

Summary
Hajdu-Cheney syndrome (HCS), a rare autosomal disorder caused by heterozygous mutations in NOTCH2, is clinically characterized by acroosteolysis, severe osteoporosis, short stature, neurological symptoms, cardiovascular defects, and polycystic kidneys. Recent studies identified that aberrant NOTCH2 signaling and consequent osteoclast hyperactivity are closely associated with the bone-related disorder pathogenesis, but the exact molecular mechanisms remain unclear. Here, we demonstrate that sustained osteoclast activity is largely due to accumulation of NOTCH2 carrying a truncated C-terminus that escapes FBW7-mediated ubiquitination and degradation. Mice with osteoclast-specific Fbw7 ablation revealed osteoporotic phenotypes reminiscent of HCS, due to elevated Notch2 signaling. Importantly, administration of Notch inhibitors in Fbw7 conditional knockout mice alleviated progressive bone resorption. These findings highlight the molecular basis of HCS pathogenesis and provide clinical insights into potential targeted therapeutic strategies for skeletal disorders associated with the aberrant FBW7/NOTCH2 pathway as observed in patients with HCS.
Keywords: Hajdu-Cheney syndrome, NOTCH2, Osteoclast, Ubiquitination, SCF E3 ubiquitin ligase, FBW7, Osteolysis, Osteoporosis, Osteoclastogenesis, NOTCH inhibitor
eTOC Blurb
Fukushima et al. demonstrated that the sustained osteoclast activity in Hajdu-Cheney syndrome (HCS) is largely due to elevated protein abundance of the C-terminus truncating NOTCH2 mutant that escapes FBW7-mediated ubiquitination and proteolysis, suggesting that the FBW7/NOTCH2 signaling pathway is a potential therapeutic target for osteolytic bone disorders, including HCS.
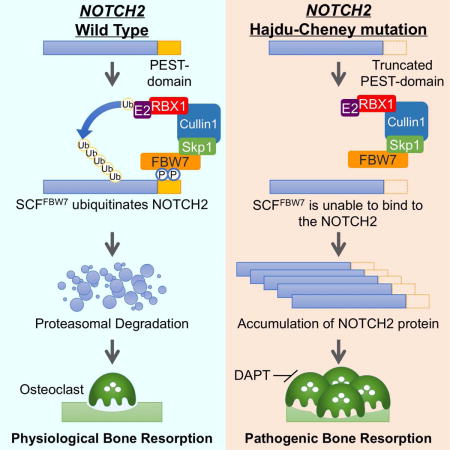
Introduction
Bone metabolism and homeostasis are regulated by the bone remodeling activities of osteoblasts and osteoclasts, and an imbalance in these activities leads to the development of various skeletal disorders (Boyle et al., 2003; Rodan and Martin, 2000). Hajdu-Cheney syndrome (HCS) [OMIM ID: 102500] is a rare autosomal dominant disorder characterized by progressive bone abnormalities, accompanied with acroosteolysis, severe osteoporosis, short stature, specific craniofacial features, wormian bones, neurological symptoms, cardiovascular defects, and polycystic kidneys (Brennan and Pauli, 2001; Zanotti and Canalis, 2014). HCS is associated with heterozygous truncating mutations in NOTCH2, encoding a single-pass transmembrane protein from the NOTCH receptor family (NOTCH1-NOTCH4 in mammals) (Isidor et al., 2011; Simpson et al., 2011).
NOTCH2 plays critical roles in cell fate determination (Kopan and Ilagan, 2009). The spatial and temporal regulation of Notch expression and modifications confer functional diversity to the pathway. NOTCH2 interaction with its single-pass transmembrane ligands, Delta/Jagged proteins, generally expressed on the cell surface of adjacent cells, triggers a multi-step proteolytic cleavage of the NOTCH protein adjacent to its transmembrane domains, partly through activation of ADAM10/17 (extracellular) and the presenilin complex of proteases (intracellular). Consequently, NOTCH2 cleavage releases the NOTCH2 intracellular domain (ICD), which translocates to the nucleus where it activates the transcription of various downstream target genes (Kopan and Ilagan, 2009).
Previous studies revealed important roles of the Notch2 pathway in bone resorption (Fukushima et al., 2008; Hilton et al., 2008; Isidor et al., 2011; Simpson et al., 2011). Suppression of all Notch receptor activities via osteoblast-specific deletion of presenilin-1 and -2 leads to age-related osteoporosis, partly due to enhanced osteoclast formation (Hilton et al., 2008). We previously showed that Notch2 depletion in osteoclast precursors impairs osteoclastogenesis, while ectopic Notch2 ICD expression facilitates osteoclast differentiation (Fukushima et al., 2008). Interestingly, knock-in mice harboring the Notch2Q2319X HCS mutant exhibited osteopenia due to enhanced osteoclast formation, without significantly affecting osteoblast number and activity (Canalis et al., 2016). This phenotype was alleviated by an antibody against Notch2 negative regulatory region (NRR) (Canalis et al., 2017). Thus, osteoclastic bone loss caused by sustained NOTCH2 activity may lead to HCS pathogenesis.
NOTCH2 mutations identified in HCS are clustered in the last exon encoding a region rich in proline-glutamate-serine-threonine (PEST sequence) (Isidor et al., 2011; Simpson et al., 2011), which is implicated in proteasome-dependent protein degradation (Rechsteiner and Rogers, 1996). These NOTCH2 mutations are either nonsense or missense variants, causing an immature truncation of NOTCH2 protein that lacks the functional PEST domain (Isidor et al., 2011; Simpson et al., 2011). Equivalent NOTCH1 mutations in the PEST sequence were previously reported in cancer (Wang et al., 2015). For instance, truncated NOTCH1 ICD contributes to the development of T-cell acute lymphoblastic leukemia (T-ALL) (Weng et al., 2004), indicating that deletion or truncation of PEST sequences confers gain of Notch function.
The ubiquitin proteasome system (UPS) plays critical roles in various cellular processes, including cell cycle progression, immune response, and metabolism by targeting protein substrates for ubiquitination (Hershko and Ciechanover, 1998). The approximately 600 E3 ubiquitin ligases encoded in the human genome confer substrate specificity in the UPS system (Li et al., 2008). The SCF (Skp1-Cullin1-F-box protein) complex belongs to the Cullin-Ring type of E3 ligases (CRL), the largest family of E3 ligases (Deshaies and Joazeiro, 2009; Petroski and Deshaies, 2005). SCF utilizes 69 variable F-box proteins as substrate receptors, thereby providing the necessary diversity for specific enzymatic reactions controlling downstream substrates protein stability (Nakayama and Nakayama, 2006; Wang et al., 2014). Among them, FBW7 is one of the best-characterized F-box proteins, which selectively target oncogenic proteins such as cyclin E, c-Myc, c-Jun, and Mcl-1 for proteasome-dependent degradation (Davis et al., 2014; Wang et al., 2014). In addition, FBW7 mutations, deletions, or epigenetic silencing is frequently observed in various cancers (Davis et al., 2014). In vivo studies using tissue-specific FBW7 ablation or knock-in mouse models confirmed FBW7 tumor suppressive function, especially in the context of leukemia and colorectal cancer (Davis et al., 2014; Wang et al., 2014).
FBW7 has recently been implicated in osteoblast and chondrocyte differentiation (Yumimoto et al., 2013), suggesting important roles of FBW7 as a modulator of skeletal development. However, the molecular link between FBW7 function and bone formation under physiological and pathophysiological conditions remains unknown. Here, we characterized NOTCH2 as a bona fide SCFFBW7 substrate and further indicated that NOTCH2 mutants of HCS escape SCFFBW7-catalyzed NOTCH2 ubiquitination and subsequent degradation. Osteoclast-specific Fbw7 knockout mice exhibited osteoporotic and acroosteolysis-like phenotypes, which are characteristic manifestations observed in patients with HCS. Furthermore, treatment of Fbw7 conditional knockout mice with Notch inhibitors relieved the observed pathological bone phenotypes, suggesting that the FBW7/NOTCH2 signaling pathway is a potential therapeutic target for osteoclastic skeletal disorders, including HCS.
Results
SCFFbw7 Controls NOTCH2 Protein Stability
NOTCH1 is an unstable protein whose stability is largely controlled by proteasome-dependent proteolysis (O’Neil and Look, 2007). However, the molecular mechanism underlying NOTCH2 stability has not been characterized. NOTCH2 protein has a short half-life compared to NOTCH1 (Figure 1A). This led us to speculate that NOTCH2 protein stability is also regulated by the ubiquitin-proteasome pathway (Liu et al., 2016). Since CRL is the largest family of E3 ubiquitin ligase and governs NOTCH1 protein stability (O’Neil et al., 2007; Thompson et al., 2007), we hypothesized that CRL negatively regulates NOTCH2 protein abundance. To identify which CRL complex interacts with NOTCH2, we performed a co-immunoprecipitation (IP) analysis. NOTCH2 specifically interacted with Cullin1, a scaffold subunit of SCF (Skp1-Cullin1-F-box protein) E3 ligase complex, but not with other Cullin family members in cells (Figure 1B). Furthermore, depletion of Cullin1 led to elevated protein abundance and stability of NOTCH2 in HeLa cells (Figures 1C and S1A), which in turn facilitated Jagged1-dependent accumulation of the NOTCH2 intracellular domain (ICD) in the nucleus (Figures S1B and S1C). Furthermore, Skp1 and Rbx1, additional components of the SCF complex, also interacted with NOTCH2 in cells (Figures S1D and S1E). Together, these data suggest a role for the SCF complex in regulating NOTCH2 protein turnover.
Figure 1. SCFFBW7 controls NOTCH2 protein stability.
