

Abstract
Synthetic lethality is an approach to study selective cell killing based on genotype. Previous work in our laboratory has shown that loss of RAD52 is synthetically lethal with BRCA2 deficiency, while exhibiting no impact on cell growth and viability in BRCA2-proficient cells. We now show that this same synthetically lethal relationship is evident in cells with deficiencies in BRCA1 or PALB2, which implicates BRCA1, PALB2 and BRCA2 in an epistatic relationship with one another. When RAD52 was depleted in BRCA1- or PALB2-deficient cells, a severe reduction in plating efficiency was observed, with many abortive attempts at cell division apparent in the double-depleted background. In contrast, when RAD52 was depleted in a BRCA1- or PALB2-wildtype background, a negligible decrease in colony survival was observed. The frequency of ionizing radiation-induced RAD51 foci formation and double-strand break-induced homologous recombination (HR) was decreased by 3- and 10-fold, respectively, when RAD52 was knocked down in BRCA1- or PALB2-depleted cells, with minimal effect in BRCA1- or PALB2-proficient cells. RAD52 function was independent of BRCA1 status, as evidenced by the lack of any defect in RAD52 foci formation in BRCA1-depleted cells. Collectively, these findings suggest that RAD52 is an alternative repair pathway of RAD51-mediated HR, and a target for therapy in cells deficient in the BRCA1– PALB2–BRCA2 repair pathway.
Keywords: BRCA1, PALB2, BRCA2, RAD52, synthetic lethality, RAD51
INTRODUCTION
Synthetic lethality as an approach to cancer therapy is an exciting new area of research that promises cancer-specific targeted therapy.1,2 Synthetic lethality arises when the simultaneous inactivation of two (or more) genes leads to cell death, whereas inactivation of only one of these genes does not affect viability. With this guiding principle, cells with a cancer-related mutation or loss of a single gene can be evaluated for a second gene target that would render the cancer cells non-viable, thereby achieving tumor-specific kill while sparing normal cells. In tumor cells, a single gene alteration can exist as a genetic null or genetic mutant, and each form of genetic alteration may not exhibit the same synthetically lethal relationship with the second target gene; therefore both forms of genetic alteration in tumors should be explored for greatest therapeutic potential.1 Depletion of candidate gene products by small interfering RNA (siRNA) has proved to be a powerful tool for investigating gene inactivation, but there is always the question of how well the depletion results in inactivation. A positive result (lethality from two gene alterations but not one) is clear, but a negative result does not exclude a synthetically lethal relationship.
A single gene alteration on its own may confer a distinct level of lethality or sickness, but in combination with a second gene depletion it may lead to a significantly greater lethal effect (that is, synthetically lethal), affording therapeutic utility. This strategy has been exploited in targeting BRCA deficiencies with promising clinical results.3 BRCA1- and BRCA2-deficient tumor cells are impaired in homologous recombination (HR) and have adapted themselves to survive without these key proteins. They are, therefore, an excellent example of a vulnerability to synthetically lethal approaches that target DNA repair pathways, such as poly (ADP-ribose) polymerase inhibitors that block single-strand break repair.4,5
Recent discoveries of alternative mediators of RAD51 function offer new potential synthetically lethal targets in BRCA-deficient tumors. RAD51 is a HR protein that forms a helical nucleoprotein filament involved in homology search and strand pairing during DNA double-strand break (DSB) repair,6 and its loss in proliferating cells results in cell lethality.7 In budding yeast, RAD52 mediates the RAD51 filament formation necessary for successful HR.8 However, in mammalian cells, BRCA2 is the predominant mediator of RAD51 filament formation.9,10 Therefore, mammalian cells with deficiencies in BRCA2 will only be viable if an alternate pathway for RAD51 recruitment to damage sites exists.
Recently, we showed that human RAD52 was the mediator of this alternate RAD51 pathway.11 RAD52 interacts with RAD51 to stimulate strand invasion and promotes homologous pairing.12 In BRCA2-defective cells, RAD52 provides essential HR function, which is observed by cell viability, gene conversion assays and RAD51 nuclear foci formation.11 Furthermore, RAD52 depletion in BRCA2-deficient cells resulted in a severe proliferation defect and extensive chromosomal fragility, thus demonstrating a synthetically lethal relationship between BRCA2 and RAD52.11
Besides operating as a mediator of RAD51 in BRCA2-deficient cells, RAD52 mediates the function of single-strand annealing (SSA), which repairs DSBs between two repetitive DNA sequences.13 RAD52 binds to single-stranded DNA and somehow aligns the repeat sequences, thereby allowing the complementary sequences to anneal. Interestingly, RAD52-mediated SSA activity is increased with loss of BRCA2, indicating an upregulation of RAD52-SSA with deficiencies in BRCA2-RAD51-directed HR repair, presumably through shunting of DSBs that are unable to undergo RAD51-HR towards the SSA repair.13,14 However, with loss of BRCA1, the opposite effect is observed. SSA is decreased in cells with BRCA1 deficiencies, demonstrating a regulatory role of BRCA1 on the RAD52–SSA pathway.13 This observation, along with the other known roles of the multi-functional BRCA1 protein in cellcycle checkpoint activity, E3 ubiquitin ligase activity and BRCA2– RAD51-mediated HR, posited the potential role of BRCA1 in directing RAD52–RAD51 HR.15 BRCA1 may directly regulate RAD52 function in RAD51-directed repair, whereas it is known that BRCA2 is entirely independent of RAD52.11
Another HR protein to consider is PALB2 (partner and localizer of BRCA2). PALB2 is immediately downstream of BRCA1, physically and functionally links BRCA1 and BRCA2 in the HR pathway,16-19 and is an established breast cancer susceptibility gene.20 Autosomal dominant mutations in PALB2 predispose to pancreatic, prostate and other cancers.21,22 Being upstream of BRCA2, it is unknown if PALB2 is involved in recruiting and regulating RAD52 in RAD51-mediated HR. Although the specific genetic interactions of BRCA1 and PALB2 with RAD52 are unclear, the working hypothesis was that BRCA1 and PALB2 are independent of RAD52, which would suggest a synthetically lethal relationship. However, it is also possible that RAD52–RAD51-driven HR could be dependent on BRCA1 or PALB2, and thus we sought to investigate the relationship between these proteins.
