

Abstract
The calcineurin inhibitor cyclosporine A suppresses the immune system but promotes hypertension, vascular dysfunction, and renal damage. Cyclosporine A decreases regulatory T cells and this contributes to the development of hypertension. However, cyclosporine A’s effects on another important regulatory immune cell subset, myeloid-derived suppressor cells (MDSCs), is unknown. We hypothesized that augmenting MDSCs would ameliorate the cyclosporine A-induced hypertension and vascular and renal injury and dysfunction and that cyclosporine A reduces MDSCs in mice. Daily interleukin-33 treatment, which increased MDSC levels, completely prevented cyclosporine A-induced hypertension and vascular and renal toxicity. Adoptive transfer of MDSCs from control mice into cyclosporine A-treated mice after hypertension was established dose-dependently reduced blood pressure and vascular and glomerular injury. Cyclosporine A treatment of aortas and kidneys isolated from control mice for 24 hours decreased relaxation responses and increased inflammation, respectively, and these effects were prevented by the presence of MDSCs. MDSCs also prevented the cyclosporine A-induced increase in fibronectin in microvascular and glomerular endothelial cells. Lastly, cyclosporine A dose-dependently reduced the number of MDSCs by inhibiting calcineurin and preventing cell proliferation, as other direct calcineurin signaling pathway inhibitors had the same dose-dependent effect. These data suggest that augmenting MDSCs can reduce the cardiovascular and renal toxicity and hypertension caused by cyclosporine A.
Keywords: calcineurin inhibitor, hypertension, experimental, inflammation, lymphocytes, T cells
INTRODUCTION
Cyclosporine A (CsA) still remains one of the primary chronic immunosuppressive drugs for use in patients with focal segmental glomerulosclerosis, membranous nephropathy, psoriasis, rheumatoid arthritis, and other autoimmune diseases despite the known potential cardiovascular and renal side effects.1 Numerous studies have reported mechanisms by which CsA promotes hypertension and vascular and renal toxicity including increased sympathetic nerve activity and decreased vascular relaxation.2–7 Significantly safer immunosuppressive drugs are on the way, but until they are routinely used in the clinic, ways to reduce the detrimental cardiovascular and renal effects of CsA are needed.
One of the mechanisms by which CsA causes hypertension and cardiorenal toxicity is by altering immune cell subsets. We and others have reported that CsA decreases anti-inflammatory and tolerogenic regulatory T cells (Tregs), but increases interleukin (IL)-17-producing T cells, which contribute to the development of hypertension, as we recently reported that augmenting Tregs or neutralizing IL-17 normalized blood pressure and prevented vascular and renal injury in CsA-treated mice.8–16 However, it is unknown whether CsA affects another anti-inflammatory regulatory immune cell, myeloid-derived suppressor cells (MDSCs).
MDSCs consist of immature myeloid cells that are precursors for granulocytes, macrophages, and dendritic cells. Their immunosuppressive function is mediated largely by an upregulation of arginase and inducible nitric oxide synthase (iNOS or NOS2) which leads to arginine catabolism and reactive oxygen species production and a subsequent decrease in T-cell proliferation.17–23 MDSCs can secrete various anti-inflammatory cytokines including IL-4, IL-10, IL-13, and transforming growth factor, and are also reported to induce Tregs under certain conditions.24–26 In conjunction with Tregs, MDSCs are the major regulatory immune cells that help quench immunity and inflammation. Shah et al. reported recently that MDSC levels increase in various mouse models of hypertension (angiotensin II, NOS inhibition, and high salt), similar to what is known to happen in other states of inflammation.27 Further augmenting MDSC levels by adoptive transfer was able to lower blood pressure in all hypertensive models while depleting MDSCs exacerbated the inflammation and hypertension caused by angiotensin II. However, it is possible that inhibition of calcineurin by CsA reduces MDSC levels, similar to the effect it has on Tregs.
We utilized two strategies to determine whether augmenting MDSC levels would be effective in reducing CsA-induced organ injury and hypertension: 1) exogenous IL-33 which is well known to increase MDSC levels,28, 29 and 2) MDSC adoptive transfer. We also determined whether CsA directly affects MDSCs. We hypothesized that treatment with either IL-33 or isolated MDSCs would attenuate the development of vascular and renal injury and hypertension induced by CsA in mice, and that calcineurin inhibition decreases MDSCs and this loss of a source of anti-inflammatory cytokines contributes to the end organ injury and hypertension caused by CsA.
MATERIALS AND METHODS
Please see the online supplement for detailed Materials and Methods.