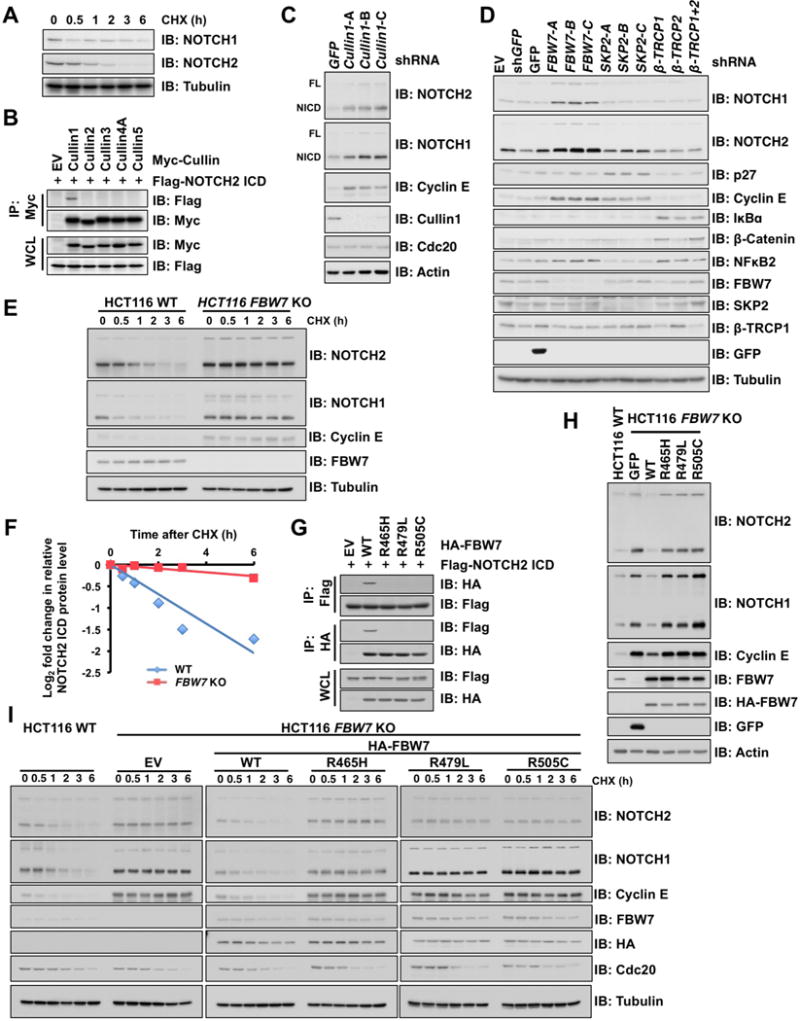
A. Immunoblot (IB) analysis of whole cell lysates (WCL) derived from HeLa cells treated with 20 μg/mL cycloheximide (CHX) and harvested at the indicated time points.
B. IB analysis of WCL and anti-Myc immunoprecipitates (IPs) derived from 293T cells transfected with Flag-NOTCH2 ICD along with empty vector (EV) or the Myc-Cullins (Cullin1, 2, 3, 4A, and 5). At 24 h post-transfection, cells were treated with proteasome inhibitor, 15 μM MG132, for 12 h before harvesting.
C. IB analysis of WCL derived from HeLa cells infected with shRNA lentiviral vectors specific for GFP or Cullin1. After infection, cells were selected with 1 μg/mL puromycin for 7 days before harvesting. FL, Full length; NICD, NOTCH intracellular domain.
D. IB analysis of WCL derived from HeLa cells infected with indicated shRNA lentiviral vectors specific for GFP, FBW7, SKP2, β-TRCP1, β-TRCP2, or β-TRCP1+2 (shRNAs against both β-TRCP1 and β-TRCP2 paralogs).
E. IB analysis of WCL derived from wild-type (WT) (FBW7+/+) and FBW7 knockout (FBW7−/−) HCT116 cells treated with 20 μg/mL cycloheximide (CHX) and harvested at the indicated time points.
F. Quantification of the band intensities in (E). NOTCH2 ICD intensity was normalized by Tubulin and to the t = 0 time point.
G. IB analysis of WCL and anti-HA IPs derived from 293T cells transfected with Flag-NOTCH2 ICD along with EV, WT, R465H, R479L, or R505C HA-FBW7. At 24 h post-transfection, cells were treated with 15 μM MG132 for 12 h before harvesting.
H. IB analysis of WCL derived from parental WT HCT116 (FBW7+/+) and FBW7 knockout HCT116 (FBW7−/−) cells infected with lentiviral vectors expressing GFP and WT, R465H, R479L, or R505C HA-FBW7.
I. IB analysis of WCL derived from parental WT HCT116 and FBW7-reconstituted FBW7 knockout HCT116 cells that are presented in (H). Cells were treated with 20 μg/mL CHX and harvested at the indicated time points.
See also Figure S1
We next investigated which F-box protein, the substrate recognition subunit of SCF, regulates NOTCH2 proteolysis. FBW7 knockdown utilizing multiple shRNAs in HeLa cells induced NOTCH2 protein accumulation, whereas no significant increase in NOTCH2 protein levels was observed in cells depleted of other related F-box proteins, including SKP2 and β-TRCP (Figure 1D). Furthermore, FBW7 depletion significantly extended the half-life of NOTCH2 (Figures 1E, 1F, and S1F) and enhanced Jagged1-dependent nuclear accumulation of NOTCH2 (Figures S1G and S1H). These results indicate that NOTCH2 accumulation may be largely due to increased protein turnover.
FBW7 WD40-repeat domain is frequently mutated in cancer, notably at hot-spot positions R465, R479, and R505 (Davis et al., 2014), which are crucial for the structural integrity of the substrate recognition pocket. Wild-type (WT), but not the WD40-mutated FBW7, interacted with NOTCH2 ICD (Figure 1G). Furthermore, re-introducing WT FBW7, but not WD40-mutated FBW7, in FBW7−/− cells led to a decrease in protein abundance of NOTCH2 (Figures 1H and S1I) due to FBW7-mediated shortened NOTCH2 protein half-life (Figures 1I, S1J and S1K). NOTCH2 mRNA levels were not significantly affected following depletion of Cullin1 and FBW7 (Figures S1L and S1M). Taken together, these data indicated that SCFFBW7 primarily regulates NOTCH2 protein stability through post-translational modification.
GSK3 Facilitates NOTCH2 Protein Degradation
Substrate recognition by FBW7 typically requires prior phosphorylation of its substrates within the consensus degron motif (Crusio et al., 2010). To identify the upstream modifying enzyme(s) governing FBW7 phosphorylation, we co-transfected NOTCH2 in 293T cells with a set of protein kinases previously reported to modulate FBW7 substrates, and assessed their effects on NOTCH2 protein abundance. Notably, ectopic expression of active GSK3, but neither a kinase-dead form of GSK3 (K85A) nor other kinases examined (Figures 2A and S2A), diminished NOTCH2 protein levels. Consistently, GSK3 knockdown in HeLa cells resulted in NOTCH2 protein accumulation (Figures 2B and S2B) without significant changes in NOTCH2 mRNA levels (Figure S2C). GSK3 was previously shown to phosphorylate NOTCH2 ICD and impact on NOTCH2 transcriptional activity (Espinosa et al., 2003). To further determine the functional impacts of GSK3-mediated NOTCH2 phosphorylation, we performed co-IP experiments following GSK3 inhibitor treatment. Inhibition of GSK3 in 293T cells largely disrupted the interaction between FBW7 with NOTCH2 (Figure 2C). Conversely, ectopic expression of GSK3 facilitated FBW7-directed NOTCH2 ubiquitination (Figure 2D), while the inhibition of endogenous GSK3 activity diminished the FBW7-mediated ubiquitination of NOTCH2 (Figure S2D). These findings indicate that GSK3 phosphorylates NOTCH2 to promote FBW7-mediated NOTCH2 ubiquitination and degradation.
Figure 2. GSK3-mediated phosphorylation of NOTCH2 promotes its subsequent degradation.
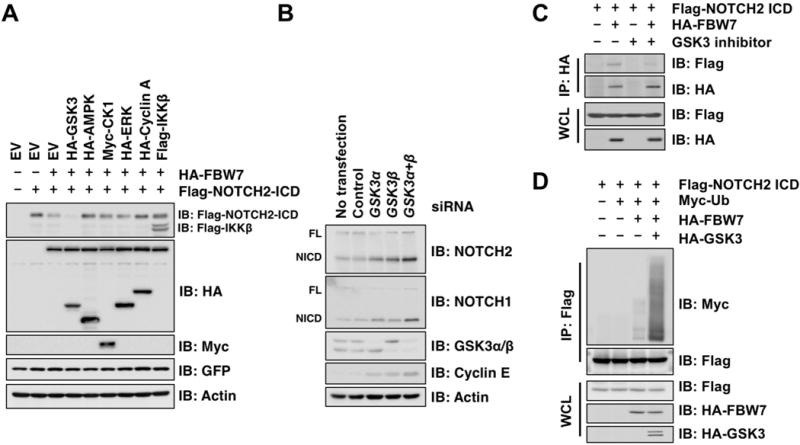
A. IB analysis of WCL derived from 293T cells transfected with Flag-NOTCH2 ICD and HA-FBW7 along with the expression plasmids of protein kinases as indicated. GFP served as an internal control for transfection efficiency.
B. IB analysis of WCL derived from HeLa cells transfected with control and indicated siRNAs specific for GSK3α and GSK3β. FL, Full length; NICD, NOTCH intracellular domain.
C. IB analysis of WCL and anti-HA IPs derived from 293T cells transfected with Flag-NOTCH2 ICD and HA-FBW7 as indicated. At 24 h post-transfection, cells were treated with GSK3 specific inhibitor, BIO (2 μM), and 15 μM MG132, for 12 h before harvesting.
D. IB analysis of WCL and anti-Flag IPs derived from 293T cells transfected with Flag-NOTCH2 ICD, Myc-ubiquitin (Myc-Ub), HA-FBW7, and HA-GSK3β as indicated. At 24 h post-transfection, cells were treated with 15 μM MG132 for 12 h before harvesting.
See also Figure S2
NOTCH2 Mutations in HCS Impair the FBW7 Phospho-Degron Motif
HCS is an ideal model to study a physiological function of NOTCH2 as C-terminal truncation mutations in HCS are shown to confer gain-of-function properties to Notch2 in mice (Canalis et al., 2017; Canalis et al., 2016; Vollersen et al., 2017). Moreover, most identified HCS NOTCH2 mutations cluster between the second nuclear localization signal (NLS) and the PEST domain, resulting in C-terminal premature truncation of NOTCH2 that lacks the PEST sequence (Figure 3A). A survey of primary sequences around the truncated region led us to identify several putative phospho-degron motifs for recognition by FBW7 (Figure 3B). To determine the molecular link between HCS NOTCH2 mutations and elevated NOTCH2 protein abundance and downstream signaling, we utilized two frequently observed HCS truncation mutations to generate NOTCH2 ICD deletion constructs (V2151L fsX4, named Δ1, and P2417I fsX6, named Δ2, hereafter). As shown in Figures 3C and 3D, both Δ1 and Δ2 mutants abrogated FBW7 binding and were subsequently protected from FBW7/GSK3-mediated degradation. Mechanistically, absence of FBW7-mediated degradation was likely due to abolishment of FBW7-mediated poly-ubiquitination of the NOTCH2 mutants (Figure 3E), leading to their extended protein half-life compared to WT-NOTCH2 (Figures 3F and S3A).
