

ABSTRACT
Shigella is unique among enteric pathogens, as it invades colonic epithelia through the basolateral pole. Therefore, it has evolved the ability to breach the intestinal epithelial barrier to deploy an arsenal of effector proteins, which permits bacterial invasion and leads to a severe inflammatory response. However, the mechanisms used by Shigella to regulate epithelial barrier permeability remain unknown. To address this question, we used both an intestinal polarized model and a human ex-vivo model to further characterize the early events of host-bacteria interactions. Our results showed that secreted Serine Protease A (SepA), which belongs to the serine protease autotransporter of Enterobacteriaceae family, is responsible for critically disrupting the intestinal epithelial barrier. Such disruption facilitates bacterial transit to the basolateral pole of the epithelium, ultimately fostering the hallmarks of the disease pathology. SepA was found to cause a decrease in active LIM Kinase 1 (LIMK1) levels, a negative inhibitor of actin-remodeling proteins, namely cofilin. Correspondingly, we observed increased activation of cofilin, a major actin-polymerization factor known to control opening of tight junctions at the epithelial barrier. Furthermore, we resolved the crystal structure of SepA to elucidate its role on actin-dynamics and barrier disruption. The serine protease activity of SepA was found to be required for the regulatory effects on LIMK1 and cofilin, resulting in the disruption of the epithelial barrier during infection. Altogether, we demonstrate that SepA is indispensable for barrier disruption, ultimately facilitating Shigella transit to the basolateral pole where it effectively invades the epithelium.
KEYWORDS: cofilin, LIM Kinases, SepA, Shigella, SPATEs
Introduction
Shigella spp. are Gram-negative enteric bacilli belonging to the Enterobacteriaceae family. Infection of the human colonic mucosa by Shigella results in an acute inflammatory disease characterized by abdominal cramps, fever, and severe diarrhea, often containing blood and mucus. Shigella ssp. is one of the leading etiological agents of diarrhea worldwide, causing 80–165 million cases and up to 600,000 deaths per year.1 Particularly, S. flexneri and S. sonnei are responsible for endemic shigellosis, also known as bacillary dysentery, in impoverished regions of the world where this disease remains a major cause of mortality among children under 5 y old.2
Shigella is a host-specific pathogen with a distinctive mode of pathogenesis that involves invasion of colonic epithelial cells through their basolateral pole.3 During infection, colonic epithelial cell invasion is orchestrated by proteins encoded in a 31-kb region of the 220-kb virulence plasmid,4,5 including: 1) proteins necessary to assemble a type III secretion system (T3SS); 2) translocators (IpaB and IpaC) and effectors (IpaD, IpgB1, IpgD and IcsB with their exclusive chaperones) secreted throughout the T3SS; and 3) 2 transcriptional activators (VirB and MxiE).6,7 However, the mechanism by which Shigella relocates from the luminal (apical) to the serosal (basolateral) pole of the colonic epithelium is poorly understood. Traditionally, the translocation event has been attributed to uptake and transport by M cells.8 S. flexneri is also capable of altering apical protein complexes, allowing them to cross the paracellular space to reach the basolateral pole, an event that also decreases barrier function.9,10 Apical protein complexes in the intestinal epithelium are composed of tight junction proteins that establish the boundary between the apical and basolateral poles of the epithelium, while maintaining cell polarity. The opening of the epithelium barrier is tightly regulated by several mechanisms, including actin dynamics. Specifically, cofilin promotes opening of the tight junction proteins by severing actin filaments, which result in increased barrier permeability.11-14 Cofilin is a ubiquitous actin-binding protein that controls the depolymerization of actin filaments by severing ‘aged’ monomers at the barbed end of the filaments.15 Members of the LIM kinase family (LIMKs), namely LIMK1 and 2 are responsible to regulated cofilin activation.15,16 Phosphorylation of cofilin by LIMKs inhibits its actin binding, severing, and depolymerizing activities. Actin dynamics via cofilin/LIMK activation during epithelial disruption, cell division, motility, and morphogenesis have been extensively studied12,14,16-20
The pathogenicity of Shigella is largely due to its ability to invade epithelial cells basolaterally. Using polarized intestinal epithelial monolayers, we have previously shown that S. flexneri interfaces at the apical pole of the epithelium, physiologically mimicking what happens in the intestine after bacterial digestion and its subsequent transition through the paracellular space to reach and invade the basolateral epithelial pole.9 SepA is the major protein secreted by S. flexneri in culture21 and is also highly secreted during infection.22,23 To date, no physiologically relevant substrates are known for SepA.24 Given that SepA has been reported to play a role S. flexneri tissue invasion,21 the aim of this study was to examine the role of Shigella SepA in epithelial disruption via cofilin/LIMK activation.
SepA is encoded outside the mxi-spa locus on the 220-kb virulence plasmid. It belongs to the serine protease autotransporters of Enterobacteriaceae (SPATEs) family, which has been implicated in a myriad of pathogenic functions, including bacterial invasion, colonization, promotion of inflammation, and disease severity.23-27 Members of this family share a common structural organization, characterized by an N-terminal sec-dependent signal peptide, followed by a “passenger domain” (the functional, secreted part of the protein), and a C-terminal β-barrel referred to as the ‘translocator” domain. The signal sequence is required for sec-dependent translocation of the nascent protein across the bacterial inner membrane, whereas the C-terminal β-barrel domain is necessary for the translocation of the passenger domain through the outer membrane. Together, these comprise the Type V secretion system. SepA has been associated with bacterial virulence not just in Shigella infections, but also in infections with Enteroaggregative Escherichia coli that also harbor this gene,23,28 thereby highlighting the potential role of SepA in bacterial invasion.
Results
Role of SepA in epithelial barrier disruption
To assess the role of SepA in tissue invasion, we used our well-established monolayer model of polarized intestinal cells29 to study apical and basolateral S. flexneri infection. When the apical epithelial pole was infected with S. flexneri, the ability to invade epithelial monolayers depended on SepA. Monolayers infected with a ΔsepA strain showed significantly reduced rates of bacterial invasion (43.67 ± 5.33%) compared with epithelial monolayers infected with wild-type strain (Fig. 1A). The avirulent, non-invasive S. flexneri BS103 strain yielded lower rates of bacterial invasion (12.67 ± 3.13%) compared with epithelial monolayers infected with wild-type strain. Transformation with pZK15, an inducible plasmid containing the sepA gene,21 complemented the ΔsepA strain and reestablished the ability to invade polarized monolayers through the apical pole, yielding values similar to epithelial monolayers infected with wild-type strain. Interestingly, transformation of the BS103 strain with pZK15 did not improve the ability to invade epithelial cells (9.0 ± 0.5%), suggesting that SepA is necessary but not sufficient to cause bacterial invasion. However, when directly infecting the basolateral pole, thus bypassing the boundaries imposed by the epithelial barrier, SepA was not required for S. flexneri invasion. Compared to wild-type strains, ΔsepA strains did not exhibit a significant decrease in bacterial invasion through the basolateral pole (Fig. 1B). However, infection with the BS103 strain showed an impaired invasion capacity that did not improve with SepA complementation (BS103+pZK15).
Figure 1.
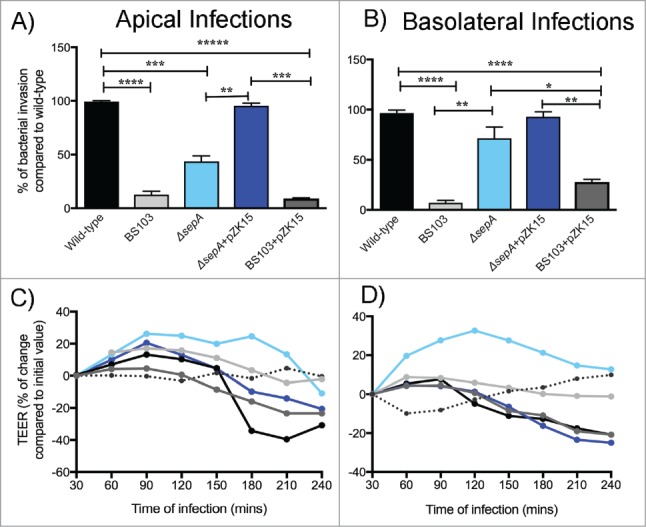
SepA facilitates bacterial invasion by opening the epithelial barrier. Bacterial invasion was assessed by infecting polarized epithelial cell monolayers either at the apical (A) or basolateral (B) poles with the following S. flexneri strains: wild-type (black), an avirulent, non-invasive BS103 (light gray), ΔsepA (light blue), ΔsepA+pZK15 (dark blue), and BS103+pZK15 (dark gray). Data are expressed as colony-forming units (CFU) per monolayer ( ± SEM) compared with wild-type. Regulation of TEER after stimulation with S. flexneri strains was also evaluated after infecting the apical (C) or basolateral (D) poles with the same strains used above. TEER values are expressed as percentage of change compared with initial values. The data are expressed as means ± SEM of triplicate samples for all conditions tested.