Animals and Treatments
Male C57Bl/6J mice (Jackson Laboratory) aged 10–18 weeks were utilized for the CsA treatment studies. Systolic blood pressures were measured by tail-cuff (IITC, Inc.) daily, at the same time each day, following 3 days of training. Following blood pressure measures, mice were injected ip with CSA (50 mg/kg/day; Alomone, Israel) or diluent (saline and DMSO, 0.2% final concentration) as described previously.9, 30–32 Some mice were also given daily i.p. injections of IL-33 (0.5 ug/mouse/day; eBioscience).28, 29
Other control and CSA-treated mice were injected i.p. with primary MDSCs isolated from male control mice on days 1, 4, and 7. Male control mice were euthanized and MDSCs were isolated from the spleens and bone marrow according to the manufacturer’s protocol (Miltenyi Biotech; #130-094-538).
Animals were anesthetized on day 8 with isoflurane and euthanized by cervical dislocation. All procedures were approved by the Institutional Animal Care and Use Committee in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Serum Creatinine
Blood was obtained by cardiac puncture under anesthesia. Serum creatinine was measured according to the manufacturer’s protocol (R&D Systems).
Vascular Reactivity
Vascular reactivity was measured in endothelium-intact aortas as described previously.31–34 Concentration-force curves were generated for the endothelium-dependent dilator acetylcholine (ACh) and the endothelium-independent dilator sodium nitroprusside following contraction with an EC70 concentration of phenylephrine (PE).
Immunoblotting
For protein analyses from whole aortic and kidney tissue, tissue lysates were processed and total protein concentration was determined as described previously.9 The blots were probed for Fibronectin (Abcam), E-selectin (Abcam), collagen (Millipore), calcineurin (Cell Signaling), and β-actin (Sigma) and were identified simultaneously (800 nm and 700 nm wavelengths, respectively) using near-infrared visualization (Odyssey System, LI-COR Biosciences). Densitometry was performed using the Odyssey software.
Histology and Morphometrical Analysis
Left kidney histological sections were prepared as described previously.9 Three blinded investigators quantitated glomerular injury according to a scale described previously.9 In brief, 15 glomeruli per tissue section were scored on a 0–4 scale, where 0=normal size, normal mesangium, and no congestion; 1=minimal hypertrophy, mesangial expansion, and/or congestion; 2=mild hypertrophy, mesangial expansion, and/or congestion; 3=moderate hypertrophy, mesangial expansion, and/or congestion; and 4=severe hypertrophy, mesangial expansion, and congestion. Mean glomerular injury index for each group was calculated by [(0 × N0)+(1 × N1)+(2 × N2)+(3 × N3)+(4 × N4)]/15, where N0-N4=number of glomeruli with scores of 0 to 4, respectively, as described previously.9, 35
Flow Cytometry
Spleens were harvested from the mice and single cell suspensions were generated as described previously.9 One million cells from the spleen were stained with anti-mouse CD11b clone M1/70 (BD Pharmingen), anti-mouse Ly-6G and Ly6C clone RB6-8C5 (BD Pharmingen), or isotype controls for 1 hour at 4°C in the dark. Other splenocytes were stained with anti-mouse CD3e (BD Pharmingen), anti-mouse CD4 (BD Pharmingen), anti-mouse CD25 clone PC61.5 (eBioscience), or isotype controls. These cells were then fixed and permeabilized with the BD CytoFix/CytoPerm™ Fixation/Permeabilization Kit (BD Pharmingen) followed by intracellular staining using an anti-mouse FoxP3 antibody (eBioscience). Flow cytometry was performed on a BD FACS Canto II and analyzed using FlowJo. CD11b+/Gr1+ MDSCs and CD3+/CD4+/CD25+/FoxP3+ lymphocytes were quantified and averaged as described previously.9, 32 Data are expressed as % of lymphocytes.
Isolated Aortas and Kidneys and Treatments
Aortas were excised from male control mice, cleaned of adipose tissue, and incubated with CsA (50 umol/L) for 24 hours in the absence and presence of 75,000 MDSCs isolated from male control mice. Segments 2 mm in length were cut from the thoracic portion, mounted in a myograph, and vascular reactivity studies were performed as above while the rest of the tissue was used for immunoblotting. Right kidneys isolated from the same control mice were treated with CsA (50 umol/L) for 24 hours in the absence and presence of 75,000 MDSCs and then immunoblotting for fibronectin and actin were performed as above.
Microvascular and Glomerular Endothelial Cells and Treatments
Murine microvascular and glomerular endothelial cells were obtained from Cell Biologics and cultured according the manufacturer’s guidelines. Endothelial cells were treated with CsA (50 umol/L) for 24 hours in the absence and presence of 75,000 MDSCs isolated from control mice. The cells were then processed for fibronectin immunoblotting as described above.