Figure 3. NOTCH2 mutations in HCS impair the FBW7 recognizable phospho-degron motif.
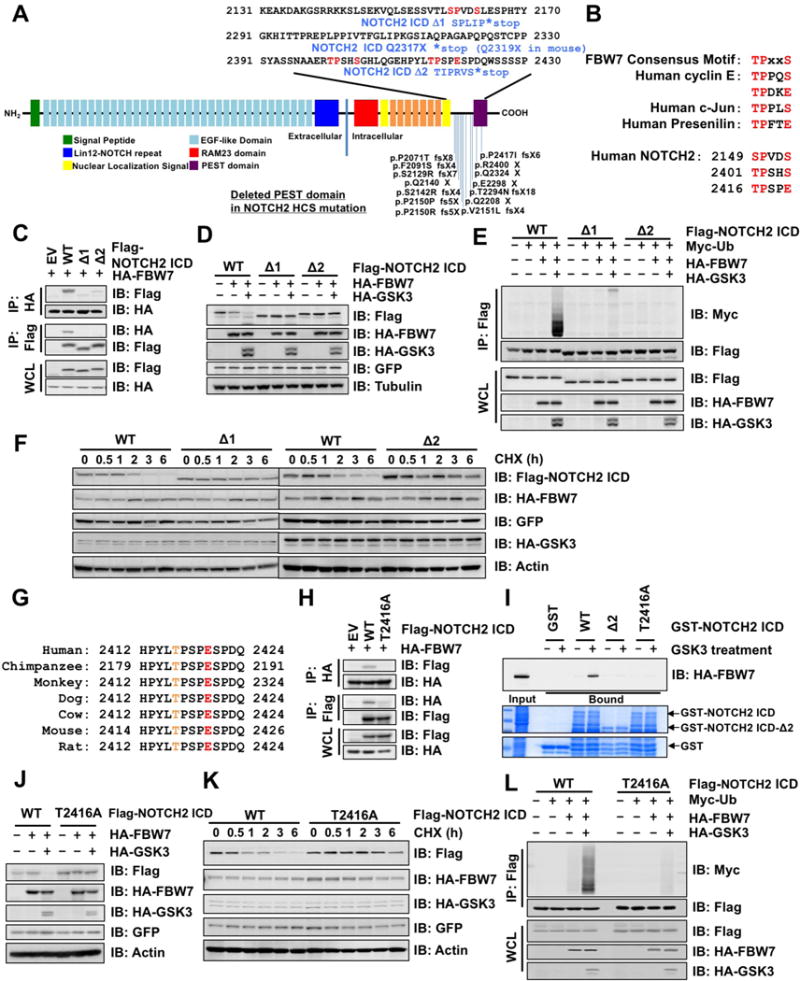
A. Domain structure of human NOTCH2 protein, positions of mutations reported in patients with HCS, and truncation mutations used in this study (namely NOTCH2 ICD Δ1 and Δ2).
B. FBW7 consensus degron motifs in human cyclin E, c-Jun, Presenilin, and NOTCH2.
C. IB analysis of WCL and anti-HA or -Flag IPs derived from 293T cells transfected with EV or indicated Flag-NOTCH2 ICD (WT, Δ1, or Δ2). At 24 h post-transfection, cells were treated with 15 μM MG132 for 12 h before harvesting.
D. IB analysis of WCL derived from 293T cells transfected with Flag-NOTCH2 ICD, HA-FBW7, and HA-GSK3β as indicated. GFP served as the internal control for transfection efficiency.
E. IB analysis of WCL and anti-Flag IPs derived from 293T cells transfected with Flag-NOTCH2 ICD (WT, Δ1, or Δ2), Myc-Ub, HA-FBW7, and GSK3β as indicated. At 24 h post-transfection, cells were treated with 15 μM MG132 for 12 h before harvesting.
F. IB analysis of WCL derived from HeLa cells transfected with Flag-NOTCH2 ICD (WT, Δ1, or Δ2), HA-FBW7, and HS-GSK3β. At 48 h post-transfection, cells were treated with 20 μg/mL CHX and harvested at the indicated time points.
G. Alignment of NOTCH2 sequences surrounding the putative FBW7 degron motif from different species.
H. IB analysis of WCL and anti-HA or -Flag IPs derived from 293T cells transfected with EV or Flag-NOTCH2 ICD (WT or T2416A) and HA-FBW7. At 24 h post-transfection, cells were treated with 15 μM MG132 for 12 h before harvesting.
I. In vitro binding of HA-FBW7 with GST-NOTCH2 ICD. Bacterially purified GST or GST-NOTCH2 ICD (WT, Δ2, or T2416A) protein treated with active GSK3 as indicated was incubated with WCL derived from 293T cells transfected with HA-FBW7. The bound HA-FBW7 protein with GST-NOTCH2 ICD was eluted and subjected to IB analysis.
J. IB analysis of WCL derived from 293T cells transfected with Flag-NOTCH2 ICD (WT or T2416A), HA-FBW7, and HA-GSK3β as indicated. GFP served as an internal control for transfection efficiency.
K. IB analysis of WCL derived from HeLa cells transfected with Flag-NOTCH2 ICD (WT or T2416A), HA-FBW7, and HA-GSK3β. At 48 h post-transfection, cells were treated with 20 μg/mL CHX and harvested at the indicated time points.
L. IB analysis of WCL and anti-Flag IPs from 293T cells transfected with Flag-NOTCH2 ICD (WT or T2416A), Myc-Ub, HA-FBW7, and GSK3β as indicated. At 24 h post-transfection, cells were treated with 15 μM MG132 for 12 h before harvesting.
See also Figure S3
Given that both truncation mutants displayed similar effects with regard to evasion from FBW7-mediated proteolysis, we set out to pinpoint the critical degron deleted in the Δ2 mutant. To this end, we found that a potential CPD motif harboring the phospho-Thr residue (T2416) was highly conserved across different species (Figure 3G). Alanine substitution of Thr2416 (T2416A) centered in the degron motif abolished its interaction with FBW7 in 293T cells (Figure 3H) and abrogated GSK3-mediated phosphorylation-dependent interaction with FBW7 in vitro (Figure 3I). Moreover, the T2416A mutant was resistant to FBW7/GSK3-mediated degradation (Figure 3J), leading to an extended half-life (Figures 3K and S3B), and reduced ubiquitination by FBW7/GSK3 (Figure 3L). We therefore conclude that FBW7 regulates NOTCH2 stability via a single degron motif including T2416 (2416TPSPES) since introducing mutations to other degron-like motifs (2149SPVDSL and 2401TPSHSP) (Figures 3A, 3B, and S3C) did not affect the interaction between FBW7 and NOTCH2 (Figure S3D), and the FBW7/GSK3-mediated NOTCH2 downregulation (Figure S3E). Taken together, these data support the model that sustained NOTCH2 activity observed in HCS is likely due to deletion of the FBW7 degron motif in NOTCH2 C-terminus region, leading to elevated NOTCH2 protein abundance in HCS.
Gain of NOTCH2 Function Promotes Osteoclastogenesis in Peripheral Blood Monocytes Derived from Patients with HCS
We further validated the physiological significance of these findings using both engineered mouse models and human pathological specimens derived from patients with HCS. We previously described a positive role of Notch2 pathway in RANKL-induced osteoclastogenesis (Fukushima et al., 2008). Similarly, sustained Notch2 activity in Hajdu-Cheney mouse model (Notch2Q2319X) has been recently shown to enhance osteoclast formation, leading to osteopenia, a characteristic of HCS (Canalis et al., 2017; Canalis et al., 2016). Considering the contribution of FBW7 in the osteopenia phenotype, we generated the human NOTCH2 Q2317X (corresponding to mouse Q2319X) mutant for biochemical analyses and showed that the Q2317X mutant failed to interact with FBW7 (Figure S4A) and exhibited diminished FBW7-mediated ubiquitination in cells (Figure S4B). These data suggested that the osteoporotic phenotypes of the HCS mice might partly result from Notch2 stabilization by escaping FBW7-dependent proteolysis. Thus, we next examined how Notch2 gain-of-function in HCS affects osteoclast differentiation.
Depletion of Notch2 in mouse bone marrow cells resulted in suppression of RANKL-induced osteoclastogenesis (Figure 4A and 4B), while re-introducing NOTCH2 with the HCS mutation (Δ2) or FBW7 degron mutation (T2416A) significantly enhanced osteoclast formation compared to WT NOTCH2 (Figures 4A and 4B). Consistently, osteoclastogenesis was significantly enhanced in peripheral blood derived from patients with HCS (Figures 4: Case #1 and S4: Case #2 and #3) compared with control subjects (Figures 4C and 4D, S4D and S4G). Semi-quantitative RT-PCR analyses revealed a comparable NOTCH2 mRNA expression in osteoclasts from both the HCS patients and control subjects. On the other hand, NFATc1, an osteoclast differentiation marker, cathepsin K, a functional bone-resolving osteoclast marker, and Hes1, a NOTCH2 target gene, were upregulated in patients (Figure 4E, S4E and S4H). Furthermore, NOTCH2 abundance in cells derived from patients with HCS was significantly higher than in cells derived from control subjects (Figure 4F, S4C, S4F and S4I). More importantly, upregulation of NOTCH2 protein was largely due to its extended half-life (Figure 4G), but not impaired FBW7 function as the half-life of another FBW7 substrate, cyclin E, is comparable between patient and control samples (Figure 4G). These data imply that sustained NOTCH2 abundance and signaling caused by the aberrant FBW7/NOTCH2 pathway may facilitate osteoclast formation in vivo.
Figure 4. Gain of NOTCH2 function promotes osteoclastogenesis in peripheral blood monocytes derived from patients with HCS.