To begin to test the hypothesis that SepA compromises intestinal barrier integrity from the apical pole, we examined the loss of epithelial barrier function by measuring the transepithelial electrical resistance (TEER).9 Apical infection of polarized intestinal monolayers with wild-type S. flexneri resulted in a significant decrease in TEER, reaching levels at which the barrier would likely be permeable to large molecules30 after 4 hours of infection (Fig. 1C). Conversely, infection with the ΔsepA strain did not affect intestinal permeability and resulted in TEER values similar to infection with the avirulent, non-invasive BS103 strain. Infection with the ΔsepA+pZK15 and BS103+pZK15 complemented strains altered barrier integrity and decreased TEER. Similar results were observed when the epithelial cell monolayers were infected basolaterally (Fig. 1D).
Effect of SepA on actin dynamic circuits in barrier disruption
Based on evidence supporting that cofilin function on actin dynamics is implicated in increased paracellular permeability of human colonic epithelial cell monolayers,31 we used an ex-vivo 3D culture model (SMI-100) to examine the interplay between SepA and cofilin activation in a more physiologically relevant setting.32 Apical infection with wild-type S. flexneri affected the columnar architecture of the SMI-100 villi, which was accompanied by the disruption of F-actin organization (Fig. 2A, top panel). Moreover, infection triggered apical accumulation of cofilin, specifically at the site of bacterial contact (Fig. 2A, middle panel), with a 5.5-fold overall increase in apical cofilin expression compared with non-infected SMI-100 (Fig. 2B). In contrast, infection with the ΔsepA strain did not disrupt the epithelial barrier and, similar to BS103-infected or mock-treated SMI-100, no effect was observed on the columnar architecture or F-actin organization. Cofilin expression levels were comparable between SMI-100 infected with ΔsepA or BS103 strains. Infection with the complemented ΔsepA+pZK15 strain restored the ability to disrupt the villi architecture and resulted in increased cofilin expression at the apical pole. Furthermore, we observed that S. flexneri SepA-bearing strains caused lesions on the intestinal barrier characterized by epithelial desquamation, which is the shedding of cells from the villi tips (Fig. 2A bottom panel), and that cofilin was highly expressed in desquamated cells (Fig. 2A).
Figure 2.
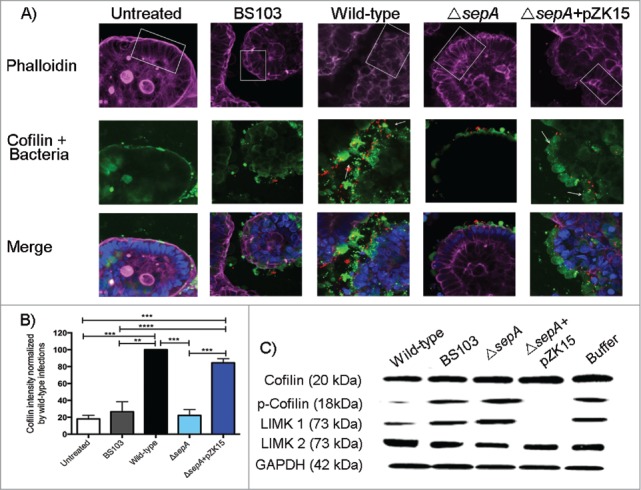
SepA regulates proteins involved in actin dynamics. (A) Z-section confocal microscopy (20X magnification) of the SMI-100 3D intestinal model after apical infection with the following S. flexneri strains: wild-type, BS103, ΔsepA, ΔsepA+pZK15, and mock-treated. The top panel shows the F-actin architecture (phalloidin in purple) of the SMI-100; white squares highlight sections showing where the differences in the columnar architecture of the epithelium are more noticeable after infection. The middle panel shows bacteria (in red) and cofilin expression (in green) on infected SMI-100; white arrows point at desquamated cells at the apical pole of the epithelium exhibiting high cofilin expression. The bottom panel shows the merged figures with the cell nuclei (in blue). (B) Intensity of cofilin expression after infection of the SMI-100 with the S. flexneri strains described above was estimated using Photoshop CS4. Values are expressed as percent of cofilin intensity compared with intensity in cells infected with wild-type S. flexneri. Values represent the mean ± SEM of 3 separate experiments. (C) Western blot analysis of coflin, p-cofilin, LIMK1 and LIMK2 expression on epithelial cell monolayers apically-infected with S. flexneri strains.
Using our in vitro model of a polarized intestinal epithelial cell monolayer, we further investigated if S. flexneri infection affected active (non-phosphorylated) vs. inactive (phosphorylated, or p-cofilin) cofilin levels. Epithelial monolayers infected apically with wild-type S. flexneri strains showed a significant decrease in p-cofilin, but total cofilin protein levels remained unchanged suggesting SepA control over cofilin activation but not over protein expression (Fig. 2C). Apical infection with the ΔsepA strain did not cause a decrease in p-cofilin levels, with similar levels compared with mock-treated monolayers or those infected with the avirulent, non-invasive S. flexneri BS103 strain (Fig. 2C). Complementation of the ΔsepA strain with the SepA inducible plasmid (ΔsepA+pZK15), reestablished the ability to dramatically reduce p-cofilin levels post-infection.
Our results support that SepA has a role in disrupting the cytoskeletal architecture of the intestinal epithelium by promoting cofilin activation upon S. flexneri infection. However, we did not find evidence supporting that SepA directly targets cofilin (Fig. S1). Since activation of cofilin is tightly regulated by members of the LIM kinase family (LIMKs), specifically LIMK1 and LIMK2,15 we investigated the possibility that SepA could regulate LIMK1 or LIMK2 activation during S. flexneri infection. Figure 2C shows that apical infection with wild-type S. flexneri caused a dramatic decrease in LIMK1 expression. Infection with ΔsepA strain or the avirulent, non-invasive BS103 strain did not affect LIMK1 expression, with protein expression levels comparable to those observed in mock-treated monolayers. Apical infection with the complemented ΔsepA+pZK15 strain caused a decrease in LIMK1 protein expression comparable to cells infected with wild-type S. flexneri (Fig. 2C). We observed SepA-dependent down regulation of p-cofilin and LIMK1 after basolateral infection with any S. flexneri strain, including the BS103 strain complemented with the sepA plasmid (BS103+pZK15) (Fig. S2). Although these results suggest that SepA targets LIMK1, we did not observe proteolysis when incubating recombinant human LIMK1 with active SepA isolated from bacterial supernatants (Fig. S3). No difference was observed in LIMK2 expression in cells infected with any of the S. flexneri strains or the mock-treated cells.
Functional consequence of epithelial disruption by SepA
Enterocyte invasion is a crucial step in Shigella pathogenesis, which is consistent with the observation that Shigella reaching the underlying lamina propria evokes an intense inflammatory response.33 Using a transwell assay, we examined whether SepA disruption of the epithelial barrier was a key requirement for neutrophil transmigration. As expected, apical infection of intestinal monolayers with wild-type S. flexneri elicited a marked migration of neutrophils across the monolayer in the basolateral-to-apical direction (Fig. 3A and B). Apical infection with the ΔsepA strain did not stimulate polymorphonuclear leukocyte (PMN) transepithelial migration, with a 55.60 ± 7.16% reduction compared with cells infected with the wild-type strain (Fig. 3A). Similar responses were observed when infecting monolayers with the avirulent, non-invasive BS103 strain or the BS103+pZK15 complemented strain with a 53.11 ± 7.03% and 38.13 ± 3.99% reduction in PMN transepithelial migration, respectively. However, apical infection with the ΔsepA+pZK15 complemented strain restored the ability to induce PMN transepithelial migration to levels comparable to infection with wild-type S. flexneri. When applying Shigella directly to the basolateral pole of the epithelium, SepA function on the barrier is not sufficient to impede PMN migration. Specifically, infection with the ΔsepA strain induced PMN transepithelial migration to the same extent as infection with wild-type or the complemented ΔsepA+pZK15 strain (Fig. 3B). Infection with the non-invasive BS103 strain showed reduced PMN transepithelial migration even when complemented with the inducible SepA plasmid (BS103+pZK15; Fig. 3B).
Figure 3.
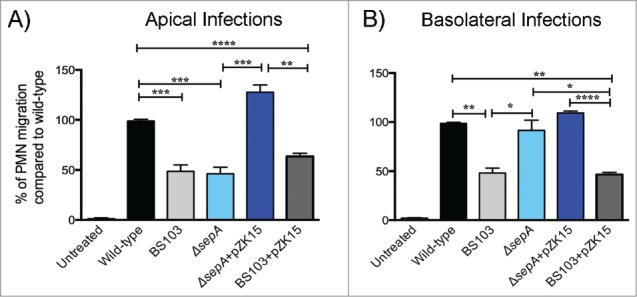
SepA facilitates neutrophil infiltration when S. flexneri infects the apical pole. Polarized epithelial cell monolayers were infected either at the (A) apical or (B) basolateral pole with the following S. flexneri strains: wild-type (black), an avirulent, non-invasive BS103 (light gray), ΔsepA (light blue), ΔsepA+pZK15 (dark blue), and BS103+pZK15 (dark gray). The data are expressed as means ± SEM of triplicate samples for all conditions tested.