Isolated MDSCs and Treatments
MDSCs were isolated from male control mice spleens and bone marrow using the Miltenyi Biotech MDSC isolation kit. GM-CSF (10 ng/mL; R&D Systems) was added to the cells for survival. MDSCs were incubated for 24 hours with different concentrations of CsA (0, 10, 25, and 50 umol/L), the calcineurin autoinhibitory peptide (CAIP; 0, 10, 50 umol/L; Calbiochem), or the selective NFAT inhibitor VIVIT (0, 10, and 25 umol/L; Calbiochem) in the absence and presence of 75,000 MDSCs isolated from male control mice. The % of live MDSCs compared to 24-hour, DMSO vehicle-treated MDSCs were then determined by flow cytometry.
Statistical Analyses
Results are presented as mean + SEM. For multiple comparisons, either a 2-way ANOVA or a repeated measures 2-way ANOVA [main effects: group (Con or CsA) and treatment (Vehicle, IL-33, or MDSCs)] was used followed by the Student’s-Newman-Keuls post hoc test. The significance level was set at 0.05. All analyses were performed using SigmaStat 3.5 software.
RESULTS
IL-33 Prevents CsA-Induced Hypertension and Vascular and Renal Toxicity
Daily CsA treatment significantly increased systolic blood pressure in mice (Figure 1A) and decreased acetylcholine-induced aortic relaxation responses (Figure 1B) while having no effect on sodium nitroprusside-induced relaxation responses (Figure 1C). Aortas and kidneys from CsA-treated hypertensive mice had significantly increased levels of the injury marker fibronectin (Figure 1D). Glomeruli from CsA-treated mice exhibited significantly more injury evidenced by hypertrophy, mesangial expansion, hypercellularity, and congestion (Figure 1E). Serum creatinine levels were also increased 2-fold in CsA-treated mice (Con=0.7014 ± 0.0794, CsA=1.4174 ± 0.0207; CsA p<0.05 vs. Con). All of these effects were associated with a significant decrease in splenic MDSC levels (Figure 1F). The gating strategy for flow cytometry analysis of splenic MDSCs is presented in Figure S1, and representative dot plots for each group are presented in Figure S2.
Figure 1.
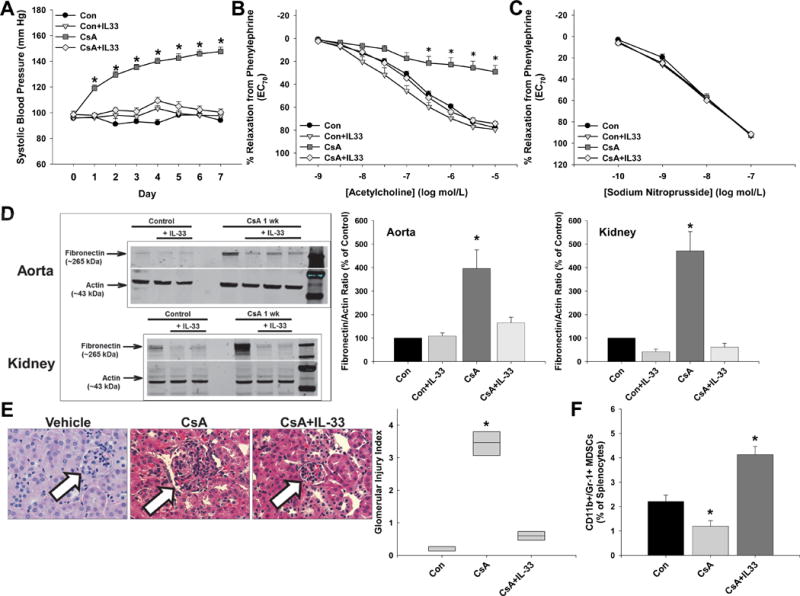
IL-33 increases MDSCs and prevents cyclosporine A-induced hypertension and vascular and renal injury. A, Systolic blood pressure was measured daily prior to ip injections of DMSO-saline vehicle (Control), cyclosporine A (CsA), and IL-33. Aortic relaxation responses to acetylcholine (B) and sodium nitroprusside (C) were measured after contraction with phenylephrine on day 8 following euthanization. D, Aortic and renal fibronectin levels were measured by immunoblotting and normalized to the loading control actin. E, Representative glomeruli and glomerular injury index score for each group. F, Splenic MDSC levels as determined by flow cytometry. Results are expressed as mean + SEM. *P<0.05 vs. Con and n=6–8 mice in each group. There were no significant group × treatment interactions as determined by 2-way ANOVA or repeated measures 2-way ANOVA.
To determine whether MDSC augmentation by IL-33 can prevent the development of CsA-induced organ injury and hypertension, daily IL-33 injections began and were administered at the same time as CsA injections. Daily IL-33 therapy prevented the CsA-induced development of hypertension (Figure 1A), endothelial dysfunction (Figure 1B and 1C), and vascular (Figure 1D) and renal injury (Figure 1D and 1E). IL-33 treatment also normalized serum creatinine levels (Con=0.7014 ± 0.0794, Con+IL-33=0.3263 ± 0.2376, CsA=1.4174 ± 0.0207, CsA+IL-33=0.7964 ± 0.0517; CsA p<0.05 vs. Con). These beneficial effects were associated with a significant increase in MDSC levels (Figures 1F and S2).