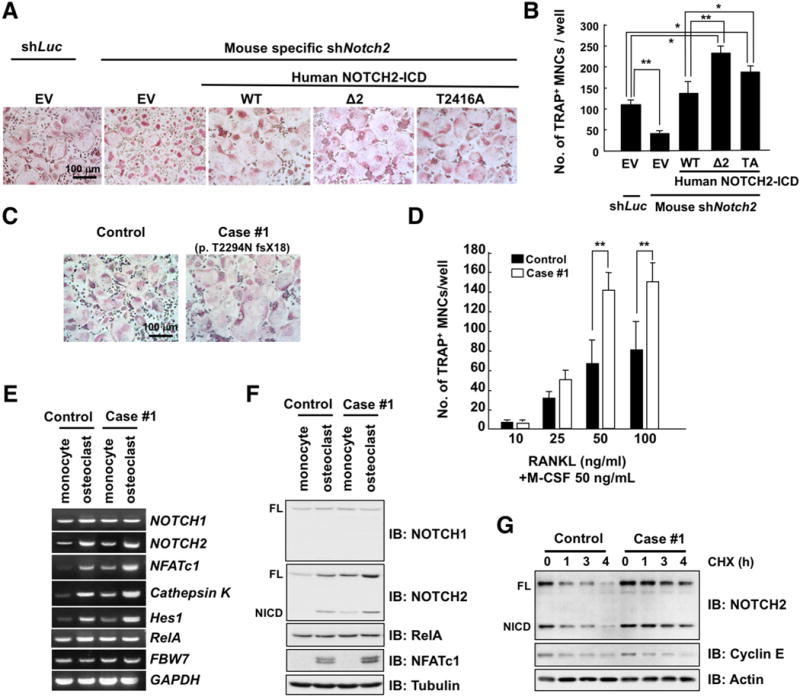
A. Microscopic analysis of osteoclasts. Mouse bone marrow cells (BMCs) were cultured in the presence of M-CSF (50 ng/mL) for 3 days. Cells were infected with shRNA lentiviral vectors specific for luciferase (Luc) or mouse Notch2 and sequentially infected with lentiviral expression vector of empty vector (EV) or human NOTCH2 ICD (WT, Δ2, or T2416A) as indicated. The infected cells were cultured with RANKL (50 ng/mL) for 3 days. Cells were fixed and stained for tartrate-resistant acid phosphatase (TRAP), a histochemical osteoclast differentiation marker. The TRAP-positive multinucleated cells (MNCs), which display cytoplasmic red staining and a minimum of three nuclei, were counted as osteoclasts. Scale bar, 100 μm.
B. TRAP-positive MNCs in each well were counted. Date represents mean ± SD, n = 3, *p < 0.05, **p < 0.01, Student’s t test.
C. Peripheral blood cells derived from a patient (Case #1) with HCS or control subject were cultured in the presence of M-CSF (50 ng/mL) for 3 days and then treated with RANKL (50 ng/mL) for 3 days. Cells were fixed and stained for TRAP. Scale bar, 100 μm.
D. Peripheral blood cells derived from a patient (Case #1) with HCS or control subject were cultured in the presence of M-CSF (50 ng/mL) for 3 days. Then the differentiated monocytes were treated with increasing concentration of RANKL for 3 days for differentiation to osteoclasts. Cells were fixed and stained for TRAP, and then TRAP-positive MNCs were counted. Data represents mean ± SD, n = 3, **p < 0.01, Student’s t test.
E. RT-PCR analysis of NOTCH1, NOTCH2, RelA, NFATc1, Cathepsin K, Hes1, FBW7, and GAPDH expression in monocytes and osteoclasts of a HCS patient or control subject.
F. IB analysis of WCL derived from monocytes and osteoclasts of a control subject or patient with HCS.
G. IB analysis of WCL derived from osteoclasts of a HCS patient or control subject, which were treated with 20 μg/mL CHX and harvested at the indicated time points.
See also Figure S4
Osteoclast-Specific Ablation of Fbw7 Results in Enhanced Bone Resorption Phenocopying HCS
Notch2Q2319X knock-in mice display osteopenia, possibly stemming from accelerated osteoclast formation, but the underlying molecular mechanism remains undefined (Canalis et al., 2016). To determine if alteration of the Fbw7/Notch2 signaling pathway affects bone homeostasis, we investigated Fbw7 physiological function in utilizing an Fbw7 knockout mouse model. Whole body depletion of Fbw7 (Fbw7−/−) is embryonic lethal therefore we generated conditional knockout (cKO) mice in which Fbw7 expression is specifically abolished in osteoclasts by crossing Fbw7F/F mice with mice expressing Cre recombinase under the control of the osteoclast-specific cathepsin K promoter (Figures S5A and S5B).
Histomorphometric analysis indicated that Fbw7 cKO mice exhibited an enhanced osteoclast formation and increased bone absorption-related matrix (Figure 5A). These phenotypes are characterized by the significant increase in osteoclast bone surface area, osteoclast number, and bone erosion (Figure 5B). However, no significant change was observed in osteoblast differentiation parameters including osteoblast bone surface area, osteoblast number (Figure 5B), and osteoblastic bone formation (Figure 5C). Notably, radiographic analyses utilizing soft X-ray and 3D microcomputed tomography (microCT) indicated significant levels of bone resorption in Fbw7 cKO mice compared with their WT littermates (Figures 5D and 5E). More importantly, compared to WT mice, Fbw7 cKO mice displayed pathological bone phenotypes such as progressive bone resorption in the tibia and hind paws (Figures 5D and 5E). Mechanical analyses of femur and tibia from control and Fbw7 cKO mice showed that bone strength of Fbw7 cKO mice decreased by 40% and 36%, respectively (Figure 5F), confirming that Fbw7 ablation significantly increased bone fragility. The microstructural indicators further revealed that bone mass was lower in Fbw7 cKO mice with increased trabecular separation which is an osteoclastic bone-resolving parameter, while no significant change was observed in bone mineral density that is a surrogate of osteoblastic calcification (Figure 5G). Altogether, these results demonstrated that the osteoclast-specific Fbw7 deletion led to osteopenia secondary to accelerated osteoporosis, a phenotype that is typically observed in patients with HCS and a HCS mouse model expressing mutant NOTCH2.
Figure 5. Osteoclast-specific genetic ablation of Fbw7 results in enhanced bone resorption that phenocopies HCS.
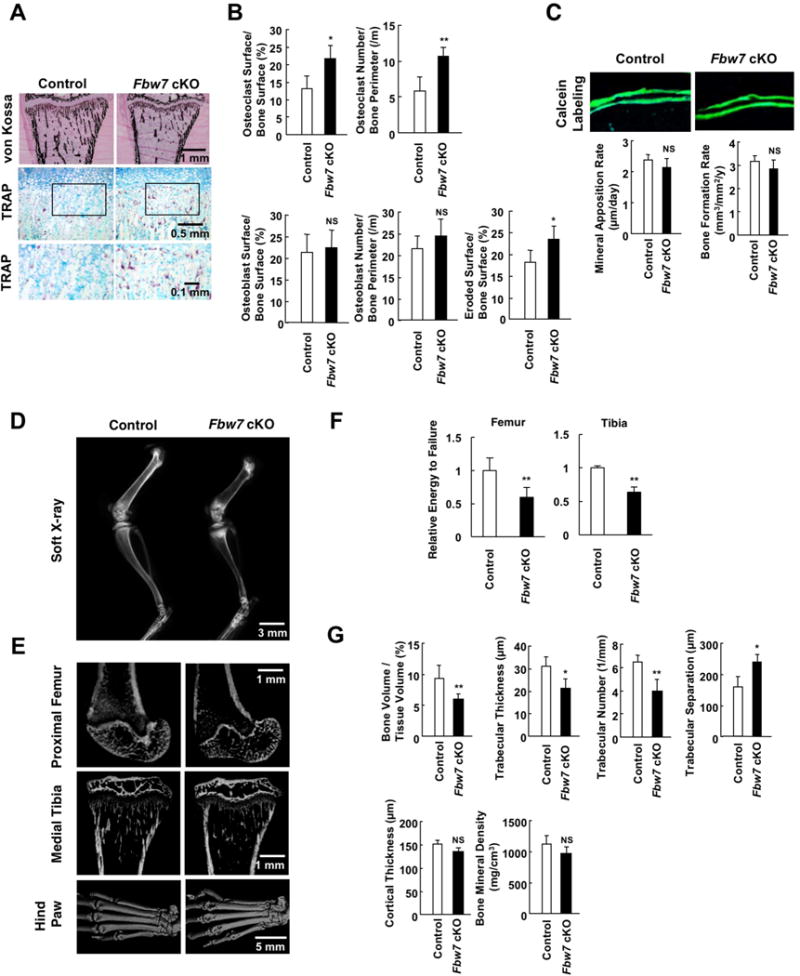
A. Histological analyses of von Kossa- (upper panel, scale bar: 1 mm) and TRAP- (middle panel, scale bar: 0.5 mm, lower panel, scale bar: 0.1 mm) stained sections of the proximal tibia from Fbw7 cKO mice (Cathepsin K-Cre/Fbw7F/F) and control mice (Fbw7F/F).
B. Histomorphometric analyses of the proximal tibia. Data represents mean ± SD, n = 5, *p < 0.05, **p < 0.01, NS, not significant, Student’s t test.
C. Calcein labeling of the tibia from Fbw7 cKO and control mice, and bone formation rate. Data represent mean ± SD, n = 5, NS, not significant, Student’s t test.
D. Soft X-ray images of the femurs. Scale bar, 3 mm.
E. Micro-CT images of the proximal femur (upper, scale bar: 1 mm), medial tibia (middle, scale bar: 1 mm), and hind paws (lower, scale bar: 5 mm).
F. Three point bending tests for assessing biomechanical bone properties of femur and tibia from Fbw7 cKO and control mice. Energy to failure (N.mm) was measured and normalized to the control mouse group. Data represent mean ± SD, n = 4-6, **p < 0.01, Student’s t test.
G. Microstructural parameters of the distal femur. Data represents mean ± SD, n = 3, *p < 0.05, **p < 0.01, NS, not significant, Student’s t test.