Structural basis of SepA protein function
To better understand the role of SepA, we resolved the crystal structure of the SepA passenger domain by X-ray diffraction to identify features that could help explain its functional activity (PDB ID: 5J44). As shown in Figure 4A, the SepA gene encodes a bipartite autotransporter protein consisting of an N-terminal signal peptide, a 110 kDa passenger domain, and a 30 kDa C-terminal transmembrane β-domain. The SepA passenger domain is the most abundant protein released into the extracellular milieu and has been found in large amounts in S. flexneri culture media21 (Fig. S4A). A single-step purification strategy using size exclusion chromatography was used to purify SepA from conditioned culture medium (Fig. S4B). The elution volume was consistent with SepA being monomeric. Multi-angle light scattering (MALS = 99 kDa ± 0.3%) further confirmed that SepA was a monomer. We obtained SepA crystals that diffracted at a 2.9 Å resolution. Molecular replacement with the pathogenic E. coli hemoglobin protease (Hpb, PBD: 1WXR) was used as a search model to obtain initial phase information. Clear omit electron density was observed for most of the protein, with the C-terminal cap region after residue 800 only becoming clear after several cycles of manual rebuilding and refinement. The final model includes residues 1–1006 with no gaps or breaks, but C-terminal residues 1007–1033 were not observed. Data collection and refinement statistics are presented in Table S1.
Figure 4.
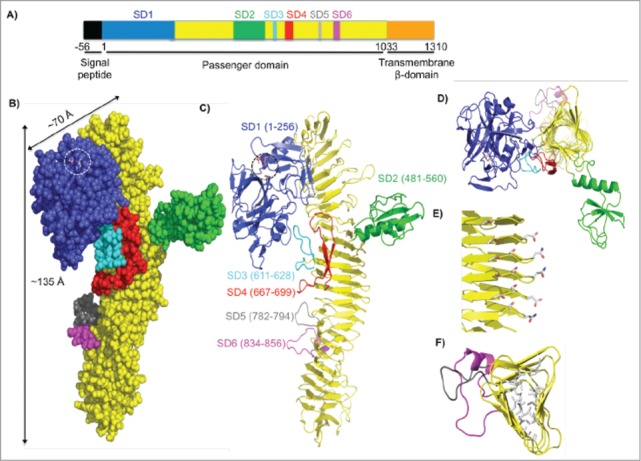
Crystal structure of the SepA passenger domain. (A) Linear schematic representation of the SepA gene. The first 56 residues indicate a signal sequence (black), which directs SepA for secretion. Residues 1–1033 comprise the passenger domain. SD1 contains the serine protease domain (blue, residues 1–256), followed by a β-helix with insertions (yellow, residues 257–1003) including SD2 (green, residues 481–560), SD3 (cyan, residues 611–628), SD4 (red, residues 667–699), SD5 (gray, residues 782–794), and SD6 (magenta, residues 834–856). The C-terminal transmembrane β–domain (orange, residues 1034–1310) folds into a pore from which the passenger domain is secreted. (B) Space-filling representation of the passenger domain using the same color scheme as in the schematic representation of SepA in A. Length and width dimensions are indicated. The Ser211-His78-Asp106 catalytic triad is highlighted and shown as a stick representation. (C) Ribbon diagram representation of the passenger domain. (D) Top view of the passenger domain. For simplicity, residues 257–480 were omitted. Representative sub-domains stem from the corners of the β–helix. (E) Amino acid stacking interactions (Thr-Ser, Ser-Ser, and Asp-Asn; residue numbers are given in Figure S2). (F) Hydrophobic amino acid stacking on the interior of the β-helix as seen through residues 700–900.
The central feature of the SepA passenger domain is a continuous 24-turn β–helix extending from residues 257–1033 (Fig. 4B and C; in yellow). Each turn is formed by 3 short β–strands, which interact with β–strands in neighboring turns, giving rise to extended β–sheets arranged in a slinky-like triangular tube (Fig. 4D). The structure is stabilized by extensive hydrogen bonding that connects the winding networks of parallel β-strands by a repetitive pattern of stacked serine, threonine, aspartic acid, and asparagine residues in discrete parts on the β–helix (Figs. 4E and S5A), and by stacked aliphatic and aromatic hydrophobic amino acid side chains in the interior of the β–helix (Fig. 4F). Both ends of the β–helix are sealed with β-hairpin caps (Fig. S5A). The β–helix is decorated with 6 subdomains (SD1-SD6, Fig. 4C), each inserted into a single loop of the β-helix. The most prominent subdomains are SD1 (256 residues, in blue) and SD2 (80 residues in green). The SD3, SD4, SD5, and SD6 subdomains lack extensive secondary structure or folded cores, and are substantially smaller than SD1 and SD2. SD3 and SD4 (cyan and red) make contact with the underside of SD1 and appear to function as the anchor points on which the protease rests upon the β-helix. SD5 and SD6 (gray and magenta), which are short loops variably present among SPATEs family members, extend from the β–helix core just below SD3 and SD4. SD1, SD3, and SD4 (but not SD2, SD5, and SD6) are common to all SPATEs family members whose structures have been determined to date (Fig. S6).
SD1 adopts a serine protease fold with a canonical catalytic Ser211-His78-Asp106 triad near the protein surface, adjacent to an apparent S1 side-chain binding specificity pocket (Fig. 5A). The overall fold of this domain is similar to those observed in other serine proteases, including bovine chymotrypsin (RMSD 2.4Å). To test the hypothesis that the catalytic triad imparts SepA SD1 with protease activity, we mutated Serine 211 to Alanine (predicted to be catalytically inactive) on the sepA gene in the pZK15 plasmid. Subsequently, we transformed ΔsepA strains with this plasmid now referred to as S211A. Both wild-type SepA and the S211A variant protein were secreted at comparable levels in liquid media from S. flexneri cultures (Fig. S7A). Moreover, the purified proteins had similar thermal stabilities (Fig. S7B). When evaluating the wild-type SepA and S211A variant enzymatic activity using the chromogenic substrate N-Suc-Ala-Ala-Pro-Phe p-NA, previously reported to be hydrolyzed by SepA,27,28 the S211A mutation reduced hydrolysis activity to background levels (Fig. 5B). To test whether SepA protease activity was required for disruption of intestinal epithelial monolayers, we evaluated the ability of the S211A strain to modulate barrier permeability using TEER. Similar to the ΔsepA strain, the S211A strain lost the ability to disrupt the epithelial monolayer (Fig. 5C) and showed a significant decrease in bacterial invasion (Fig. 5D). Finally, we determined the effect of S211A infection on LIMK1 modulation. Infection with S211A did not decrease LIMK1 protein levels when compared with infection with the wild-type or the ΔsepA+pZK15 complemented strains (Fig. 5E). These results show that the ability to cause barrier disruption relies on S. flexneri SepA protease activity on actin regulators, which may ultimately facilitate the paracellular passage of Shigella to the basolateral epithelial pole.
Figure 5.
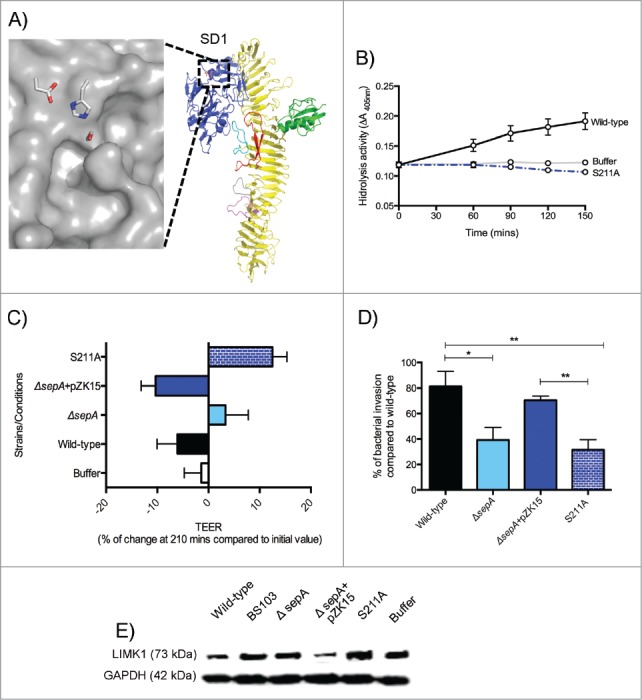
SepA SD1. (A) Surface representation (in gray) of the active site catalytic triad (Ser211-His78-Asp106). The location of the putative SD1 specificity pocket is indicated by an asterisk. (B) Protease cleavage assay with the wild-type SepA protein (black), S211A (dotted blue), and the buffer control (gray). (C) Regulation of TEER after 210 min stimulation with S. flexneri is abolished when infecting with S211A strain (blue with pattern). (D) Polarized cell monolayers were apically-infected with S. flexneri strains. Data are expressed as standardized colony-forming units (CFU) per monolayer ( ± SEM) compared with CFU in wild-type infection. (E) Western blot analysis of LIMK1 expression.