RESULTS
RAD52 depletion is synthetically lethal with deficiencies in BRCA1 or PALB2
Plating efficiency experiments with the MCF7 cell line were performed to measure the lethal effect of the loss of RAD52 in BRCA1- and PALB2-depleted cells. We observed that colony survival after acute depletion of BRCA1 or RAD52 alone showed moderate clonogenic defects (Figure 1a, left). However, significant survival defects were observed when both proteins were depleted simultaneously (Figure 1a, left). Furthermore, abortive attempts at cell division were evident with numerous colonies of < 50 cells, as shown in the representative photomicrograph (Figure 1a, right), which is most apparent with the concurrent depletion of BRCA1 and RAD52. PALB2 and RAD52, when simultaneously depleted, exhibited similar significant reduction in plating efficiency and increased number of abortive colonies (Figure 1b). Of note, BRCA1 or PALB2 deficiencies alone are not essential in the context of a transformed cell, which is noted through the adequate plating efficiency of these cells. These results were confirmed in U2OS cells subjected to similar conditions, demonstrating that lethality is maximal in the cells with double depletion of BRCA1 or PALB2 with RAD52 (Figure 1c).
Figure 1.
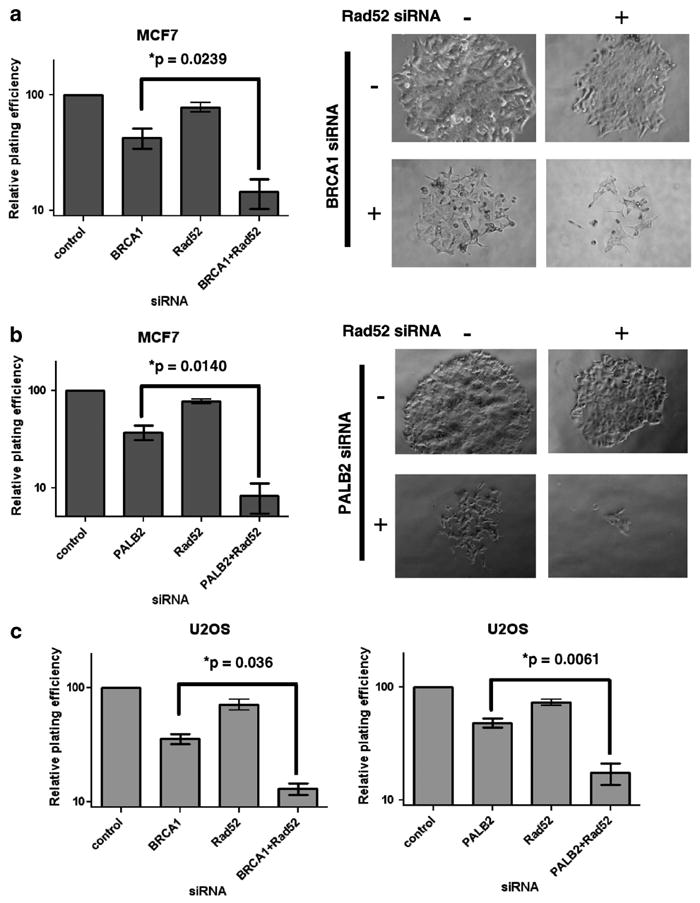
RAD52 exhibits synthetic lethality with BRCA1 and PALB2. (a) (Left) Plating efficiency (clonogenic survival) of MCF7 cells that were cultured as previously described in Feng et al.,11 and were depleted of BRCA1, RAD52 or both proteins by commercially available siRNAs (siBRCA1: siGENOME SMARTpool, Dharmacon, Lafayette, CO, USA; siRAD52: ID no. 142431, Ambion, Austin, TX, USA; control: negative control siRNA cat. no. 1027310, Qiagen, Valencia, CA, USA), and transfected into cells by Nucleofector (Lonza, Basel, Switzerland) according to the manufacturers’ recommendations. Plating efficiency was measured in MCF7 cells by seeding in triplicate 100, 400, 800, 1600, 3200 and 6400 cells into six-well plates. Cells were plated 72 h after transfection with the labeled siRNAs. Colonies (> 50 cells) were counted by fixing and staining with crystal violet (0.05%) 14–21 days after seeding. The results shown are from three independent experiments with error bars representing s.e.m. Differences between cells transfected with BRCA1 siRNA alone compared with both BRCA1 and RAD52 siRNA are significant (P=0.0239), as determined by an unpaired t-test. (Right) Photomicrographs are of representative colonies 14 days after plating. (b) (Left) Plating efficiency of MCF7 cells following siRNA depletion of PALB2, RAD52 or both proteins as in (a) (siPALB2: siGENOME SMARTpool, Dharmacon). Differences between cells transfected with PALB2 siRNA alone compared with both PALB2 and RAD52 siRNA are significant (P=0.0140), as determined by an unpaired t-test. (Right) Photomicrographs are of representative colonies 14 days after plating (as in a). (c) Plating efficiency of U2OS cells that were cultured in the same conditions, as previously described for MCF7 cells. U2OS cells were transfected with the labeled siRNA oligos (as in a, b) and plated 72 h later. Colonies were counted by fixing and staining, as described above. Differences between cells transfected with BRCA1 siRNA alone compared with both BRCA1 and RAD52 siRNA are significant (P=0.036), as determined by an unpaired t-test, in addition to cells transfected with PALB2 siRNA alone compared with both PALB2 and RAD52 siRNA (P=0.0061).
