAPE-PLD, we tested the selectivity of this effect by evaluating whether the compound might influence the activity of other enzymes that are characterized by zinc-dependent catalysis. However, 18 (50 μM) had no inhibitory effect on three such enzymes: carbonic anhydrase (CA) II, neutral endopeptidase and angiotensin-converting enzyme (ACE) (Table S5). Additionally, the compound (300 μM) did not affect (i) isofunctional, but zinc-independent enzyme activities present in cytosolic and microsomal fractions of mouse brain or small intestine (Figure S5), or (ii) lipid hydrolase activities involved in FAE metabolism, including human fatty acid amide hydrolase (FAAH)-1 (Figure S6a) and human N-acylethanolamine acid amidase (NAAA) (Figure S6b). Finally, we assessed whether 18 might be used to probe the functions of NAPE-PLD in intact cells. Human HEK-293 cells, which constitutively express the enzyme, were incubated with the compound (50 μM) for 4 hours and NAPE and FAE levels were measured in lipid extracts by liquid chromatography-mass spectrometry (LC-MS). Exposure to 18 resulted in a substantial accumulation of non-metabolized NAPE species (Figure S7a). At these time points, and in the absence of any other stimulation, the endogenous levels of oleoylethanolamide (OEA) and palmitoylethanolamide (PEA) remained unmodified, while significant changes were observed in stearoylethanolamide (SEA) (Figure S7b).
In conclusion, the present report describes the identification of the quinazoline sulfonamide derivative, 18 (ARN19874), as the first inhibitor of intracellular NAPE-PLD activity.
Supplementary Material
Acknowledgments
This work was partially supported by the NIH grant DA012413 (to D. P.). D. Piomelli ideated and directed the project; B. C. designed, performed and analyzed in vitro experiments; E. D., D. P. and P. T. designed and performed chemical syntheses; M. M., S. R. and M. M. ideated and performed in silico experiments; N. R. designed and performed cell experiments; P. M. and G. G. generated of one protein mutant and provided crystallographic information; T. B. performed chemical syntheses; D. P. wrote the manuscript along with M. M. and B. C..
Footnotes
Electronic Supplementary Information (ESI) available: experimental and in silico procedures, synthesis and characterization for new compounds. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Petersen G, Pedersen AH, Pickering DS, Begtrup M, Hansen HS. Chemistry and physics of lipids. 2009;162:53–61. doi: 10.1016/j.chemphyslip.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 2.Magotti P, Bauer I, Igarashi M, Babagoli M, Marotta R, Piomelli D, Garau G. Structure. 2015;23:598–604. doi: 10.1016/j.str.2014.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang J, Okamoto Y, Morishita J, Tsuboi K, Miyatake A, Ueda N. The Journal of biological chemistry. 2006;281:12325–12335. doi: 10.1074/jbc.M512359200. [DOI] [PubMed] [Google Scholar]
- 4.Ueda N, Okamoto Y, Morishita J. Life sciences. 2005;77:1750–1758. doi: 10.1016/j.lfs.2005.05.018. [DOI] [PubMed] [Google Scholar]
- 5.Brooks HB, Geeganage S, Kahl SD, Montrose C, Sittampalam S, Smith MC, Weidner JR. Assay Guidance Manual. Bethesda (MD): 2004. [Google Scholar]
- 6.Cadas H, Gaillet S, Beltramo M, Venance L, Piomelli D. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1996;16:3934–3942. doi: 10.1523/JNEUROSCI.16-12-03934.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Copeland RA. Methods Biochem Anal. 2005;46:1–265. [PubMed] [Google Scholar]
- 8.Pontis S, Ribeiro A, Sasso O, Piomelli D. Crit Rev Biochem Mol Biol. 2016;51:7–14. doi: 10.3109/10409238.2015.1092944. [DOI] [PubMed] [Google Scholar]
- 9.Wang X, Miyares RL, Ahern GP. J Physiol. 2005;564:541–547. doi: 10.1113/jphysiol.2004.081844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coulon D, Faure L, Salmon M, Wattelet V, Bessoule JJ. Biochimie. 2012;94:75–85. doi: 10.1016/j.biochi.2011.04.023. [DOI] [PubMed] [Google Scholar]
- 11.Natarajan V, Schmid PC, Schmid HH. Biochimica et biophysica acta. 1986;878:32–41. doi: 10.1016/0005-2760(86)90341-3. [DOI] [PubMed] [Google Scholar]
- 12.Chapman KD, Moore TS., Jr Plant physiology. 1993;102:761–769. doi: 10.1104/pp.102.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sandoval JA, Huang ZH, Garrett DC, Gage DA, Chapman KD. Plant physiology. 1995;109:269–275. doi: 10.1104/pp.109.1.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vermehren C, Hansen HS, Clausen-Beck B, Frokjaer S. Int J Pharm. 2003;254:49–53. doi: 10.1016/s0378-5173(02)00681-6. [DOI] [PubMed] [Google Scholar]
- 15.Shangguan T, Pak CC, Ali S, Janoff AS, Meers P. Biochimica et biophysica acta. 1998;1368:171–183. doi: 10.1016/s0005-2736(97)00151-x. [DOI] [PubMed] [Google Scholar]
- 16.Basu A, Prence E, Garrett K, Glew RH, Ellingson JS. Archives of biochemistry and biophysics. 1985;243:28–34. doi: 10.1016/0003-9861(85)90770-2. [DOI] [PubMed] [Google Scholar]
- 17.Okamoto Y, Morishita J, Wang J, Schmid PC, Krebsbach RJ, Schmid HH, Ueda N. Biochem J. 2005;389:241–247. doi: 10.1042/BJ20041790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leishman E, Mackie K, Luquet S, Bradshaw HB. Biochimica et biophysica acta. 2016;1861:491–500. doi: 10.1016/j.bbalip.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Margheritis E, Castellani B, Magotti P, Peruzzi S, Romeo E, Natali F, Mostarda S, Gioiello A, Piomelli D, Garau G. ACS Chem Biol. 2016;11:2908–2914. doi: 10.1021/acschembio.6b00624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Supuran CT. Bioorg Med Chem Lett. 2010;20:3467–3474. doi: 10.1016/j.bmcl.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 21.Falsini M, Squarcialupi L, Catarzi D, Varano F, Betti M, Di Cesare Mannelli L, Tenci B, Ghelardini C, Tanc M, Angeli A, Supuran CT, Colotta V. J Med Chem. 2017;60:6428–6439. doi: 10.1021/acs.jmedchem.7b00766. [DOI] [PubMed] [Google Scholar]
- 22.Supuran CT, Scozzafava A, Casini A. Med Res Rev. 2003;23:146–189. doi: 10.1002/med.10025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.