SD2 lies opposite of SD1 on the β-helix (Fig. 6A), branching from one corner of the β-helix and forming a discrete α+β domain with a central 3-stranded, anti-parallel β–sheet. This central fold is characteristic of small subdomains present in certain chitinases and chitin deacetylases. Among these, structural similarity was greatest for the carbohydrate-binding module families 5 and 12, as defined by the glyco-database (www.cazy.org) (Fig. 6B). The residues conserved across carbohydrate-binding module families 5 and 12 (Fig. 6C) also are found in SepA SD2, primarily in the hydrophobic core. No function has been ascribed to SD2 in any bacterial passenger domain. To test whether SD2 was required for SepA-dependent virulence, we substituted the sequence encoding the entire SD2 domain (residues 482–555) with one encoding a single glycine residue on the sepA gene in the pZK15 plasmid. We then proceeded to transform this plasmid, referred to as ΔSD2, into ΔsepA strain. Similar to the protease-inactive SepA variant (S211A), SepA ΔSD2 was efficiently secreted from recombinant S. flexneri. SepA ΔSD2 also retained wild-type thermal stability and protease activity (Fig. S7C). Moreover, we found that SD2 was not required to modulate epithelial barrier permeability nor for bacterial invasion, with values comparable to those observed in wild-type and ΔsepA+pZK15 S. flexneri strains (Fig. S7D and E). Thus, in our in vitro model, SD2 does not seem to be essential for barrier function disruption or for the invasiveness observed in SepA-harboring strains.
Figure 6.

SepA SD2. (A) SD2 highlighted in a ribbon representation of SepA. (B) Side-by-side comparison of minimal SepA SD2 (residues 491–541, green) with chitinase C, a carbohydrate-binding module 5 family member from S. griseus (residues 32–79, blue), and chitin deacetylase, a carbohydrate-binding module family 12 member from V. parahaemolyticus (residues 333–379, orange). Overlay of the 3 truncated proteins and superimposition of conserved residues from the protein alignment onto SepA. (C) CLUSTALW alignment of the 3 aforementioned amino acid sequences. Conserved residues are shown in black and semi-conserved residues in gray.
Discussion
Shigella spp. is one of the leading etiological agents of diarrhea worldwide, and is unique among enteric pathogens, as it invades colonic epithelium from the basolateral pole. Here, we report that a protein secreted by Shigella, namely SepA, is essential for disruption of epithelial barrier integrity during infection. SepA has been previously associated with virulence not only in Shigella infections but also in Enteroaggregative Escherichia coli infections causing severe diarrhea and illness in children in Mali.25 In this study, we show the structural basis of SepA protein function and the mechanism of action of SepA during S. flexneri infection.
Using a polarized intestinal monolayer mimicking what occurs in vivo during infection, we demonstrate that SepA is crucial for S. flexneri invasion of intestinal cells only when the bacteria faces the apical pole of the epithelium. Conversely, a previous report failed to observe differences in bacterial invasion when infecting intestinal cells monolayers with ΔsepA.21 This discrepancy is likely to be explained based on the differences of the model used: Benjelloun-Touimi et al. performed experiments on non-confluent HeLa cells while our experiments were performed in fully confluent, polarized colon cell lines (T84 and HCT8). Our observations suggest that the functioning of SepA might be dependent on spatial and temporal events during infection (i.e. at early stages of infection when bacteria encounter the apical pole of the epithelial barrier).3,10
Supporting this hypothesis, we found that SepA is responsible for disruption of the polarized epithelial barrier during Shigella infection. Prior studies attributed the barrier dysfunction during in vitro Shigella infection to the translocation of PMN from the basolateral pole to the apical pole where bacteria reside.10 Moreover, the same group of investigators showed that such barrier dysfunction was accompanied by an increase in Shigella virulence.34 Since then, we have demonstrated that in the absence of PMN, Shigella itself can cause barrier dysfunction,9 and for the first time we now report that SepA plays a critical role in this phenomenon. The fact that the same barrier defects are achieved by infecting either basolaterally or apically suggest that SepA might engage an ubiquitous receptor and/or target an unique protein that is evenly distributed throughout the epithelial cells. It is also possible that SepA targets a variety of receptors and/or proteins depending on where the bacteria resides, or that targets of SepA are diverse and can be found both at the apical or basolateral poles of the epithelium. Supporting the later, another class II SPATEs called Pic -also found in Shigella- has been shown to target an array of proteins during infection.24,35-37 However, more studies are needed to elucidate these possibilities.
Noteworthy, transepithelial migration of PMN to the site of infection driven by epithelial release of IL-8 has been shown to contribute to massive epithelial damage during S. flexneri infection.38 Here, we showed that the PMN transepithelial migration induced during apical infection is contingent on bacterial invasion, which in turn is dependent on SepA disruption of the epithelial barrier. Strains lacking SepA, which are unable to cause barrier disruption and bacteria invasion from the apical pole, exhibited a reduced ability to trigger PMN transepithelial migration. However, when directly infecting the basolateral pole of the epithelium, SepA function on the barrier was not sufficient to deter PMN transepithelial migration, confirming a temporal-spatial requirement of SepA during S. flexneri infection. Although the effects of SepA on bacterial invasion and PMN transepithelial migration during infection are indirect (via epithelial permeability), SepA activity is still fundamental for S. flexneri pathogenesis.
Our results of the 3D reconstructed epithelial tissue model are consistent with prior observations23 that demonstrate SepA-dependent morphological alterations in the intestinal epithelium. Particularly, we observed lesions on the intestinal epithelium largely characterized by cell desquamation, as well as a marked disruption of the columnar architecture- a hallmark of shigellosis. Mechanistically, we further discovered that SepA orchestrates the apical accumulation of cofilin during S. flexneri infection. As above mentioned, cofilin plays a fundamental role in actin-dynamics.39-41 Cofilin can be activated at the plasma membrane by hydrolysis of PtdIns(4,5) P2, which sequesters the actin-binding residues of cofilin. In the cytosol, cofilin is activated by either high pH levels or by de-phosphorylation of the actin-binding residues.39 Here, we observed that following S. flexneri infection there was a SepA-dependent increase in cofilin activation (a.k.a. dephosphorylation) in the cytosol. Such dephosphorylation is essential for the actin-severing and -depolymerization activity of this protein.41,42
We further found that the significant increase in activate cofilin was accompanied by a decrease in LIMK1 expression, a protein known to negatively regulate cofilin by phosphorylation of its Ser3 residue. Thus, our results indicate that SepA plays a role in actin dynamics during infection by orchestrating the activation of cofilin via LIMK1 downregulation. Activation of cofilin has been shown to increase epithelial permeability by redistributing actin and tight junction proteins.12,15 Moreover, loss of LIMK1 and constitutive expression of cofilin has been shown to affect cell adhesion and compaction in the differentiating cell layers of psoriatic patients.43 In these patients, cofilin activation was associated with inflamed skin lesions where epithelial desquamation is abundant.43 In light of these reports, we believe that the accumulation of cofilin observed in cells shedding from the infected SMI-100 also support the loss of cell adhesion promoting intestinal lesions upon S. flexneri infection. It is also possible that at the plasma membrane, additional activation of cofilin might be important to disrupt the epithelial barrier during Shigella infection. In this regard, recent reports have shown that actin foci associated with Shigella infection require signaling, which is dependent on PtdIns(4,5) P2.44 We therefore speculate that hydrolysis of PtdIns(4,5) P2 by phospholipase C could release unphosphorylated cofilin at the plasma membrane.
We further determined the structure of SepA to gain insight into the function of this molecule during S. flexneri infection. SepA is composed of a long β–helix domain with grafted subdomains in a radial fashion. Formation of similar β–helix structures in other SPATEs family members have been hypothesized for assisting in the ATP-independent translocation of the passenger domain across the bacterial cell outer membrane.45-47 Like other members of the class-II SPATEs family, SepA contains a SD2 that has structural homology to chitinase/chitin binding domain-containing proteins, but whether these domains actually bind carbohydrates remains unknown. Given the role of chitinase-like molecules in bacterial adhesion, invasion, and consequent inflammation in the colon, we posit that SD2 could function in assisting SepA recognition of its receptor/target on the cell surface of colonic cells. However, complete deletion of SD2 neither impacts the proteolytic activity of SepA nor the ability of S. flexneri to decrease TEER or to invade epithelial cells. Further experimentation, is required to more precisely address the potential role of the SD2 on virulence.