RAD52 depletion in a BRCA1-deficient background produces a severe defect in RAD51 focus formation
HR function appears to be required for cell survival as RAD51-deficient cells are lethal.7 Unrepaired DSBs arising in S-phase in the absence of HR are potentially lethal due to deleterious genomic rearrangements. Given the loss of viability after depletion of RAD52 in a BRCA1-deficient background, we hypothesized this lethal effect was due to defective RAD51-directed HR. The RAD51 protein can be detected as subnuclear foci, which represent their recruitment and participation in the DNA repair process by HR. The ability to form RAD51 nuclear foci correlates with a cell’s ability to undergo HR, and therefore is a useful proxy measure of HR.23,24
Both BRCA1 and RAD52 are known to associate with RAD51 and have a key role in the repair of DSBs by HR.11,24 We and others have previously shown the effect of BRCA1 on RAD51 foci formation after exposure to ionizing radiation (IR).23 In the BRCA1-mutant human breast cancer cell line HCC1937, the formation of IR-induced RAD51 foci is defective relative to a BRCA1-complemented cell and compared with the MCF7 cell line with wild-type BRCA1.23 To investigate the effect of RAD52 depletion in a BRCA1 proficient and deficient setting, HCC1937 cells were stably reconstituted with BRCA1-wildtype or empty vector (Figure 2a, left).23 In HCC1937 cells depleted of RAD52 (Figure 2a, left), those complemented with BRCA1-wildtype showed rescue of RAD51 foci formation after IR damage (Figure 2a, right) compared with HCC1937 cells containing the empty vector, which showed a severe defect in RAD51 foci. These results suggest that in the absence of BRCA1, RAD52 is a key mediator of RAD51 foci formation, whereas in a BRCA1-proficient context RAD52 contributes minimally to RAD51 function.
Figure 2.
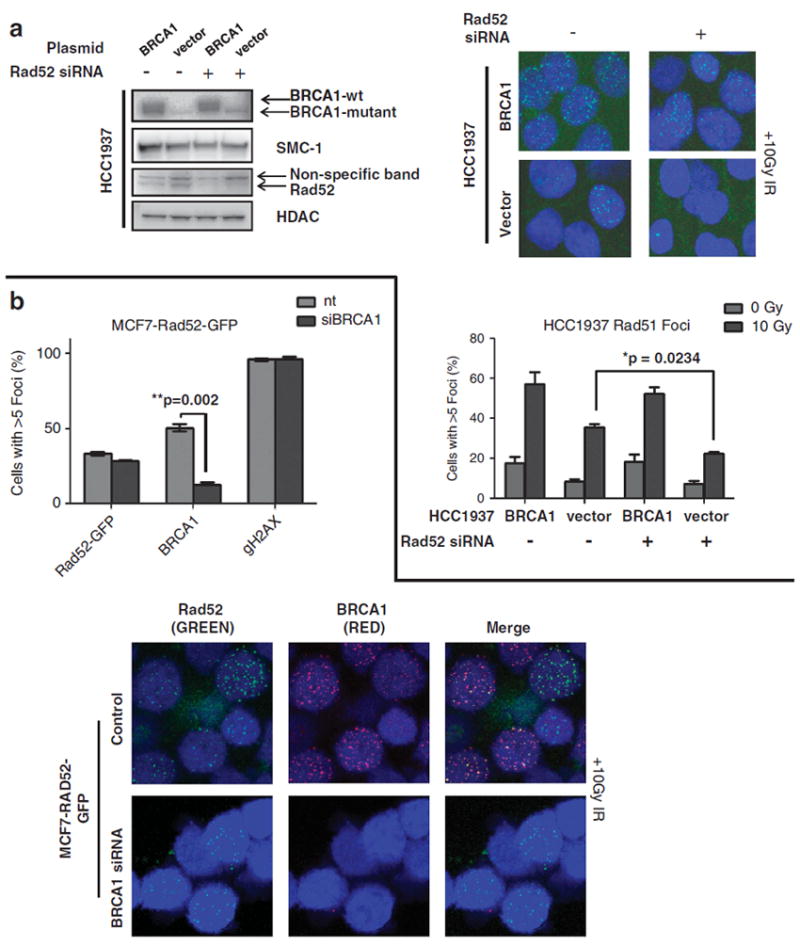
RAD51 foci formation in BRCA1-deficient cells is dependent on RAD52, but RAD52 foci formation is not dependent on BRCA1. (a) HCC1937 cells, complemented with a pcDNA3 expression vector containing wildtype BRCA1 or empty control, as previously reported,35 were transfected with the indicated siRNAs and immunoblotted with the indicated antibodies (anti-BRCA1: OP92, Calbiochem, La Jolla, CA, USA; anti-Rad52: SC-8350, Santa Cruz, Santa Cruz, CA, USA; anti-SMC1: A300-055A, Bethyl, Montgomery, TX, USA; anti-HDAC: #06-720, Upstate/Millipore, Billerica, MA, USA). Confocal images of IR-induced RAD51 foci in HCC1937 cells that were stably transfected with wildtype BRCA1 or a vector control.35 Cells were then transiently transfected with RAD52 siRNA or a control siRNA, then seeded in eight-chamber tissue culture slides (Fisher, Pittsburgh, PA, USA) and incubated overnight. Cells were fixed in 4% paraformaldehyde at room temperature for 15 min, followed by a block and permeabilization step with 10% BGS and 0.5% Triton-X for 1 h. Commercially available antibodies against RAD51 (Ab-1, Santa Cruz) were used for staining, as previously described.11 Images were obtained using a Carl Zeiss confocal laser scanning microscope (Carl Zeiss Microscopy, LLC, Thornwood, NY, USA) and processed using ImageJ and Adobe Photoshop software. For foci quantification 200 nuclei were counted, with cells forming > 5 foci scored as positive. The percentage of cells with RAD51 foci is shown (mean and s.e. from three independent experiments). Differences between HCC1937 cells stably transfected with an empty vector and then control siRNA compared with empty vector cells transfected with RAD52 siRNA are significant (P=0.0234), as determined by an unpaired t-test. (b) MCF7 cells were transfected with GFP-tagged RAD52 and a pool of cells were stably selected with G418 (Mediatech, Inc., Manassus, VA, USA) at 1 mg/ml concentration. These MCF7-RAD52-GFP cells were transfected with control or BRCA1 siRNA. After 10 Gy of IR, cells were fixed and stained with commercially available antibodies against BRCA1 (SC-6954, Santa Cruz) and phosphorylated Ser-139 H2AX (05-636, Millipore, Billerica, MA, USA). Confocal images of RAD52-GFP, BRCA1 and gamma-H2AX (gH2AX) foci were captured and scored. The percentage of cells with RAD52 and BRCA1 foci is shown (mean and s.e. from three independent experiments). Differences in BRCA1 foci between control and BRCA1 siRNAtransfected cells were significant (P=0.002), as determined by an upaired t-test.
To address the potential confounding factor associated with the growth adaptation of cells to BRCA1 deficiency, MCF7 cells were acutely depleted of BRCA1 and/or RAD52 by siRNA (Supplementary Figure 1A), and then subjected to the identical RAD51 foci formation assay. Depletion of BRCA1 alone resulted in a moderate suppression of IR-induced RAD51 foci (Supplementary Figure 1B), and RAD52 depletion alone produced almost no effect if wild-type BRCA1 was retained. However, the simultaneous depletion of BRCA1 and RAD52 resulted in a severe defect in RAD51 foci after exposure to 10 Gy of IR (Supplementary Figure 1B). Taken together, these data show that RAD52 supports RAD51 function in a BRCA1-deficient background.
RAD52 function does not depend on BRCA1
Human RAD52 and RAD51 interact and synergistically enhance in vitro reactions of DNA recombination.25,26 The ability of these two proteins to colocalize in subnuclear foci offers in vivo evidence of their role in DNA repair.11 To understand the role of RAD52 and its relationship with BRCA1 in mediating RAD51-dependent HR, MCF7 cells were transfected with green fluorescent protein (GFP)-tagged RAD52 and a pool of MCF7-RAD52-GFP cells were stably generated. These MCF7-RAD52-GFP cells were then depleted of BRCA1 by siRNA and subsequently subjected to IR (10 Gy), where RAD52, BRCA1 and gamma-H2AX foci in these cells were quantified (Figure 2b). Thirty-four percent of the cells that were transfected with a control siRNA contained RAD52 foci. RAD52 foci were detected in a similar percentage of cells (29%) transfected with BRCA1 siRNA. This is in contrast to BRCA1 foci formation, where 51% of cells subjected to control siRNA contained BRCA1 foci, whereas a greater than threefold decrease in BRCA1 foci formation was observed in cells transfected with BRCA1 siRNA, with only 13% of these cells expressing BRCA1 subnuclear foci (Figure 2b). These data demonstrate that RAD52 function occurs independently of BRCA1 status in human cells.