See also Figure S5
Fbw7-Deleted Osteoclast Precursors Display Elevated Capacity of Osteoclast Differentiation
To determine the correlation between pathological manifestations of Fbw7 cKO mice and Fbw7 physiological function, we evaluated the efficiency of osteoclast formation using bone marrow cells obtained from Fbw7 cKO and control mice. Notably, RANKL-induced osteoclast formation was increased in Fbw7 cKO bone marrow cells compared to control cells (Figures 6A and 6B). Furthermore, Notch2 was elevated only in osteoclasts, but not other bone marrow-derived cell types, from the osteoclast-specific Fbw7 cKO mice, which also exhibited elevation in osteoclast differentiation markers, c-fos and NFATc, and the activation of NFκB2 pathway occurring in differentiated osteoclasts (Figures 6C and S6A). Furthermore, a cohort of Notch2 downstream target genes was induced, including Hes1, NFATc1, and cathepsin K (Figures S6B-S6D).
Figure 6. Fbw7-depleted osteoclast precursors display elevated capacity of osteoclast differentiation.
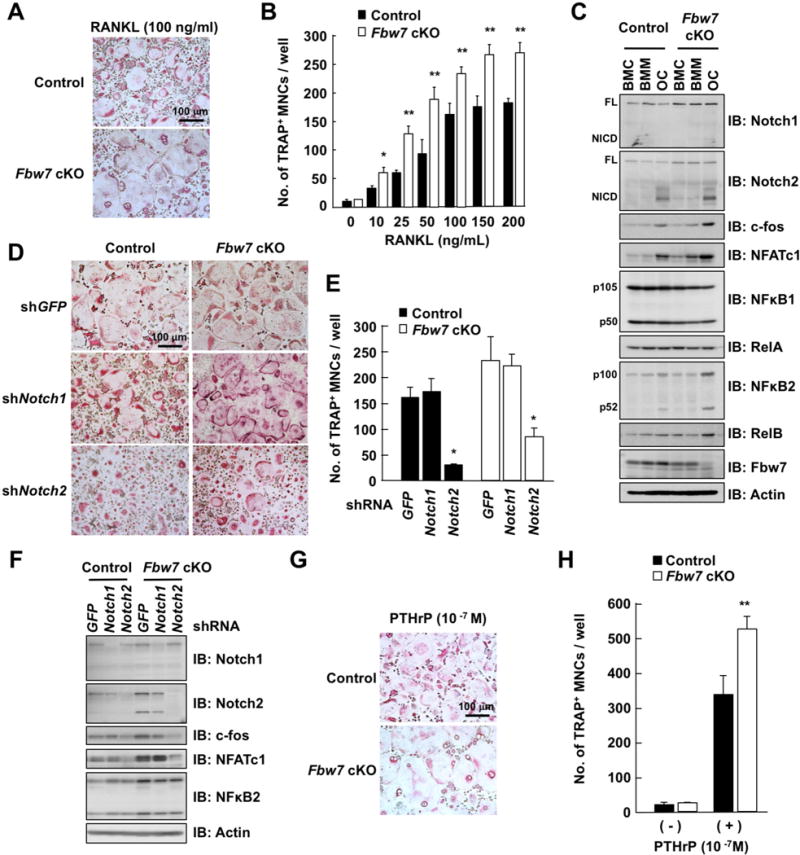
A. BMCs derived from Fbw7 cKO or control mice were cultured in the presence of M-CSF (50 ng/mL) for 3 days. The culture of differentiated bone marrow macrophages (BMM) was treated with 100 ng/mL RANKL for 3 days for differentiation into osteoclasts (OC). Cells were fixed and stained for TRAP, and TRAP-positive MNCs were counted. Scale bar: 100 μm.
B. BMM in (A) were treated with increasing concentration of RANKL for 3 days for differentiation to osteoclasts. Cells were fixed and stained for TRAP, and TRAP-positive MNCs were counted. Data represents mean ± SD. The presented data was obtained from three animals (n = 3) of each genotype, and plotted the average of three animal data with six-sample number per mouse, *p < 0.05, **p < 0.01, Student’s t test.
C. IB analysis of WCL derived from BMC, BMM, and OC cultured in (A).
D. Microscopic view of osteoclasts. Mouse BMCs were cultured in the presence of M-CSF (50 ng/mL) for 3 days. Cells were infected with shRNA lentiviral vectors specific for GFP, Notch1, or Notch2. The infected cells were cultured with RANKL (50 ng/mL) for 3 days for differentiation into osteoclasts. Cells were fixed and stained for TRAP. Scale bar: 100 μm.
E. Osteoclasts cultured in (D) were fixed and stained for TRAP, and the TRAP-positive MNCs were counted. Data represent mean ± SD, n = 3, *p < 0.05, Student’s t test.
F. IB analysis of WCL derived from osteoclasts cultured in (D).
G. Representative microscopic view of osteoclast formation in co-culture of BMM and osteoblasts in the presence of PTHrP (10−7 M). MNCs were fixed and stained for TRAP. Scale bar: 100 μm.
H. TRAP-positive MNCs were counted. Data represents mean ± SD, n = 3, **p < 0.01, Student’s t test.
See also Figure S6
Previous studies suggested that Notch1 activity is also critical in bone remodeling (Bai et al., 2008). Therefore, to distinguish Notch1 and Notch2 physiological roles, we depleted either Notch1 or Notch2 in both Fbw7 cKO and control bone marrow cells. Interestingly, Notch2 depletion suppressed RANKL-induced osteoclastogenesis and expression of osteoclast markers c-fos and NFATc, whereas Notch1 depletion had no significant effect in osteoclast differentiation in this experimental setting (Figure 6D–6F). These results support a critical role for NOTCH2 in RANKL-induced osteoclastogenesis. Moreover, the addition of PTHrP, which stimulates RANKL secretion from osteoblasts, significantly promoted osteoblast-supported osteoclastogenesis in Fbw7 cKO bone marrow cells (Figure 6G and 6H). Taken together, our data imply that Notch2 stabilization in osteoclasts leads to the elevated bone resolving phenotypes observed in Fbw7 cKO mice.
Notch Inhibitor Treatment Suppresses Bone Resorption in Vivo
A recent study reported that Notch inhibitor, N-[N-(3,5-difluorophenylacetate)-L-alanyl]-(S)-phenylglycine t-butyl ester (DAPT), effectively suppresses osteoarthritis (OA) progression in animal models (Hosaka et al., 2013). Since the observed bone loss phenotypes in Fbw7 cKO mice were largely due to the aberrant Notch2 signaling, we next explored whether Notch inhibitor treatment is effective to reverse the observed pathological bone dysfunction in Fbw7 cKO mice. Treatment of bone marrow cells derived from Fbw7 cKO and control mice with DAPT, γ-secretase inhibitor (GSI-1), and zoledronic acid (widely used for osteoporosis treatment in the clinic) efficiently inhibited osteoclast formation (Figures 7A–7C and S7). However, all three agents were relatively less effective in suppressing the growth of osteoclasts derived from Fbw7 cKO, which might be due to a relatively higher Notch2 abundance. Radiographic analyses further demonstrated that intraperitoneal injection of DAPT in Fbw7 cKO mice suppressed bone resorption and improved bone-resorptic parameters. Comparable effects were achieved with GSI-1 and zoledronic acid treatments (Figures 7D and 7E). These pharmacological approaches indicated that inhibiting Notch2 signaling led to a reversal of pathogenic bone-loss phenotypes in Fbw7 cKO mice (Figure 7F).
Figure 7. Notch inhibitor treatment suppresses bone resorption in vivo.
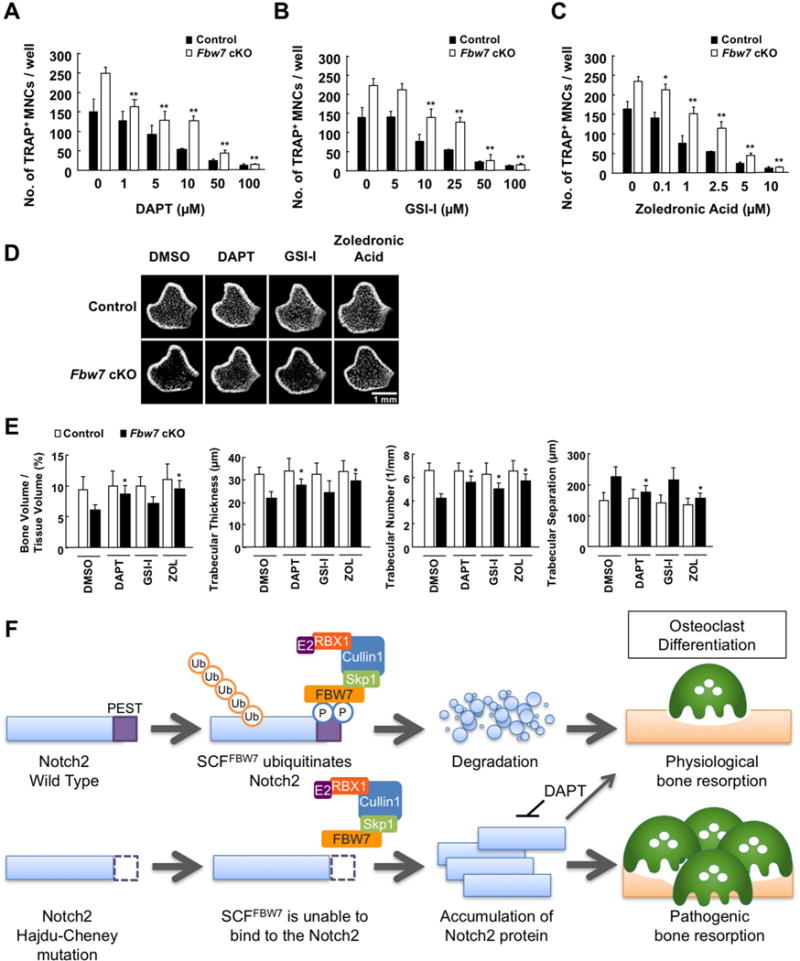
A-C. BMCs derived from Fbw7 cKO or control mice were cultured in the presence of M-CSF (50 ng/mL) for 3 days for differentiation into BMMs, and further cultured with 100 ng/mL RANKL for 3 days for differentiation into osteoclasts. Cells were fixed and stained for TRAP. At 3 days prior to fixation, cells were treated with increasing concentration of DAPT (A), GSI-1 (B), or zoledronic acid (C). The TRAP-positive MNCs, which display cytoplasmic red staining and a minimum of three nuclei, were counted as osteoclasts. Data represents mean ± SD, n = 3, **p < 0.01, Student’s t test.