Among the other subdomains, a serine protease catalytic domain (or SD1) is found in all SPATEs members. Protease substrates are known for some SPATEs family members. For example, E. coli O157:H7 EspP cleaves the complement factors C3/C3b and C5 presumably as part of an immune evasion strategy48 and pathogenic E. coli Hpb cleaves hemoglobin presumably as a source of iron for bacterial metabolism.49 We found that mutation of the SD1 active site nucleophile (S211A) was essential for SepA's proteolytic activity and its ability to disrupt the epithelial barrier, consequently hindering S. flexneri invasion. Epithelial permeability, LIMK1 downregulation, and bacterial invasion are all abolished when infecting with the S211A strain at the apical pole. Both LIMK1 and cofilin can be regulated by proteolysis,50,51 and both are downstream of protease-activated cell surface receptors,52-54 but additional studies are required to determine whether SepA SD1 acts through these pathways. Finally, we found that obliteration of the proteolytic activity did not prevent the secretion of the SepA passenger domain, suggesting that an additional protein is responsible for the cleavage of the linker region, as seen in other SPATEs.45
In conclusion, we propose a mechanism of action for SepA (Fig. 7) in which at early stages of infection, S. flexneri secretes SepA to the extracellular milieu where it interacts with the apical pole of the intestinal epithelium. Whether SepA is internalized, binds to a receptor, or cleaves proteins at the surface of the cell remains uncertain. Regardless, our study shows that the serine protease activity of SepA causes downregulation of LIMK1, resulting in increased cofilin activation. Active cofilin then prompts depolymerization and severing of actin, ultimately disrupting the epithelium barrier. As shown by our group, a permissive epithelial barrier allows bacteria to breach basolateral pole of the epithelium.9 Once at the basolateral pole, Shigella deploys the entire T3SS artillery for intracellular invasion,3 evoking massive PMN infiltration and inflammation.33 In sum, we report the fundamental role of SepA in spatial and temporal events leading to S. flexneri pathogenesis.
Figure 7.
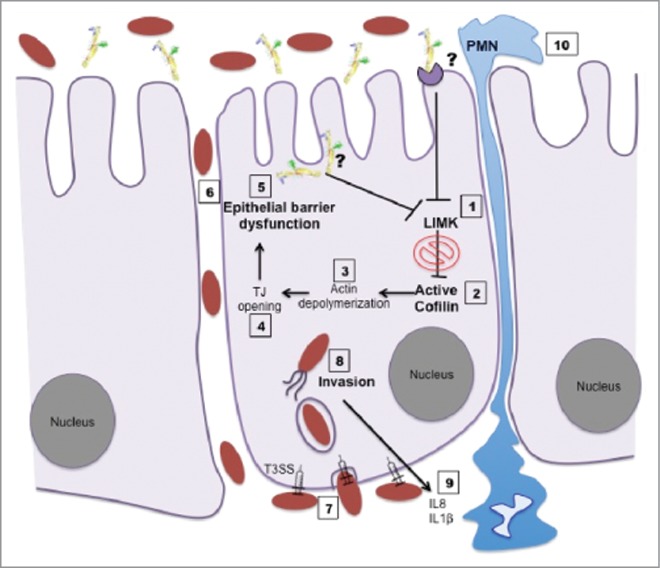
Schematic model of the function of SepA during S. flexneri infection.
Materials and methods
Bacterial strains
A total of 10 S. flexneri strains were used in this study (Table S2). Bacterial cells were transformed with pmCherry or GFP (pKC561) fluorescent plasmids that were kindly provided by the Leong Lab.55 Both ΔsepA and ΔsepA+pZK15 strains were obtained from the Maurelli laboratory.21 The serine protease inactive SepA (S211A) was created by the substitution the sequence encoding the serine active site nucleophile at position 211 to sequence encoding an alanine on the sepA gene in the pZK15 plasmid, which was used for complementation. The ΔsepA strain that was transformed with the pZK15 plasmid carrying the S211A mutation were referred to as the S211A strain. For the generation of ΔSD2, the sequence encoding the putative carbohydrate-binding domain (residues 482–555) on the pZK15 plasmid was replaced by a sequence encoding a single glycine using overlapping PCR. The ΔsepA strain that was transformed with the pZK15 plasmid carrying the SD2 deletion was referred to as the ΔSD2 strain.
Bacterial cultures were maintained at −80°C in 20% glycerol stocks. Bacteria were streaked onto Tryptic Soy Agar with 0.2% of Red Congo and plates were incubated at 37°C for 12–18 hrs. For infections, one colony was selected from the fresh plate and was incubated overnight in Tryptic Soy Broth, with antibiotics (Ampicillin [100 mg/mL] or Chloramphenicol [20 mg/mL) and IPTG when required. The next morning, the bacterial cells were diluted 1:100 in fresh Tryptic Soy Broth and incubated for 2 ½ hrs, using antibiotics and IPTG when required. Subsequently, the bacteria were prepared by washing 2X with Hank's balance salt solution or HBBS+ (Thermo Fisher Scientific, 14025092) and re-suspended in the same buffer.
Tissue culture
For all of the experiments, we used an in vitro model of a polarized intestinal epithelial cell monolayer, as described previously by us.33,56 Briefly, human intestinal epithelial cells (T84 or HCT8) were maintained in Dubelcco's Modified Eagle's medium/F-12 supplemented with HEPES (15 mM; pH 7.5) and 10% fetal bovine serum. To obtain polarized monolayers, cells were grown in 24 mm (used for PMN transepithelial migration, invasion and barrier function assays) or 75 mm (used for protein lysate collection) collagen-coated permeable polycarbonate filters or trans-wells (Corning, CLS3413–48EA). Cells were incubated at 37°C with 5% CO2. Monolayers were used when cellular confluence and differentiation was achieved, usually after 7–8 d of inoculation. All tissue culture reagents were obtained from Invitrogen.
Invasion assays
After equilibration with HBSS+ for 20–30 mins at 37°C, epithelial cell monolayers were apically- or basolaterally-infected to estimate the percentage of bacterial invasion compared with wild-type infection as described previously.29 Briefly, after 1 ½ hrs of infection, the monolayers were washed and placed in warm plates with 1 ml of HBSS+ and Gentamicin (50 ug/mL). In parallel, 150 μl of HBSS+ with the same concentration of Gentamicin was also applied to the apical pole of the epithelial cell monolayers and incubated for 3 hrs. After this incubation, the cells were washed and the epithelial cell monolayers were lysed by incubating in Triton X (1%) for 1 hr. A serial dilution of each infected epithelial cell monolayer was made to determine the colony forming units (CFU) per condition. Similarly, a serial dilution from the bacteria inoculum used to infect the monolayers was made. CFU per condition were standardized by dividing the CFU obtained from the monolayers by the CFU obtained from the bacteria used for infection. Results were normalized by values obtained with wild-type infection, which represented the maximum value (100%) of bacterial invasion.
Transepithelial electrical resistance (TEER) measurements
To determine currents, transepithelial potentials, and resistance, a commercially available voltage clamp (Bioengineering Dept., University of Iowa) was used. Through the use of Ohm's law (PD (mV)/100 mA•cm2 = Resistance), tissue resistance and transepithelial current can be determined. Intestinal cell monolayers were equilibrated with HBSS+ for 20–30 mins. Subsequently, monolayers were infected apically with S. flexneri strains and resistance was measured every 30 mins during infection as described previously.9
Protein lysate collection and western blots
For protein harvest, the monolayers (HCT8 or T-84) were apically- or basolaterally-infected at a multiplicity of infection (MOI) of 100 for 1 hr. Following infection, cells were washed and harvested on ice in a lysis buffer containing the following: 1% Triton X-100, 100 mM NaCl, 10 mM HEPES, 2 mM EDTA, 4 mM Na3VO4, 40 mM NaF, 200 mM PMSF, and a protease inhibitor cocktail (Roche, 11836153001). Lysates were centrifuged at 14,000 rpm at 4°C for 30 mins and the supernatants containing the cytosol-associated proteins were collected. Proteins were electrophoresed on a 4–20% precast polyacrylamide gel (BioRad, 4561093) and transferred to nitrocellulose membranes.
The following antibodies were used: Total cofilin (Abcam, ab42824), p-cofilin (Santa Cruz BioTechnologies, sc-271923), LIMK1 (Santa Cruz BioTechnologies, sc-28370), and LIMK 2 (Abcam, ab9766). The antibodies were used according to the manufacturer's specifications. The polyclonal antibody against SepA was kindly obtained from the Dr. James Nataro laboratory. HRP-labeled secondary antibodies (Invitrogen, A27036 and A28177) and enhanced chemoluminescence (Thermo Fisher, 34580) were used to detect the protein bands. Buffer-treated monolayers were included as controls. All experiments were performed at least 3 times.
EpiIntestinal tissue model (SMI-100)
The tissue models were developed at MatTek and kindly provided for this study. The SMI-100 tissue models have been developed to serve as an efficient human model system for microbial interaction studies. Briefly, human intestinal stem cells are grown at the air liquid interface on fibroblasts and endothelial cells on a permeable filter support that allows the study of apical epithelial infections. This tissue model includes enterocytes, Paneth cells, M cells, tuft cells, and intestinal stem cells in a highly differentiated, polarized epithelium (more at: http://mattek.com/epiIntestinal/datasheet). The SMI-100 are stratified, thereby exhibiting structured columnar-shaped basal cells, villi-like structure, brush borders, and functional tight junctions. SMI-100 were maintained in DMEM for 1–2 d. Following equilibration, SMI-100 were infected with fluorescent S. flexneri strains (at OD600 = 0.1, Table S2) for 4 hrs at 37°C. Four hours was sufficient to observe bacterial invasion of epithelial cells in our initial optimization experiments (data not shown).