RAD52 depletion impairs HR in BRCA1-deficient cells
To study how the severe defect in forming RAD51 foci in BRCA1- and RAD52-deficient cells affected functional HR activity, a gene conversion assay was performed with the pDR-GFP reporter.27 In this assay, successful HR restores GFP expression, quantifiable by flow cytometry as shown in Figure 3a.
Figure 3.
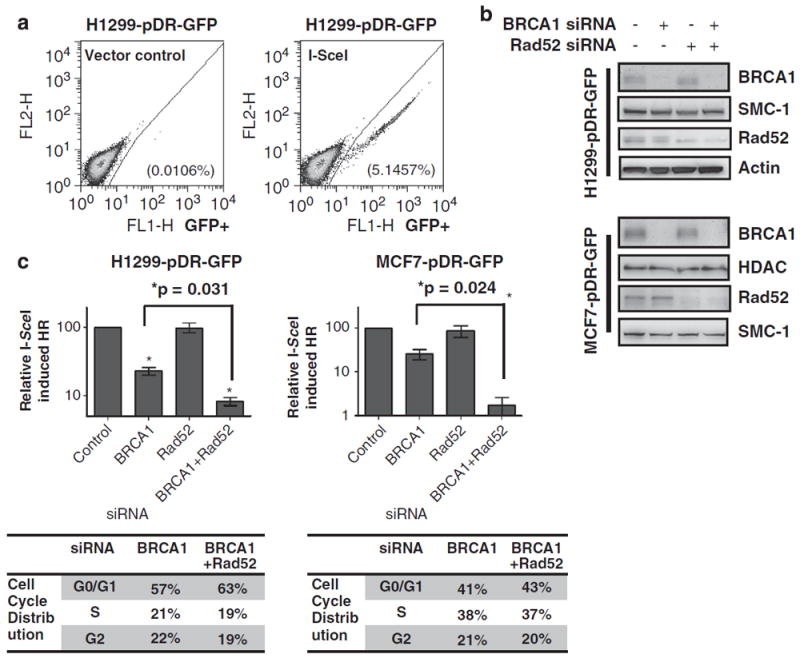
RAD52 is required for homology-directed gene conversion in BRCA1-depleted cells. (a) Flow cytometry of H1299 cells cultured as previously described,28 containing the recombination substrate, pDR-GFP,27 transfected with the I-SceI endonuclease plasmid or with vector alone by Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s recommendations and analyzed for the percentage of green fluorescent cells. At 24 h after transfection with respective siRNAs, cells with the pDR-GFP construct stably integrated were transfected with pCMV-I-SceI to induce DSBs. Cells were collected 72 h later for analysis using a FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). (b) Immunoblots of H1299-pDR-GFP and MCF7-pDR-GFP cells that were transfected with the indicated combinations of siRNA. (c) The frequency of I-SceI break-induced recombination (HR) measured by flow cytometry in H1299-pDR-GFP and MCF7-pDR-GFP transfected with the labeled siRNAs. The bars represent the results of three or more independent experiments; error bars show s.e.m. Differences between cells transfected with BRCA1 siRNA alone compared with both BRCA1 and RAD52 siRNA are significant (P=0.031, P=0.024 for H1299-pDR-GFP and MCF7-pDR-GFP cells, respectively), as determined by an unpaired t-test. A summary of the cell cycle distribution shows similar rates between the labeled siRNA conditions; see Supplementary Figure 2 for detailed methods and data.
H1299 and MCF7 cell lines with the pDR-GFP substrate chromosomally integrated (referred to as H1299-pDR-GFP and MCF7-pDR-GFP) have been previously used to measure HR.11,28 When RAD52 was knocked down in BRCA1-depleted cells (Figure 3b), a 10- and 30-fold reduction in I-SceI-induced recombination was observed in H1299-pDR-GFP and MCF7-pDRGFP cells, respectively (Figure 3c). However, in BRCA1-proficient cells RAD52 knockdown alone had no appreciable effect on the cumulative HR frequency compared with control (Figure 3c). Cell cycle profiling was performed to assess whether RAD52 depletion affects HR rates through S/G2 cell cycle phase shifts. Similar cell cycle distributions were observed between the depletion of BRCA1 alone compared with the double depletion of BRCA1 and RAD52, thereby demonstrating that RAD52 directly affects HR frequency and not through cell cycle re-distribution (Figure 3c, Supplementary Figure 2A). A second RAD52 siRNA target sequence was utilized and a similar phenotype was observed, thereby minimizing the possibility of off-target siRNA effects (Supplementary Figure 3A). Overall, these data were consistent with the RAD51 foci data, where RAD52 depletion only had a significant effect in BRCA1-deficient cells.