D. Micro-CT images of the distal femurs derived from Fbw7 cKO or control mice that were intraperitoneally injected with DMSO, DAPT (5 mg/kg), GSI-1 (5 mg/kg), or zoledronic acid (ZOL) (250 μg/kg) every four days for 8 weeks. Scale bar: 1 mm.
E. Microstructural parameters of the distal femurs analyzed in (D). Data represents mean ± SD, n = 5, *p < 0.05, Student’s t test.
See also Figure S7
Discussion
In this study, we determined that the SCFFBW7 E3 ubiquitin ligase plays crucial roles in controlling bone homeostasis. Although NOTCH2 mutations have been considered as a cause of HCS (Isidor et al., 2011; Simpson et al., 2011), molecular mechanisms underlying how NOTCH2 variations lead to skeletal abnormalities remain unclear. Here we show that the C-terminus truncated region of NOTCH2 contains a consensus FBW7 degron motif, which mechanistically explains NOTCH2 stabilization and activation in osteoclasts in HCS. We demonstrated that bone marrow cells derived from patients with HCS have an enhanced capacity of osteoclastogenesis partly due to sustained NOTCH2 activity, confirming that misregulation of NOTCH2 proteolysis leads to HCS bone resorptic features. Osteoclast-specific Fbw7 knockout mice display severe bone-loss phenotype, characteristic in HCS patients with severe osteoporosis. The observed phenotypes were partly due to abnormal NOTCH2 accumulation in osteoclasts as additional depletion of Notch2, but not Notch1, rescued these phenotypes. These data imply that FBW7/NOTCH2 signaling plays crucial roles in bone homeostasis and that dysfunction of this signaling pathway could lead to various skeletal disorders.
While Notch has been shown to regulate osteoblast differentiation, knowledge of Notch function in osteoclast differentiation is relatively limited. We previously reported that Notch2 function is essential in the process of osteoclast differentiation (Fukushima et al., 2008). In recent mouse studies, LyzM-Cre mediated myeloid-specific Notch1−/−Notch2−/−Notch3−/− triple knockout facilitates osteoblast formation and concomitantly impairs osteoclastogenesis (Bai et al., 2008). Other recent studies demonstrated that the Notch2 HCS mutant mice faithfully recapitulate the features of HCS (Canalis et al., 2016; Vollersen et al., 2017). Additionally, a recent case study reported that the administration of denosumab, a RANKL antagonist, increases bone mineral density in a patient with HCS (Adami et al., 2016). This finding further confirms the notion that the manifestation of HCS with NOTCH2 mutation is due to osteoclast hyper-activation (Adami et al., 2016).
Here, we identified a molecular mechanism underlying how NOTCH2 HCS mutations lead to hyper osteoclast activity by dysregulation of FBW7-mediated Notch2 degradation. Similarly, in T-ALL cells, the truncated NOTCH1 protein escapes proteasome-dependent degradation directed by the SCFFBW7 E3 ubiquitin ligase complex (O’Neil et al., 2007; Thompson et al., 2007). In addition, tissue-specific genetic-engineered mice with FBW7 deletion or expressing a deficient FBW7 frequently develop cancers with accumulation of Notch protein (Crusio et al., 2010; Davis et al., 2014). Consistent with these in vivo cancer models, we also observed that osteoclast-specific Fbw7 inactivation promotes Notch2 accumulation, leading to hyper osteoclastogenesis. Therefore, these animal studies suggest that NOTCH2 post-translational modification is a potential therapeutic target for treating patients with osteoporosis.
In clinical practice, bisphosphonate, which suppresses osteoclast formation, is commonly used for treating patients with HCS (Canalis and Zanotti, 2014; Galli-Tsinopoulou et al., 2012). Recent studies indicate that NOTCH2-dependent osteoclastic bone resorption results in HCS development (Canalis et al., 2017; Canalis et al., 2016; Isidor et al., 2011; Simpson et al., 2011; Vollersen et al., 2017). Notably, the administration of anti-NOTCH2 NRR antibody in the HCS mouse reverses the osteopenia phenotype (Canalis et al., 2017). This study using NOTCH2 neutralization antibody supports our strategy that inhibition of osteoclastogenesis via administration of Notch inhibitors as well as bisphosphonate may become a first-line treatment for HCS. Notably, our study demonstrates the efficacy of a specific Notch signaling inhibitor, DAPT, against excess bone resorption in Fbw7 cKO mice. Interestingly, a recent study on osteoarthritis (OA) showed that an intra-articular injection of DAPT significantly suppresses joint-destruction in (Col2a1)-CreERT/Rbpjfl/fl OA mice (Hosaka et al., 2013). In addition, DAPT treatment of mice overexpressing TNFα that causes inflammatory bone resorption also effectively suppresses systemic osteoporotic phenotype (Zhang et al., 2014). Since DAPT efficiently inhibited the observed bone absorption in the osteoclast-specific Fbw7 conditional knockout mice, Notch signaling could be a potential target for treating various bone absorptic disorders. In addition to Notch receptor inhibition, targeting Notch ligands could be an alternative approach to suppress Notch activation. We previously reported that Jagged1, the Notch ligand implicated in metastatic bone destruction, activates Notch2 signaling pathway in osteoclasts (Fukushima et al., 2008), suggesting that Jagged1 antagonists could be potential therapeutics. However, TGFβ or PTHrP activates Jagged1 in the osteoclast supportive environment, which might cause an unexpected Jagged1 activation in the setting of Notch activating genetic disorders (Fukushima et al., 2008; Nakao et al., 2007).
In summary, we demonstrated that the NOTCH2/FBW7 signaling plays crucial roles in osteoclast differentiation, and misregulation of the NOTCH2 proteolytic pathway leads to hyper bone absorption. Consistently, we demonstrated that the osteoclast-specific inactivation of Fbw7 in mice results in systemic osteoporosis. Our study implies that the loss of FBW7 function leads to skeletal dysfunction and aberrant cell cycle regulation. Given that various genetic bone abnormalities are reported in clinic, and age-related osteoporosis is becoming one of the most common public health issues, this study provides insights for the development of therapeutics for patients with HCS and other genetic bone-loss disorders.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCES SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hiroyuki Inuzuka (hinuzuka@tohoku.ac.jp).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
All cell lines listed in the Key Resources Table were maintained in DMEM medium supplemented with 10% fetal bovine serum (FBS), 100 units of penicillin and 100 μg/mL streptomycin. Wild-type (FBW7+/+) and FBW7−/− HCT116 and DLD1 cells were kind gifts from Dr. Bert Vogelstein (Rajagopalan et al., 2004). After cells were infected with various lenti-viral shRNA or FBW7-expressing lenti-viral vectors, the resulting cells were treated with 1 μg/ml puromycin for 7 days to eliminate the non-infected cells before the subsequent biochemical assays. Mouse bone marrow cells (1 × 107 cells/ml) were cultured in 96-well plates in the presence of M-CSF (50 ng/mL) in α-MEM medium supplemented with 10% fetal bovine serum (FBS), 100 units of penicillin and 100 μg/mL streptomycin. After 3-day culture, all non-adherent cells were removed. Remaining adherent cells were considered pre-osteoclasts and were further cultured for 3 days with RANKL (50 ng/mL). Cells were fixed with 3.7% formaldehyde and stained for tartrate-resistant acid phosphatase (TRAP), an osteoclast marker. TRAP-positive multinucleated cells (MNCs) (more than three nuclei) were counted as osteoclasts. Data shown are the number of osteoclasts per culture well (values are mean ± SD, n = 3 individual experiments).
Fbw7 Conditional Knockout Mice
Floxed Fbw7 (Fbw7F/F) mice and cathepsin K-Cre (CtsK-Cre) mice were described previously (Nagai et al., 2013; Onoyama et al., 2007). Osteoclast-specific Fbw7 knockout (CtsK-Cre/Fbw7F/F) mice were generated by crossing Fbw7F/F mice with CtsK-Cre mice. For genotyping, genomic DNA was isolated from bone marrow macrophages and osteoclasts derived from control (Fbw7F/F), heterozygous knockout (CtsK-Cre/Fbw7+/F), and homozygous knockout (CtsK-Cre/Fbw7F/F) mice. Floxed or wild-type (WT) Fbw7 alleles were identified by PCR using the following primers: Floxed 1: 5′-CCTATAGGGAATTATGTTATTT-3′, Floxed 2: 5′-CTCACAGCCAAGTTATTCTGTT-3′. All animal experiments were conducted in compliance with the Institute of Laboratory Animal Research Guide for the Care and Use of Laboratory Animals and approved by the University Committee on Use and Care of Animals at the Tohoku University.
METHOD DETAILS
Plasmids
Flag-NOTCH2 ICD and GST-NOTCH2 ICD expression plasmids were constructed by subcloning the NOTCH2 ICD cDNA into pCMV-Flag and pGEX4T-1, respectively. The NOTCH2 ICD and GSK3β mutants were generated by Quik-Change XL Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA) according to manufacturer’s instruction. HA-AMPK was a gift from Dr. Toshiyuki Kobayashi (Juntendo University, Japan). Human Jagged1 plasmid was a gift from Dr. Linheng Li (Stower Institute for Medical Research) (Li et al., 1998).
Immunoblots and Immunoprecipitation
Cells were lysed in EBC buffer (50 mM Tris pH 7.5, 120 mM NaCl, 0.5% NP-40) supplemented with protease inhibitors (cOmplete Mini protease inhibitor cocktail, Sigma) and phosphatase inhibitors (PhosSTOP phosphatase inhibitor tablets, Sigma). The protein concentrations of the lysates were measured using the Bio-Rad protein assay reagent (Bio-Rad, Hercules, CA, USA). Same amounts of whole cell lysates were resolved by SDS-PAGE and immunoblotted with indicated antibodies. For immunoprecipitation, 1 mg lysates were incubated with the indicated antibody-conjugated agarose beads for 4 h. Immunoprecipitates were washed five times with NETN buffer (20 mM Tris, pH 8.0, 100 mM NaCl, 1 mM EDTA and 0.5% NP-40) before being resolved by SDS-PAGE and immunoblotted with indicated antibodies.
In Vitro Binding Assays
Prior to the in vitro binding assay, indicated bead-bound GST-NOTCH2 ICD proteins were incubated with purified, recombinant active GSK3 (NEB #P2040, with kinase reaction buffer as a negative control) in the presence of ATP at 30°C for 30 min. Next, the kinase reaction products of GST-NOTCH2 ICD were incubated with whole cell lysates derived from 293T cells transfected with HA-FBW7 for 12 h at 4°C. The bead-bound protein complex was washed 5 times and eluted by addition of 2 × SDS-PAGE sample buffer. Eluates were resolved by SDS-PAGE and probed with anti-HA antibody.