Confocal microscopy
Infected and mock-treated SMI-100 were fixed with 4% formaldehyde in 5% sucrose DPBS for 1 hr. Cells were then permeabilized with 0.5% Triton-X100 (in DPBS) for 10 mins at 37°C. Then, the primary cofilin antibody was added (Abcam, ab42824) and incubated for 1 hr. Subsequently, the following secondary antibodies were added: Alexa Fluor® 488 or Alexa Fluor® 594 (Thermo Fisher Scientific, A-11034 or R37119, respectively) followed by conjugated phalloidin Alexa Fluor® 647 (Thermo Fisher Scientific, A22287). Imaging was performed using a Nikon A1R-A1 confocal microscope. Image acquisition was performed with NIS-Elements imaging software (Nikon) followed by analyses using Volocity (Perkin Elmer). Densitometry was analyzed by Photoshop CS4 and presented as a relative intensity compared with infection with wild-type S. flexneri. Images of at least 3 experiments were included in the analysis.
PMN transepithelial migration
Polymorphonuclear leukocytes (PMNs) were isolated from whole blood obtained from normal healthy human volunteers by venipuncture. Whole blood was anti-coagulated with acid citrate/dextrose.57 The PMN transepithelial migration assays were performed as described previously.33 Briefly, following apical or basolateral infection of epithelial cell monolayers with the S. flexneri strains, 1 × 106 of freshly isolated PMNs were added to the basolateral pole and incubated for 3 hrs at 37°C. PMNs that migrated across the model epithelium were quantified by detecting the neutrophil-specific azurophil granule marker myeloperoxidase (MPO), as described previously.58
Statistical analysis
Data are shown as means ± standard error of the mean (SEM). All statistical analyses were performed using Prism v6 software. The 2-tailed unpaired student's t-test was used to calculate significant differences between treatment conditions. Asterisks are used to denote p-values: ** <0.01; *** <0.001; and ****<0.0001.
SepA isolation and crystallization
S. flexneri containing the inducible pZK15 plasmid,21 which encodes the full-length SepA, were grown in Tryptic Soy Broth at 37°C with shaking (200 RPM) to an OD600 of 0.6. Subsequently, bacteria were induced with IPTG (0.5 mM) for 6–8 hrs at 30°C with shaking. The supernatant was collected, filtered (0.22 µm filter), and concentrated 1000-fold by centrifugation on a Centricon®70 plus filter unit (Millipore, MA). The concentrated supernatant was then loaded onto a HiLoad 16/60 Superdex 200 gel filtration column (GE healthcare, MA) that was pre-equilibrated with PBS. Peak fractions of the passenger domain were concentrated to 10 mg/ml and entered into crystallization trials using the sitting drop vapor diffusion technique.
The best, initial SepA crystals diffracted to 5Å with synchrotron radiation and were grown by mixing 1μl of SepA with 1μl of 10% PEG 8K and 20% ethylene glycol 0.1MES/IMID pH 6.5 (condition D2 from the Morpheus Crystallization screen)59 at room temperature. Improved crystals were rapidly found by using an iterative process described previously, which involved mixing Morpheus condition D2 with other sparse matrix crystallization screens in 96-well format.60 These Triangular crystals of maximum dimensions 0.1 × 0.2 × 0.2 mm were grown by mixing 85% (v/v) of Morpheus condition D2 with 15% (v/v) of Crystal Screen I (Hampton Research, HR2–910–06) condition 23, which specifically contained 0.1M HEPES pH 7.5, 0.2M MgCl2, and 30% PEG 400. Crystals appeared after 1 day and were flash-frozen directly from the mother liquor. SepA had a 2.9Å maximum diffraction and was in the P3221 space group with unit cell dimensions a = b = 143.52 Å and c = 269.25 Å, with 2 molcules in the asymmetric unit.
Data collection, processing and structure determination
High-resolution X-ray diffraction data sets were recorded at the LRL-CAT 31-ID beamline Advanced Photon Source (APS) in Chicago. Images were processed using Mosflm version 7.1.1. and scaled with SCALA from the CCP4 suite.61 A pruned search model was prepared by aligning the SepA amino acid sequence with the E. coli hemoglobin protease sequence,49 PDB accession code: 1WXR, before analysis with the SCULPTOR program.62 Initial phases were obtained by molecular replacement using PHASER.63,64 Phases were improved using RESOLVE65 with 2-fold non-crystallographic symmetry real-space averaging. Composite omit maps65 and prime-and-switch maps were used to build the model. The initial model was built using AutoBuild66 and improved by cycles of manual building and refinement using COOT67 and PHENIX REFINE,68 respectively. Of the 2 SepA molecules present in the asymmetric unit (Figure S2B), molecule A displayed stronger electron density overall compared with molecule B. Residues 885–1006 in molecule B were particularly difficult to build due to weak electron density. Therefore, residues in this region have markedly higher B-factor values than the rest of the protein.
In the unit cell, the molecules pack in an unusual fashion giving rise to a helical arrangement where one of the molecules in the asymmetric unit contributes to one helix and the other to the other helix, with the 2 helices interlocking (Fig. S2C). To determine if SepA is a monomeric molecule, multi-angle light scattering with size exclusion chromatography analysis was performed using a Dawn Heleos-II instrument equilibrated with PBS and operated according to the manufacturer's instructions. DALI69 was used for identification of structural similarity with other proteins.
Enzymatic assays
To test the proteolytic activity of the wild-type, S211A, and ΔSD2 SepA, we used a reported substrate for this protein, a p-Nitroanilide peptide (Suc-Ala-Ala-Pro-Phe-pNA), as described previously (28).
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This study used resources provided by the Advanced Photon Source, a US. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357. We acknowledge the use of the Lilly Research Laboratories Collaborative Access Team (LRL-CT) beamline at Sector 31 of the Advanced Photon Source provided by Eli Lilly Company. We thank Zachary Maben for assistance with preparation of samples for the synchrotron.
Funding
This work was supported by: AM-C by the Charles A. King Trust Postdoctoral Research Fellowship Program and the Faculty Diversity Scholar Program at UMASS Medical School; EB by the Carlsberg Foundation; HCR and YZ by National Institutes of Health (NIH) Grants: AI113333, DK068181, and DK043351; BAM, AM-C and KLM by NIH Grants: DK56754 and DK33506; LJS and JRB by NIH AI-38996.
Author contributions
Study conception and design: BAM, AM-C, LJS, JRB, EB, KLM.
Acquisition of data: AM-C, JRB, LJS, EB, KLM, JT, HCR, YZ, SA.
Analysis and interpretation of data: AM-C, JRB, BAM, LJS, EB, KLM, JT, HCR, YZ, SA.
Drafting of manuscript: AM-C, BAM, JRB, LJS.