RAD52 stimulates HR in the absence of PALB2
PALB2 provides a link between BRCA1 and BRCA2 in the DNA repair pathway of HR, and is important for successful HR that is mediated by RAD51.16,18,19 Given the observation that RAD52 is an alternative mediator of RAD51-dependent HR in the background of BRCA1 or BRCA2 loss, PALB2 was another logical protein to evaluate in testing the hypothesis that the entire BRCA1–PALB2–BRCA2 pathway is dependent on RAD52 as a backup pathway. Similar to the previous experiment with BRCA1, the formation of RAD51 foci was examined in MCF7 cells after transfection of PALB2 siRNA, RAD52 siRNA or both. After treatment with 10 Gy of IR, PALB2 depletion produced an approximately twofold reduction in cells forming RAD51 foci, from 40 to 22% (Figure 4a). RAD52 depletion alone had almost no effect on RAD51 foci, whereas after double depletion of PALB2 and RAD52 only 6% of cells contained RAD51 foci (Figure 4a), thus revealing a severe deficiency in RAD51 recruitment.
Figure 4.
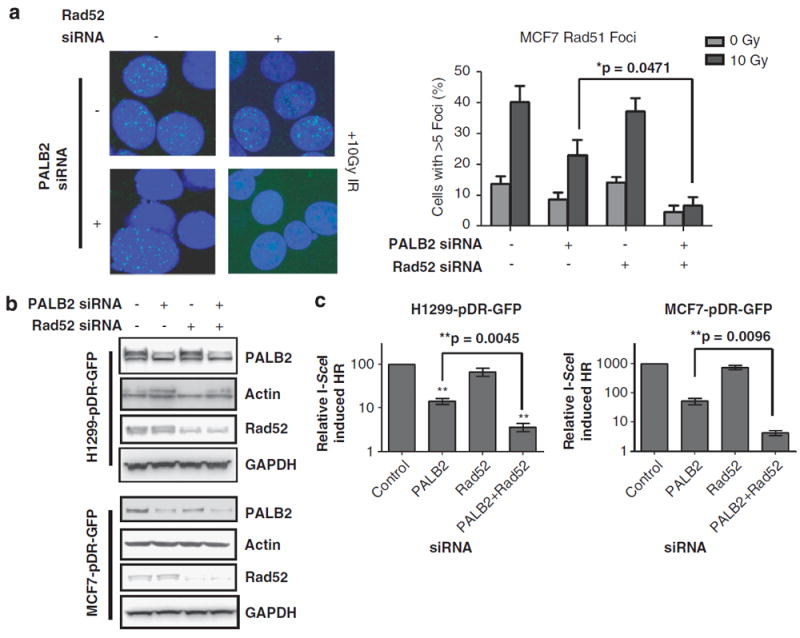
RAD52 is required for RAD51-mediated HR in PALB2-depleted cells. (a) Confocal images of IR-induced RAD51 foci in MCF7 cells following siRNA depletion of PALB2, RAD52 or both proteins. The percentage of cells with RAD51 foci is shown (mean and s.e.m from three independent experiments). Differences between cells transfected with PALB2 siRNA alone compared with both PALB2 and RAD52 siRNA are significant (P=0.0471), as determined by an unpaired t-test. (b) Immunoblots of H1299-pDR-GFP and MCF7-pDR-GFP cells transfected with the labeled combinations of control, PALB2 and RAD52 siRNA. (c) The frequency of I-SceI break-induced recombination (HR) measured by flow cytometry in H1299-pDR-GFP and MCF7-pDR-GFP cells transfected with the indicated siRNAs. The bars represent the results of three or more independent experiments; error bars show s.e.m. Differences between cells transfected with PALB2 siRNA alone compared with both PALB2 and RAD52 siRNA are significant (P=0.0045, P=0.0096 for H1299-pDR-GFP and MCF7-pDR-GFP cells, respectively), as determined by an unpaired t-test.
Supporting these results, MCF7-pDR-GFP and H1299-pDR-GFP cells that were depleted of both PALB2 and RAD52 (Figure 4b) exhibited the most severe reduction in I-SceI-induced HR (Figure 4c). However, similar to BRCA1-proficient cells, in PALB2-proficient cells RAD52 knockdown alone had no appreciable effect on the frequency of I-SceI-induced homology-mediated repair compared with control (Figure 4c). In addition, RAD52 depletion alone compared with PALB2 and RAD52 double depletion did not affect S/G2 cell cycle distribution, suggesting that RAD52 is directly affecting the rates of HR (Supplementary Figure 2B). A second RAD52 siRNA target sequence in combination with PALB2 depletion demonstrated a similar phenotype and minimized the possibility of off-target siRNA effects (Supplementary Figure 3B). Taken together, these results showed that in the absence of PALB2 RAD52 is capable of mediating RAD51-dependent HR.