Subcellular Fractionation
HeLa or HCT116 cells (1×107) were collected in 200 μl ice-cold buffer A (10 mM HEPES pH7.9, 10 mM KCl, 0.1 mM EDTA) containing protease inhibitors. Cells were left to swell on ice for 10 min, lysed by addition of 10 μl 1% NP-40, left for 5 min at RT, and vortexed vigorously. The lysates were centrifuged at 2000 rpm for 15 min at 4 °C. The supernatant was taken from the tube and cleared by centrifugation at 90,000 rpm for 30 min (cytosolic fraction). Pellets obtained after the first spin were washed with 100 μl of Buffer A, centrifuged at 3000 rpm for 1.5 min, and resuspended in Buffer C (20 mM HEPES, pH7.9, 0.4 M NaCl, 1 mM EDTA). After vigorous shake for 15 min at 4 °C, the extracts were cleared by centrifugation at 14,000 rpm for 20 min (nuclear fraction).
Peripheral Blood Sample Preparation
Peripheral blood was collected from a patient with HCS as described previously (Narumi et al., 2013). The samples from healthy volunteers were collected from individuals of the same age and sex-group as the patient. Informed consent was obtained from the patient in accordance with the guidelines of the ethics committee of Tohoku University School of Medicine (Approval No.26-51). Venous blood was collected by venipuncture in lithium heparin-containing blood collecting tubes (Nipro, Osaka, Japan) from both the patient and control. Blood samples of the patient and his matched control were taken on the same day and time and were processed within 3 h. Twenty milliliters of blood was kept separate for determination of the leucocyte composition. Peripheral blood monocytes (PBMCs) were isolated from whole blood by density gradient. Heparinized whole blood was diluted in a 1:1 concentration with Hank’s Balanced Salt Solution (Wako, Osaka, Japan). Diluted blood was layered on top of HistoPaque 1077 (Sigma) and centrifuged without brake at 1000 g for 30 min at room temperature. The interface was carefully collected and washed twice with α-MEM. Collected PBMCs were cultured for 3 days in the presence of M-CSF (100 ng/mL) in α-MEM. The cells were further cultured for 4 days with RANKL (100 ng/mL) in the presence of M-CSF (30 ng/mL).
Histologic Preparation
Bone analyses including histomorphometric and micro-radiographic examinations were performed as described previously (Maruyama et al., 2010; Soysa et al., 2010). Briefly, mice were intraperitoneally injected with 15 mg/kg of calcein twice with an interval of 5 days and were euthanized 24 h after the second injection. Fixed bones were embedded in mixtures of methyl methacrylate (MMA) and 2 hydroxyethyl methacrylate (GMA) resins as described previously (Jimi et al., 2004). Sagittal sections (4 μm) of the long bones were prepared. These sections were then stained according to von Kossa with a modified Van Gieson method to clarify the mineralized tissue. To detect osteoclasts, sections were stained for tartrate-resistant acid phosphatase (TRAP) using TRAP/ALP Stain Kit (Wako) according to manufacture’s instruction and counterstained with methyl green. Osteoclasts were designated as the TRAP-positive (TRAP+) multinucleated cells (MNCs) containing more than three nuclei located on the bone surface. Histomorphometric analyses were performed by using a light microscope with a micrometer and an image analyzer (Osteoplan II; Carl Zeiss Japan, Tokyo, Japan). The quantitative histomorphometric analyses were performed in a blinded fashion.
Radiologic Assessment
Bones were fixed in PBS-buffered glutaraldehyde (0.25%)-formalin (4%) fixative (pH 7.4) for 2 days at 4°C and washed with PBS for further studies. Radiographs of the femora and tibiae were taken by soft x-ray (model CMB-2; SOFTEX Co. Ltd., Tokyo, Japan). Images of tibia and three-dimensional (3D) reconstruction images of hind paw were obtained by focal micro-computed tomography (μCT) (SkyScan 1176, TOYO Corporation, Tokyo, Japan). Bone morphometric parameters and bone mineral density (BMD) were analyzed using CTan software (SkyScan).
Mechanical Analyses of Bone Strength
Strength of femora and tibiae from control and Fbw7 cKO mice was assessed by a three-point bending test according to the method of Jamsa (Jamsa et al., 1998). The tests were conducted on the Autograph Precision Universal Tester (Shimadzu AG-Xplus) at a span of 5 mm and a load speed of 2 mm/min. The energy to failure (N.mm) was calculated and normalized to the control mouse group.
Immunofluorescence Staining and Microscopy
HeLa cells grown on chamber slides (Iwaki, 5732-008) were fixed in 4% paraformaldehyde/PBS for 15 min, permeabilized with 0.5% Triton X-100/PBS for 5 min, and incubated in blocking buffer (5% FBS/TBST) for 1 h. The cells were stained with a primary antibody (anti-NOTCH2 D76A6 at 1:1200, or anti-Jagged1 TS1.15H at 1:400) in 0.05% BSA/TBST overnight at 4°C. Cells were washed four times for 5 min with TBST and incubated with secondary Alexa Fluor 568-conjugated goat anti-rabbit antibody and Alexa Fluor 488-conjugated donkey anti-rat antibody (Invitrogen at 1:1500 each), and with 4,6-diamidino-2-phenylindole (DAPI) for 1 h at room temperature. Cover glasses (0.12–0.17 mm, 24 × 24 mm) were then mounted on slides with PermaFluor Aqueous Mounting Medium (Lab Vision). The slides were examined with a confocal microscope (Fluoview FV10i; Olympus, Tokyo, Japan).
RT-PCR Analyses
RNA was extracted using TRIzol reagent, and the reverse transcription reaction was performed using SUPER SCRIPT III reverse transcriptase (Invitrogen, Carlsbad, CA, USA). The real-time RT-PCR reaction was performed with SYBR Select Master Mix and ABI 7500 Real-Time PCR System. All procedures were performed according to the manufacturer’s instructions. Primers used in this study are listed in Table S1.
QUANTIFICATION AND STATISTICAL ANALYSIS
Western blot band intensities were quantified by ImageJ software (NIH). All quantitative data are presented as the mean ± SD of at least three independent experiments and were analyzed by Student’s t-test for between group differences. p < 0.05 was considered statistically significant.
Supplementary Material
Highlights.
NOTCH2 Hajdu-Cheney mutants escape FBW7-mediated ubiquitination and proteolysis.
Elevated NOTCH2 and osteoclast activities in the peripheral blood of HCS patients.
Osteoclast-specific ablation of Fbw7 in mice results in osteoporotic phenotypes.
Notch inhibitor treatment in Fbw7 cKO mice relieves progressive bone loss.
Acknowledgments
We thank the patients, their families, and physicians for their cooperation, Dr. Osamu Suzuki (Tohoku University) for an instruction of a three-point bending test, Dr. Kiyoko Sameshima (Minami Kyushu National Hospital) for an arrangement of blood sampling from a patient, Dr. Yueyong Liu for critical technical assistance, and Dr. Brian J. North for careful proof-reading of the manuscript. This work was supported by JSPS Kakenhi grants (17H04396 and 26462829 to H.F., 26253092 to S.F., 16H05548 to A.Y., and 16H055290 and 16K15811 to H.I.), NIH R01 grants (GM094777 and CA177910 to W.W.), and American Cancer Society Research Scholar grants to W.W. and H.I.
Footnotes
Author Contributions
H.F. conceived and performed the experiments with assistance from K.S., A.W., S.H., T.K., D.O., S.S., M.A., K.N., and H.I. A.Y., K.O., S.F., E.J., A.B., K.I.N, K.N., Y.A., W.W., H.I., and H.F. guided and supervised the study. H.F., W.W., and H.I. wrote the manuscript. All authors commented on the manuscript.
Competing Financial Interests
None to declare.