References
- [1].Bowen A. Chapter 3: Infectious Diseases Related to Travel. CDC; 2016 [updated Retrieved 22 June 2016. [Google Scholar]
- [2].Kotloff KL, Blackwelder WC, Nasrin D, Nataro JP, Farag TH, van Eijk A, Adegbola RA, Alonso PL, Breiman RF, Faruque AS, et al.. The Global Enteric Multicenter Study (GEMS) of diarrheal disease in infants and young children in developing countries: epidemiologic and clinical methods of the case/control study. Clin Infect Dis 2012; 55(Suppl 4):S232-45; PMID:23169936; https://doi.org/ 10.1093/cid/cis753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mounier J, Vasselon T, Hellio R, Lesourd M, Sansonetti PJ. Shigella flexneri enters human colonic Caco-2 epithelial cells through the basolateral pole. Infect Immun 1992; 60(1):237-48; PMID:1729185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Buchrieser C, Glaser P, Rusniok C, Nedjari H, D'Hauteville H, Kunst F, Sansonetti P, Parsot C. The virulence plasmid pWR100 and the repertoire of proteins secreted by the type III secretion apparatus of Shigella flexneri. Mol Microbiol 2000; 38(4):760-71; PMID:11115111; https://doi.org/ 10.1046/j.1365-2958.2000.02179.x [DOI] [PubMed] [Google Scholar]
- [5].Sansonetti PJ, Kopecko DJ, Formal SB. Involvement of a plasmid in the invasive ability of Shigella flexneri. Infect Immun 1982; 35(3):852-60; PMID:6279518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sasakawa C, Komatsu K, Tobe T, Suzuki T, Yoshikawa M. Eight genes in region 5 that form an operon are essential for invasion of epithelial cells by Shigella flexneri 2a. J Bacteriol 1993; 175(8):2334-46; PMID:8385666; https://doi.org/ 10.1128/jb.175.8.2334-2346.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Parsot C, Menard R, Gounon P, Sansonetti PJ. Enhanced secretion through the Shigella flexneri Mxi-Spa translocon leads to assembly of extracellular proteins into macromolecular structures. Mol Microbiol 1995; 16(2):291-300; PMID:7565091; https://doi.org/ 10.1111/j.1365-2958.1995.tb02301.x [DOI] [PubMed] [Google Scholar]
- [8].Sansonetti PJ. Molecular and cellular mechanisms of invasion of the intestinal barrier by enteric pathogens. The paradigm of Shigella. Folia Microbiol (Praha) 1998; 43(3):239-46; PMID:9717250; https://doi.org/ 10.1007/BF02818608 [DOI] [PubMed] [Google Scholar]
- [9].Sakaguchi T, Kohler H, Gu X, McCormick BA, Reinecker HC. Shigella flexneri regulates tight junction-associated proteins in human intestinal epithelial cells. Cell Microbiol 2002; 4(6):367-81; PMID:12067320; https://doi.org/ 10.1046/j.1462-5822.2002.00197.x [DOI] [PubMed] [Google Scholar]
- [10].Perdomo JJ, Gounon P, Sansonetti PJ. Polymorphonuclear leukocyte transmigration promotes invasion of colonic epithelial monolayer by Shigella flexneri. J Clin Invest 1994;93(2):633-43; PMID:7906696; https://doi.org/ 10.1172/JCI117015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ivanov AI, McCall IC, Parkos CA, Nusrat A. Role for actin filament turnover and a myosin II motor in cytoskeleton-driven disassembly of the epithelial apical junctional complex. Mol Biol Cell 2004; 15(6):2639-51; PMID:15047870; https://doi.org/ 10.1091/mbc.E04-02-0163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nagumo Y, Han J, Bellila A, Isoda H, Tanaka T. Cofilin mediates tight-junction opening by redistributing actin and tight-junction proteins. Biochem Biophys Res Commun 2008; 377(3):921-5; PMID:18952063; https://doi.org/ 10.1016/j.bbrc.2008.10.071 [DOI] [PubMed] [Google Scholar]
- [13].Shen L, Turner JR. Actin depolymerization disrupts tight junctions via caveolae-mediated endocytosis. Mol Biol Cell 2005; 16(9):3919-36; PMID:15958494; https://doi.org/ 10.1091/mbc.E04-12-1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shiobara T, Usui T, Han J, Isoda H, Nagumo Y. The reversible increase in tight junction permeability induced by capsaicin is mediated via cofilin-actin cytoskeletal dynamics and decreased level of occludin. PLoS One 2013; 8(11):e79954; PMID:24260326; https://doi.org/ 10.1371/journal.pone.0079954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mizuno K. Signaling mechanisms and functional roles of cofilin phosphorylation and dephosphorylation. Cellular signalling 2013; 25(2):457-69; PMID:23153585; https://doi.org/ 10.1016/j.cellsig.2012.11.001 [DOI] [PubMed] [Google Scholar]
- [16].Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, Nishida E, Mizuno K. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 1998; 393(6687):809-12; PMID:9655398; https://doi.org/ 10.1038/31735 [DOI] [PubMed] [Google Scholar]
- [17].Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, et al.. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 1998; 393(6687):805-9; PMID:9655397; https://doi.org/ 10.1038/31729 [DOI] [PubMed] [Google Scholar]
- [18].Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K, Narumiya S. Signaling from rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 1999; 285(5429):895-8; PMID:10436159; https://doi.org/ 10.1126/science.285.5429.895 [DOI] [PubMed] [Google Scholar]
- [19].Zhang L, Luo J, Wan P, Wu J, Laski F, Chen J. Regulation of cofilin phosphorylation and asymmetry in collective cell migration during morphogenesis. Development 2011; 138(3):455-64; PMID:21205790; https://doi.org/ 10.1242/dev.046870 [DOI] [PubMed] [Google Scholar]
- [20].Nishita M, Tomizawa C, Yamamoto M, Horita Y, Ohashi K, Mizuno K. Spatial and temporal regulation of cofilin activity by LIM kinase and Slingshot is critical for directional cell migration. Journal of Cell Biology 2005; 171(2):349-59; PMID:16230460; https://doi.org/ 10.1083/jcb.200504029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Benjelloun-Touimi Z, Sansonetti PJ, Parsot C. SepA, the major extracellular protein of Shigella flexneri: autonomous secretion and involvement in tissue invasion. Mol Microbiol 1995; 17(1):123-35; PMID:7476198; https://doi.org/ 10.1111/j.1365-2958.1995.mmi_17010123.x [DOI] [PubMed] [Google Scholar]
- [22].Pieper R, Fisher CR, Suh MJ, Huang ST, Parmar P, Payne SM. Analysis of the Proteome of Intracellular Shigella flexneri Reveals Pathways Important for Intracellular Growth. Infection and Immunity 2013; 81(12):4635-48; PMID:24101689; https://doi.org/ 10.1128/IAI.00975-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Coron E, Flamant M, Aubert P, Wedel T, Pedron T, Letessier E, Galmiche JP, Sansonetti PJ, Neunlist M. Characterisation of early mucosal and neuronal lesions following Shigella flexneri infection in human colon. PLoS One 2009; 4(3):e4713; PMID:19274103; https://doi.org/ 10.1371/journal.pone.0004713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ruiz-Perez F, Wahid R, Faherty CS, Kolappaswamy K, Rodriguez L, Santiago A, Murphy E, Cross A, Sztein MB, Nataro JP. Serine protease autotransporters from Shigella flexneri and pathogenic Escherichia coli target a broad range of leukocyte glycoproteins. Proc Natl Acad Sci U S A 2011; 108(31):12881-6; PMID:21768350; https://doi.org/ 10.1073/pnas.1101006108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Boisen N, Scheutz F, Rasko DA, Redman JC, Persson S, Simon J, Kotloff KL, Levine MM, Sow S, Tamboura B, et al.. Genomic characterization of enteroaggregative Escherichia coli from children in Mali. J Infect Dis 2012; 205(3):431-44; PMID:22184729; https://doi.org/ 10.1093/infdis/jir757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Vijayakumar V, Santiago A, Smith R, Smith M, Robins-Browne RM, Nataro JP, Ruiz-Perez F. Role of class 1 serine protease autotransporter in the pathogenesis of Citrobacter rodentium colitis. Infect Immun 2014; 82(6):2626-36; PMID:24711562; https://doi.org/ 10.1128/IAI.01518-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ayala-Lujan JL, Vijayakumar V, Gong M, Smith R, Santiago AE, Ruiz-Perez F. Broad spectrum activity of a lectin-like bacterial serine protease family on human leukocytes. PLoS One 2014; 9(9):e107920; PMID:25251283; https://doi.org/ 10.1371/journal.pone.0107920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Benjelloun-Touimi Z, Si Tahar M, Montecucco C, Sansonetti PJ, Parsot C. SepA, the 110 kDa protein secreted by Shigella flexneri: two-domain structure and proteolytic activity. Microbiology 1998; 144 (Pt 7):1815-22; PMID:9695914; https://doi.org/ 10.1099/00221287-144-7-1815 [DOI] [PubMed] [Google Scholar]
- [29].McCormick BA, Colgan SP, Delp-Archer C, Miller SI, Madara JL. Salmonella typhimurium attachment to human intestinal epithelial monolayers: transcellular signalling to subepithelial neutrophils. J Cell Biol 1993; 123(4):895-907; PMID:8227148; https://doi.org/ 10.1083/jcb.123.4.895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Madara JL. Regulation of the movement of solutes across tight junctions. Annu Rev Physiol 1998; 60:143-59; PMID:9558458; https://doi.org/ 10.1146/annurev.physiol.60.1.143 [DOI] [PubMed] [Google Scholar]