DISCUSSION
The studies presented here demonstrate that RAD52 is synthetically lethal with BRCA1 and PALB2, specifically through the RAD51-HR pathway. RAD52 maintains RAD51-dependent HR function in response to DSB in the absence of BRCA1 or PALB2. RAD51 foci formation, DSB-induced direct repeat gene conversion and plating efficiency experiments all show the important role of RAD52 in mediating RAD51-HR for cells deficient in BRCA1 or PALB2. In addition, RAD52 function, as assessed through RAD52 foci formation, is independent of BRCA1. We know that BRCA1 exhibits a multi-functional role in the DNA damage response, including the initiation of single-strand resection, which is needed for SSA repair. However, BRCA1 does not control RAD52 function in RAD51-directed repair. The implication is that the engagement of RAD52 in HR to prevent cell death is independent of the BRCA1- dependent resection at DSBs. There are mechanisms of resection that do not require the BRCA1-CtIP connection, which are the subject of active investigations. Stalled replication intermediates, which are thought to be the potentially toxic intermediates that are rescued by HR, may have already generated single-stranded DNA for replication protein A binding, which is sufficient to engage the RAD52 mechanism of HR. Overall, these studies demonstrate that the function of RAD52 in RAD51-HR repair is independent of the entire BRCA1–PALB2–BRCA2 pathway of HR.
The mechanistic role of human RAD52 in RAD51-dependent repair of DSBs is still under investigation. Yeast Rad52 mediates Rad51-dependent strand exchange events25 and may perform the annealing step in second end capture,29 yet these biochemical functions of RAD52 were not readily evident in mammalian cells. Our work now shows that human RAD52 has a vital role in RAD51 filament formation, inferred by the formation of RAD51 foci, especially in cells deficient in the BRCA1–PALB2–BRCA2 pathway. This novel observation goes beyond the previously described role of human RAD52 in SSA.30 RAD51 has been implicated in the repair of multiple different lesion types, including DSBs, oneended DSBs generated by replication fork stalls and postreplication daughter strand gap repair (Supplementary Figure 4). Our work confirms that repair of these lesions relies upon the BRCA1–PALB2–BRCA2 pathway, with RAD52 functioning in an alternative pathway that mediates RAD51-directed repair when deficiencies exist in BRCA1, PALB2 or BRCA2 (Supplementary Figure 4), although a cooperative role for RAD52 and BRCA2 in mediating RAD51 function cannot be definitively excluded.31,32
RAD52 is necessary for colony growth of BRCA1- or PALB2-deficient cells, thereby suggesting that RAD52 depletion could halt the proliferation of BRCA1- or PALB2-defective cancer cells while sparing normal cells. The significant loss of HR in the double depleted cells suggests that this synthetically lethal effect is due to the loss of RAD51 HR function mediated through the BRCA1– PALB2–BRCA2 complex, and not because of any other known function of BRCA1 (for example, SSA regulation that also involves RAD52, E3 ubiquitin ligase function, transcriptional activity, etc.). No known mutations or inactivation of RAD52 in human tumors has been documented, making deliberate inactivation of RAD52 a potential targeted therapeutic approach that exploits this relationship in BRCA1 and PALB2 defective tumors.
An interesting area of future inquiry would be to examine the ‘BRCAness’33 of sporadic cancers and assess whether RAD52 silencing would confer tumor-specific kill, as the BRCAness profile of epithelial ovarian cancers has been correlated to sensitivity to poly (ADP-ribose) polymerase inhibitors and platinum-based chemotherapy.33 Furthermore, functional assays of RAD51 function using ex-vivo irradiation of clinical breast cancer samples34 have revealed a pool of tumors with sporadic BRCA deficiencies that can enlarge the patient population that would benefit from the therapeutic depletion of RAD52 (among other synthetically lethal strategies). In summary, we have established that RAD52 depletion can selectively target tumor cells deficient in any component of the BRCA1–PALB2–BRCA2 pathway, and is one additional method to personalize and improve cancer therapy in this population of tumors.
Supplementary Material
Acknowledgments
This work was supported in part by the Howard Hughes Medical Institute Medical Research Training Fellowship, the Radiological Society of North America Research Medical Student Grant (BHL) and by grants from the National Cancer Institute and the Susan G. Komen for the Cure (SNP).