References
- Adami G, Rossini M, Gatti D, Orsolini G, Idolazzi L, Viapiana O, Scarpa A, Canalis E. Hajdu Cheney Syndrome; report of a novel NOTCH2 mutation and treatment with denosumab. Bone. 2016;92:150–156. doi: 10.1016/j.bone.2016.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai S, Kopan R, Zou W, Hilton MJ, Ong CT, Long F, Ross FP, Teitelbaum SL. NOTCH1 regulates osteoclastogenesis directly in osteoclast precursors and indirectly via osteoblast lineage cells. The Journal of biological chemistry. 2008;283:6509–6518. doi: 10.1074/jbc.M707000200. [DOI] [PubMed] [Google Scholar]
- Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- Brennan AM, Pauli RM. Hajdu–Cheney syndrome: evolution of phenotype and clinical problems. American journal of medical genetics. 2001;100:292–310. doi: 10.1002/1096-8628(20010515)100:4<292::aid-ajmg1308>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Canalis E, Sanjay A, Yu J, Zanotti S. An Antibody to Notch2 Reverses the Osteopenic Phenotype of Hajdu-Cheney Mutant Male Mice. Endocrinology. 2017;158:730–742. doi: 10.1210/en.2016-1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canalis E, Schilling L, Yee SP, Lee SK, Zanotti S. Hajdu Cheney Mouse Mutants Exhibit Osteopenia, Increased Osteoclastogenesis, and Bone Resorption. The Journal of biological chemistry. 2016;291:1538–1551. doi: 10.1074/jbc.M115.685453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canalis E, Zanotti S. Hajdu-Cheney syndrome: a review. Orphanet journal of rare diseases. 2014;9:200. doi: 10.1186/s13023-014-0200-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crusio KM, King B, Reavie LB, Aifantis I. The ubiquitous nature of cancer: the role of the SCF(Fbw7) complex in development and transformation. Oncogene. 2010;29:4865–4873. doi: 10.1038/onc.2010.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ, Welcker M, Clurman BE. Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer cell. 2014;26:455–464. doi: 10.1016/j.ccell.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annual review of biochemistry. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- Espinosa L, Ingles-Esteve J, Aguilera C, Bigas A. Phosphorylation by glycogen synthase kinase-3 beta down-regulates Notch activity, a link for Notch and Wnt pathways. The Journal of biological chemistry. 2003;278:32227–32235. doi: 10.1074/jbc.M304001200. [DOI] [PubMed] [Google Scholar]
- Fukushima H, Matsumoto A, Inuzuka H, Zhai B, Lau AW, Wan L, Gao D, Shaik S, Yuan M, Gygi SP, et al. SCF(Fbw7) modulates the NFkB signaling pathway by targeting NFkB2 for ubiquitination and destruction. Cell reports. 2012;1:434–443. doi: 10.1016/j.celrep.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima H, Nakao A, Okamoto F, Shin M, Kajiya H, Sakano S, Bigas A, Jimi E, Okabe K. The association of Notch2 and NF-kappaB accelerates RANKL-induced osteoclastogenesis. Molecular and cellular biology. 2008;28:6402–6412. doi: 10.1128/MCB.00299-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli-Tsinopoulou A, Kyrgios I, Giza S, Giannopoulou EZ, Maggana I, Laliotis N. Two-year cyclic infusion of pamidronate improves bone mass density and eliminates risk of fractures in a girl with osteoporosis due to Hajdu-Cheney syndrome. Minerva endocrinologica. 2012;37:283–289. [PubMed] [Google Scholar]
- Gao D, Inuzuka H, Tan MK, Fukushima H, Locasale JW, Liu P, Wan L, Zhai B, Chin YR, Shaik S, et al. mTOR drives its own activation via SCF(betaTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Molecular cell. 2011;44:290–303. doi: 10.1016/j.molcel.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annual review of biochemistry. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Hilton MJ, Tu X, Wu X, Bai S, Zhao H, Kobayashi T, Kronenberg HM, Teitelbaum SL, Ross FP, Kopan R, et al. Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nature medicine. 2008;14:306–314. doi: 10.1038/nm1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosaka Y, Saito T, Sugita S, Hikata T, Kobayashi H, Fukai A, Taniguchi Y, Hirata M, Akiyama H, Chung UI, et al. Notch signaling in chondrocytes modulates endochondral ossification and osteoarthritis development. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:1875–1880. doi: 10.1073/pnas.1207458110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inuzuka H, Gao D, Finley LW, Yang W, Wan L, Fukushima H, Chin YR, Zhai B, Shaik S, Lau AW, et al. Acetylation-dependent regulation of Skp2 function. Cell. 2012;150:179–193. doi: 10.1016/j.cell.2012.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW, et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011;471:104–109. doi: 10.1038/nature09732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isidor B, Lindenbaum P, Pichon O, Bezieau S, Dina C, Jacquemont S, Martin-Coignard D, Thauvin-Robinet C, Le Merrer M, Mandel JL, et al. Truncating mutations in the last exon of NOTCH2 cause a rare skeletal disorder with osteoporosis. Nature genetics. 2011;43:306–308. doi: 10.1038/ng.778. [DOI] [PubMed] [Google Scholar]
- Jamsa T, Jalovaara P, Peng Z, Vaananen HK, Tuukkanen J. Comparison of three-point bending test and peripheral quantitative computed tomography analysis in the evaluation of the strength of mouse femur and tibia. Bone. 1998;23:155–161. doi: 10.1016/s8756-3282(98)00076-3. [DOI] [PubMed] [Google Scholar]
- Jimi E, Aoki K, Saito H, D’Acquisto F, May MJ, Nakamura I, Sudo T, Kojima T, Okamoto F, Fukushima H, et al. Selective inhibition of NF-kappa B blocks osteoclastogenesis and prevents inflammatory bone destruction in vivo. Nature medicine. 2004;10:617–624. doi: 10.1038/nm1054. [DOI] [PubMed] [Google Scholar]
- Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Milner LA, Deng Y, Iwata M, Banta A, Graf L, Marcovina S, Friedman C, Trask BJ, Hood L, et al. The human homolog of rat Jagged1 expressed by marrow stroma inhibits differentiation of 32D cells through interaction with Notch1. Immunity. 1998;8:43–55. doi: 10.1016/s1074-7613(00)80457-4. [DOI] [PubMed] [Google Scholar]
- Li W, Bengtson MH, Ulbrich A, Matsuda A, Reddy VA, Orth A, Chanda SK, Batalov S, Joazeiro CA. Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle’s dynamics and signaling. PloS one. 2008;3:e1487. doi: 10.1371/journal.pone.0001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Shen JX, Wen XF, Guo YX, Zhang GJ. Targeting Notch degradation system provides promise for breast cancer therapeutics. Critical reviews in oncology/hematology. 2016 doi: 10.1016/j.critrevonc.2016.05.010. [DOI] [PubMed] [Google Scholar]
- Maruyama T, Fukushima H, Nakao K, Shin M, Yasuda H, Weih F, Doi T, Aoki K, Alles N, Ohya K, et al. Processing of the NF-kappa B2 precursor p100 to p52 is critical for RANKL-induced osteoclast differentiation. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2010;25:1058–1067. doi: 10.1359/jbmr.091032. [DOI] [PubMed] [Google Scholar]
- Nagai Y, Osawa K, Fukushima H, Tamura Y, Aoki K, Ohya K, Yasuda H, Hikiji H, Takahashi M, Seta Y, et al. p130Cas, Crk-associated substrate, plays important roles in osteoclastic bone resorption. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2013;28:2449–2462. doi: 10.1002/jbmr.1936. [DOI] [PubMed] [Google Scholar]
- Nakao A, Fukushima H, Kajiya H, Ozeki S, Okabe K. RANKL-stimulated TNFalpha production in osteoclast precursor cells promotes osteoclastogenesis by modulating RANK signaling pathways. Biochemical and biophysical research communications. 2007;357:945–950. doi: 10.1016/j.bbrc.2007.04.058. [DOI] [PubMed] [Google Scholar]
- Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nature reviews. Cancer. 2006;6:369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- Narumi Y, Min BJ, Shimizu K, Kazukawa I, Sameshima K, Nakamura K, Kosho T, Rhee Y, Chung YS, Kim OH, et al. Clinical consequences in truncating mutations in exon 34 of NOTCH2: report of six patients with Hajdu-Cheney syndrome and a patient with serpentine fibula polycystic kidney syndrome. American journal of medical genetics. Part A. 2013;161A:518–526. doi: 10.1002/ajmg.a.35772. [DOI] [PubMed] [Google Scholar]
- O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters R, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. The Journal of experimental medicine. 2007;204:1813–1824. doi: 10.1084/jem.20070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neil J, Look AT. Mechanisms of transcription factor deregulation in lymphoid cell transformation. Oncogene. 2007;26:6838–6849. doi: 10.1038/sj.onc.1210766. [DOI] [PubMed] [Google Scholar]
- Onoyama I, Tsunematsu R, Matsumoto A, Kimura T, de Alboran IM, Nakayama K, Nakayama KI. Conditional inactivation of Fbxw7 impairs cell-cycle exit during T cell differentiation and results in lymphomatogenesis. The Journal of experimental medicine. 2007;204:2875–2888. doi: 10.1084/jem.20062299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nature reviews. Molecular cell biology. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- Rajagopalan H, Jallepalli PV, Rago C, Velculescu VE, Kinzler KW, Vogelstein B, Lengauer C. Inactivation of hCDC4 can cause chromosomal instability. Nature. 2004;428:77–81. doi: 10.1038/nature02313. [DOI] [PubMed] [Google Scholar]
- Rechsteiner M, Rogers SW. PEST sequences and regulation by proteolysis. Trends in biochemical sciences. 1996;21:267–271. [PubMed] [Google Scholar]
- Rodan GA, Martin TJ. Therapeutic approaches to bone diseases. Science. 2000;289:1508–1514. doi: 10.1126/science.289.5484.1508. [DOI] [PubMed] [Google Scholar]
- Simpson MA, Irving MD, Asilmaz E, Gray MJ, Dafou D, Elmslie FV, Mansour S, Holder SE, Brain CE, Burton BK, et al. Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nature genetics. 2011;43:303–305. doi: 10.1038/ng.779. [DOI] [PubMed] [Google Scholar]
- Soysa NS, Alles N, Weih D, Lovas A, Mian AH, Shimokawa H, Yasuda H, Weih F, Jimi E, Ohya K, et al. The pivotal role of the alternative NF-kappaB pathway in maintenance of basal bone homeostasis and osteoclastogenesis. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2010;25:809–818. doi: 10.1359/jbmr.091030. [DOI] [PubMed] [Google Scholar]
- Thompson BJ, Buonamici S, Sulis ML, Palomero T, Vilimas T, Basso G, Ferrando A, Aifantis I. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. The Journal of experimental medicine. 2007;204:1825–1835. doi: 10.1084/jem.20070872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollersen N, Hermans-Borgmeyer I, Cornils K, Fehse B, Rolvien T, Triviai I, Jeschke A, Oheim R, Amling M, Schinke T, et al. High bone turnover in mice carrying a pathogenic Notch2-mutation causing Hajdu-Cheney syndrome. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2017 doi: 10.1002/jbmr.3283. [DOI] [PubMed] [Google Scholar]
- Wang K, Zhang Q, Li D, Ching K, Zhang C, Zheng X, Ozeck M, Shi S, Li X, Wang H, et al. PEST domain mutations in Notch receptors comprise an oncogenic driver segment in triple-negative breast cancer sensitive to a gamma-secretase inhibitor. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21:1487–1496. doi: 10.1158/1078-0432.CCR-14-1348. [DOI] [PubMed] [Google Scholar]
- Wang Z, Liu P, Inuzuka H, Wei W. Roles of F-box proteins in cancer. Nature reviews. Cancer. 2014;14:233–247. doi: 10.1038/nrc3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- Yumimoto K, Matsumoto M, Onoyama I, Imaizumi K, Nakayama KI. F-box and WD repeat domain-containing-7 (Fbxw7) protein targets endoplasmic reticulum-anchored osteogenic and chondrogenic transcriptional factors for degradation. The Journal of biological chemistry. 2013;288:28488–28502. doi: 10.1074/jbc.M113.465179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotti S, Canalis E. Notch1 and Notch2 expression in osteoblast precursors regulates femoral microarchitecture. Bone. 2014;62:22–28. doi: 10.1016/j.bone.2014.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Hilton MJ, Anolik JH, Welle SL, Zhao C, Yao Z, Li X, Wang Z, Boyce BF, Xing L. NOTCH inhibits osteoblast formation in inflammatory arthritis via noncanonical NF-kappaB. The Journal of clinical investigation. 2014;124:3200–3214. doi: 10.1172/JCI68901. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.