- [31].Wang D, Naydenov NG, Feygin A, Baranwal S, Kuemmerle JF, Ivanov AI. Actin-Depolymerizing Factor and Cofilin-1 Have Unique and Overlapping Functions in Regulating Intestinal Epithelial Junctions and Mucosal Inflammation. Am J Pathol 2016; 186(4):844-58; PMID:26878213; https://doi.org/ 10.1016/j.ajpath.2015.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Foulke-Abel J, In J, Kovbasnjuk O, Zachos NC, Ettayebi K, Blutt SE, Hyser JM, Zeng XL, Crawford SE, Broughman JR, et al.. Human enteroids as an ex-vivo model of host-pathogen interactions in the gastrointestinal tract. Exp Biol Med (Maywood) 2014; 239(9):1124-34; PMID:24719375; https://doi.org/ 10.1177/1535370214529398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].McCormick BA, Siber AM, Maurelli AT. Requirement of the Shigella flexneri virulence plasmid in the ability to induce trafficking of neutrophils across polarized monolayers of the intestinal epithelium. Infect Immun 1998;66(9):4237-43; PMID:9712773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Perdomo OJ, Cavaillon JM, Huerre M, Ohayon H, Gounon P, Sansonetti PJ. Acute inflammation causes epithelial invasion and mucosal destruction in experimental shigellosis. J Exp Med 1994; 180(4):1307-19; PMID:7931064; https://doi.org/ 10.1084/jem.180.4.1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Henderson IR, Czeczulin J, Eslava C, Noriega F, Nataro JP. Characterization of pic, a secreted protease of Shigella flexneri and enteroaggregative Escherichia coli. Infect Immun 1999; 67(11):5587-96; PMID:10531204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Parham NJ, Srinivasan U, Desvaux M, Foxman B, Marrs CF, Henderson IR. PicU, a second serine protease autotransporter of uropathogenic Escherichia coli. FEMS Microbiol Lett 2004; 230(1):73-83; PMID:14734168; https://doi.org/ 10.1016/S0378-1097(03)00862-0 [DOI] [PubMed] [Google Scholar]
- [37].Harrington SM, Sheikh J, Henderson IR, Ruiz-Perez F, Cohen PS, Nataro JP. The Pic protease of enteroaggregative Escherichia coli promotes intestinal colonization and growth in the presence of mucin. Infect Immun 2009; 77(6):2465-73; PMID:19349428; https://doi.org/ 10.1128/IAI.01494-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sansonetti PJ, Arondel J, Huerre M, Harada A, Matsushima K. Interleukin-8 controls bacterial transepithelial translocation at the cost of epithelial destruction in experimental shigellosis. Infect Immun 1999; 67(3):1471-80; PMID:10024597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].van Rheenen J, Condeelis J, Glogauer M. A common cofilin activity cycle in invasive tumor cells and inflammatory cells. J Cell Sci 2009;122(Pt 3):305-11; PMID:19158339; https://doi.org/ 10.1242/jcs.031146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Andrianantoandro E, Pollard TD. Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol Cell 2006; 24(1):13-23; PMID:17018289; https://doi.org/ 10.1016/j.molcel.2006.08.006 [DOI] [PubMed] [Google Scholar]
- [41].Huang TY, DerMardirossian C, Bokoch GM. Cofilin phosphatases and regulation of actin dynamics. Curr Opin Cell Biol 2006;18(1):26-31; PMID:16337782; https://doi.org/ 10.1016/j.ceb.2005.11.005 [DOI] [PubMed] [Google Scholar]
- [42].Niwa R, Nagata-Ohashi K, Takeichi M, Mizuno K, Uemura T. Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell 2002;108(2):233-46; PMID:11832213; https://doi.org/ 10.1016/S0092-8674(01)00638-9 [DOI] [PubMed] [Google Scholar]
- [43].Honma M, Benitah SA, Watt FM. Role of LIM kinases in normal and psoriatic human epidermis. Mol Biol Cell 2006; 17(4):1888-96; PMID:16467374; https://doi.org/ 10.1091/mbc.E05-12-1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Tran Van Nhieu G, Kai Liu B, Zhang J, Pierre F, Prigent S, Sansonetti P, Erneux C, Kuk Kim J, Suh PG, Dupont G, et al.. Actin-based confinement of calcium responses during Shigella invasion. Nat Commun 2013; 4:1567; PMID:23463010; https://doi.org/ 10.1038/ncomms2561 [DOI] [PubMed] [Google Scholar]
- [45].Dautin N. Serine protease autotransporters of enterobacteriaceae (SPATEs): biogenesis and function. Toxins (Basel) 2010; 2(6):1179-206; PMID:22069633; https://doi.org/ 10.3390/toxins2061179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Junker M, Besingi RN, Clark PL. Vectorial transport and folding of an autotransporter virulence protein during outer membrane secretion. Mol Microbiol 2009; 71(5):1323-32; PMID:19170888; https://doi.org/ 10.1111/j.1365-2958.2009.06607.x [DOI] [PubMed] [Google Scholar]
- [47].Renn JP, Junker M, Besingi RN, Braselmann E, Clark PL. ATP-independent control of autotransporter virulence protein transport via the folding properties of the secreted protein. Chem Biol 2012; 19(2):287-96; PMID:22209629; https://doi.org/ 10.1016/j.chembiol.2011.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Orth D, Ehrlenbach S, Brockmeyer J, Khan AB, Huber G, Karch H, Sarg B, Lindner H, Würzner R. EspP, a serine protease of enterohemorrhagic Escherichia coli, impairs complement activation by cleaving complement factors C3/C3b and C5. Infect Immun 2010; 78(10):4294-301; PMID:20643852; https://doi.org/ 10.1128/IAI.00488-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Otto BR, Sijbrandi R, Luirink J, Oudega B, Heddle JG, Mizutani K, Park SY, Tame JR. Crystal structure of hemoglobin protease, a heme binding autotransporter protein from pathogenic Escherichia coli. J Biol Chem 2005; 280(17):17339-45; PMID:15728184; https://doi.org/ 10.1074/jbc.M412885200 [DOI] [PubMed] [Google Scholar]
- [50].Tomiyoshi G, Horita Y, Nishita M, Ohashi K, Mizuno K. Caspase-mediated cleavage and activation of LIM-kinase 1 and its role in apoptotic membrane blebbing. Genes Cells 2004; 9(6):591-600; PMID:15189451; https://doi.org/ 10.1111/j.1356-9597.2004.00745.x [DOI] [PubMed] [Google Scholar]
- [51].Barone E, Mosser S, Fraering PC. Inactivation of brain Cofilin-1 by age, Alzheimer's disease and gamma-secretase. Biochim Biophys Acta 2014; 1842(12 Pt A):2500-9; PMID:25315299; https://doi.org/ 10.1016/j.bbadis.2014.10.004 [DOI] [PubMed] [Google Scholar]
- [52].Pandey D, Goyal P, Bamburg JR, Siess W. Regulation of LIM-kinase 1 and cofilin in thrombin-stimulated platelets. Blood 2006; 107(2):575-83; PMID:16219803; https://doi.org/ 10.1182/blood-2004-11-4377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Borensztajn K, Peppelenbosch MP, Spek CA. Coagulation Factor Xa inhibits cancer cell migration via LIMK1-mediated cofilin inactivation. Thromb Res 2010; 125(6):e323-8; PMID:20347121; https://doi.org/ 10.1016/j.thromres.2010.02.018 [DOI] [PubMed] [Google Scholar]
- [54].Leonard A, Marando C, Rahman A, Fazal F. Thrombin selectively engages LIM kinase 1 and slingshot-1L phosphatase to regulate NF-kappaB activation and endothelial cell inflammation. Am J Physiol Lung Cell Mol Physiol 2013; 305(9):L651-64; PMID:24039253; https://doi.org/ 10.1152/ajplung.00071.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Vingadassalom D, Campellone KG, Brady MJ, Skehan B, Battle SE, Robbins D, Kapoor A, Hecht G, Snapper SB, Leong JM. Enterohemorrhagic E. coli requires N-WASP for efficient type III translocation but not for EspFU-mediated actin pedestal formation. PLoS Pathog 2010; 6(8):e1001056; PMID:20808845; https://doi.org/ 10.1371/journal.ppat.1001056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kohler H, Rodrigues SP, McCormick BA. Shigella flexneri Interactions with the Basolateral Membrane Domain of Polarized Model Intestinal Epithelium: Role of Lipopolysaccharide in Cell Invasion and in Activation of the Mitogen-Activated Protein Kinase ERK. Infect Immun 2002; 70(3):1150-8; PMID:11854195; https://doi.org/ 10.1128/IAI.70.3.1150-1158.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Madara JL, Colgan SP, Nusrat C, Delp C, Parkos CA. A simple approach to measurement of electrical parameters of cultured epithelial monolayers: use in assessing neutrophil epithelial interactions. Journal of Tissue Culture Methods 1992; 14:209-13; https://doi.org/ 10.1007/BF01409013 [DOI] [Google Scholar]
- [58].McCormick BA, Fernandez MI, Siber AM, Maurelli AT. Inhibition of Shigella flexneri-induced transepithelial migration of polymorphonuclear leucocytes by cadaverine. Cell Microbiol 1999; 1(2):143-55; PMID:11207548; https://doi.org/ 10.1046/j.1462-5822.1999.00014.x [DOI] [PubMed] [Google Scholar]
- [59].Gorrec F. The MORPHEUS protein crystallization screen. J Appl Crystallogr 2009; 42(Pt 6):1035-42; PMID:22477774; https://doi.org/ 10.1107/S0021889809042022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Birtley JR, Curry S. Crystallization of foot-and-mouth disease virus 3C protease: surface mutagenesis and a novel crystal-optimization strategy. Acta Crystallogr D Biol Crystallogr 2005; 61(Pt 5):646-50; PMID:15858279; https://doi.org/ 10.1107/S0907444905007924 [DOI] [PubMed] [Google Scholar]
- [61].Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al.. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 2011; 67(Pt 4):235-42; PMID:21460441; https://doi.org/ 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Bunkoczi G, Read RJ. Improvement of molecular-replacement models with Sculptor. Acta Crystallogr D Biol Crystallogr 2011; 67(Pt 4):303-12; PMID:21460448; https://doi.org/ 10.1107/S0907444910051218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr 2007; 40(Pt 4):658-74; PMID:19461840; https://doi.org/ 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr 2007; 63(Pt 1):32-41; PMID:17164524; https://doi.org/ 10.1107/S0907444906045975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr 2000; 56(Pt 8):965-72; PMID:10944333; https://doi.org/ 10.1107/S0907444900005072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung LW, Read RJ, Adams PD. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr D Biol Crystallogr 2008; 64(Pt 1):61-9; PMID:18094468; https://doi.org/ 10.1107/S090744490705024X [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 2004; 60(Pt 12 Pt 1):2126-32; PMID:15572765; https://doi.org/ 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- [68].Afonine PV, Mustyakimov M, Grosse-Kunstleve RW, Moriarty NW, Langan P, Adams PD. Joint X-ray and neutron refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 2010; 66(Pt 11):1153-63; PMID:21041930; https://doi.org/ 10.1107/S0907444910026582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res 2010; 38(Web Server issue):W545-9; PMID:20457744; https://doi.org/ 10.1093/nar/gkq366 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.