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Chan DA, Giaccia AJ. Harnessing synthetic lethal interactions in anticancer drug discovery. Nat Rev Drug Discov. 2011;10:351–364. doi: 10.1038/nrd3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iglehart JD, Silver DP. Synthetic Lethality — A New Direction in Cancer-Drug Development. N Engl J Med. 2009;361:189–191. doi: 10.1056/NEJMe0903044. [DOI] [PubMed] [Google Scholar]
- 3.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 4.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 5.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 6.Baumann P, West SC. Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem Sci. 1998;23:247–251. doi: 10.1016/s0968-0004(98)01232-8. [DOI] [PubMed] [Google Scholar]
- 7.Sonoda E, Sasaki MS, Buerstedde JM, Bezzubova O, Shinohara A, Ogawa H, et al. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J. 1998;17:598–608. doi: 10.1093/emboj/17.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shinohara A, Ogawa T. Stimulation by Rad52 of yeast Rad51-mediated recombination. Nature. 1998;391:404–407. doi: 10.1038/34943. [DOI] [PubMed] [Google Scholar]
- 9.Holloman WK. Unraveling the mechanism of BRCA2 in homologous recombination. Nat Struct Mol Biol. 2011;18:748–754. doi: 10.1038/nsmb.2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharan SK, Morimatsu M, Albrecht U, Lim DS, Regel E, Dinh C, et al. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature. 1997;386:804–810. doi: 10.1038/386804a0. [DOI] [PubMed] [Google Scholar]
- 11.Feng Z, Scott SP, Bussen W, Sharma GG, Guo G, Pandita TK, et al. From the Cover Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc Natl Acad Sci USA. 2011;108:686–691. doi: 10.1073/pnas.1010959107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McIlwraith MJ, Van Dyck E, Masson JY, Stasiak AZ, Stasiak A, West SC. Reconstitution of the strand invasion step of double-strand break repair using human Rad51 Rad52 and RPA proteins. J Mol Biol. 2000;304:151–164. doi: 10.1006/jmbi.2000.4180. [DOI] [PubMed] [Google Scholar]
- 13.Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol Cell Biol. 2004;24:9305–9316. doi: 10.1128/MCB.24.21.9305-9316.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PloS Genet. 2008;4:e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang WY, Twu CW, Chen HH, Jan JS, Jiang RS, Chao JY, et al. Plasma EBV DNA clearance rate as a novel prognostic marker for metastatic/recurrent nasopharyngeal carcinoma. Clin Cancer Res. 2010;16:1016–1024. doi: 10.1158/1078-0432.CCR-09-2796. [DOI] [PubMed] [Google Scholar]
- 16.Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci USA. 2009;106:7155–7160. doi: 10.1073/pnas.0811159106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell. 2006;22:719–729. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 18.Zhang F, Fan Q, Ren K, Andreassen PR. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol Cancer Res. 2009;7:1110–1118. doi: 10.1158/1541-7786.MCR-09-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, et al. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol. 2009;19:524–529. doi: 10.1016/j.cub.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39:165–167. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324:2–17. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Erkko H, Xia B, Nikkila J, Schleutker J, Syrjakoski K, Mannermaa A, et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature. 2007;446:316–319. doi: 10.1038/nature05609. [DOI] [PubMed] [Google Scholar]
- 23.Zhang J, Willers H, Feng Z, Ghosh JC, Kim S, Weaver DT, et al. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol Cell Biol. 2004;24:708–718. doi: 10.1128/MCB.24.2.708-718.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, et al. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997;88:265–275. doi: 10.1016/s0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- 25.New JH, Sugiyama T, Zaitseva E, Kowalczykowski SC. Rad52 protein stimulates DNA strand exchange by Rad51 and replication protein. Nature. 1998;391:407–410. doi: 10.1038/34950. [DOI] [PubMed] [Google Scholar]
- 26.Benson FE, Baumann P, West SC. Synergistic actions of Rad51 and Rad52 in recombination and DNA repair. Nature. 1998;391:401–404. doi: 10.1038/34937. [DOI] [PubMed] [Google Scholar]
- 27.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homologydirected repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi W, Ma Z, Willers H, Akhtar K, Scott SP, Zhang J, et al. Disassembly of MDC1 foci is controlled by ubiquitin-proteasome-dependent degradation. J Biol Chem. 2008;283:31608–31616. doi: 10.1074/jbc.M801082200. [DOI] [PubMed] [Google Scholar]
- 29.Sugiyama T, New JH, Kowalczykowski SC. DNA annealing by RAD52 protein is stimulated by specific interaction with the complex of replication protein A and single-stranded DNA. Proc Natl Acad Sci USA. 1998;95:6049–6054. doi: 10.1073/pnas.95.11.6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singleton MR, Wentzell LM, Liu Y, West SC, Wigley DB. Structure of the singlestrand annealing domain of human RAD52 protein. Proc Natl Acad Sci USA. 2002;99:13492–13497. doi: 10.1073/pnas.212449899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kojic M, Zhou Q, Fan J, Holloman WK. Mutational analysis of Brh2 reveals requirements for compensating mediator functions. Mol Microbiol. 2011;79:180–191. doi: 10.1111/j.1365-2958.2010.07440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qing Y, Yamazoe M, Hirota K, Dejsuphong D, Sakai W, Yamamoto KN, et al. The epistatic relationship between BRCA2 and the other RAD51 mediators in homologous recombination. PloS Genet. 2011;7:e1002148. doi: 10.1371/journal.pgen.1002148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Konstantinopoulos PA, Spentzos D, Karlan BY, Taniguchi T, Fountzilas E, Francoeur N, et al. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J Clin Oncol. 2010;28:3555–3561. doi: 10.1200/JCO.2009.27.5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Willers H, Taghian AG, Luo CM, Treszezamsky A, Sgroi DC, Powell SN. Utility of DNA repair protein foci for the detection of putative BRCA1 pathway defects in breast cancer biopsies. Mol Cancer Res. 2009;7:1304–1309. doi: 10.1158/1541-7786.MCR-09-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee JS, Collins KM, Brown AL, Lee CH, Chung JH. hCds1-mediated phosphorylation of BRCA1 regulates the DNA damage response. Nature. 2000;404:201–204. doi: 10.1038/35004614. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.