Adoptive Transfer of MDSCs Ameliorates CsA-Induced Hypertension and Vascular and Renal Injury
To determine whether MDSCs could reduce blood pressure after mice were already hypertensive (treatment study), we performed adoptive transfer of MDSCs on days 1, 4, and 7 following CsA injections. MDSC adoptive transfer dose-dependently decreased systolic blood pressure (Figure 2A). Injections of 1 million MDSCs partially lowered systolic blood pressure, while injections of 2 million MDSCs completely normalized systolic blood pressure. One million MDSCs partially improved aortic relaxation responses to acetylcholine in CsA-treated mice (data not shown), while 2 million MDSCs restored relaxation responses to control levels (Figure 2B). There were no effects on sodium nitroprusside-induced relaxation responses (Figure 2C). There was a dose-dependent decrease in aortic fibronectin levels with 2 million MDSCs completely inhibiting the CsA-induced increase (Figure 2D). The same dose-dependent effects were observed for glomerular injury (Figure 2E). Adoptive transfer of 2 million MDSCs had no effect on splenic Treg levels, which remained decreased in CsA-treated mice (Figure 2F). The gating strategy for flow cytometry analysis of splenic Tregs is presented in Figure S1.
Figure 2.
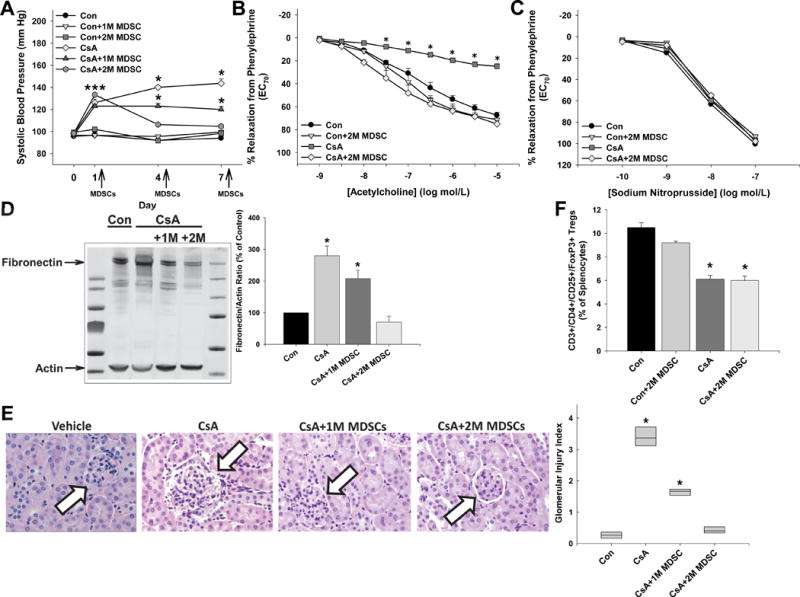
MDSC adoptive transfer eradicates the hypertension, endothelial dysfunction, and vascular and renal injury in cyclosporine A-treated mice. A, Systolic blood pressure was measured prior to injections on days 1, 4, and 7 of treatment in mice given DMSO-saline vehicle (CON), cyclosporine A (CsA), and 1 million (1M) or 2 million (2M) MDSCs. Aortic relaxation responses to acetylcholine (B) and sodium nitroprusside (C) following contraction to phenylephrine were measured on day 8 following euthanization. D, Aortic fibronectin levels were measured by immunoblotting and normalized to the loading control actin. E, Representative glomeruli and glomerular injury index score for each group. F, Splenic regulatory T cell levels as determined by flow cytometry. Results are expressed as mean + SEM. *P<0.05 vs. Con and n=6–8 mice in each group. There were no significant group × treatment interactions as determined by 2-way ANOVA or repeated measures 2-way ANOVA.
MDSCs Prevent the Direct Vascular, Renal, and Endothelial Cell Effects of CsA
Incubation of isolated aortas from control mice with CsA for 24 hours caused a significant decrease in acetylcholine-mediated relaxation responses, and this was improved partially by the presence of MDSCs (Figure 3A). In these aortas, CsA significantly increased levels of the inflammation marker E-selectin, and this was prevented completely by the presence of MDSCs (Figure 3B). Incubation of isolated kidneys obtained from control mice with CsA for 24 hours significantly increased fibronectin levels, and this was prevented by the presence of MDSCs (Figure 3C). Lastly, CsA treatment of microvascular and glomerular endothelial cells for 24 hours significantly increased fibronectin levels, and this was prevented completely by the presence of MDSCs (Figure 3D).
Figure 3.
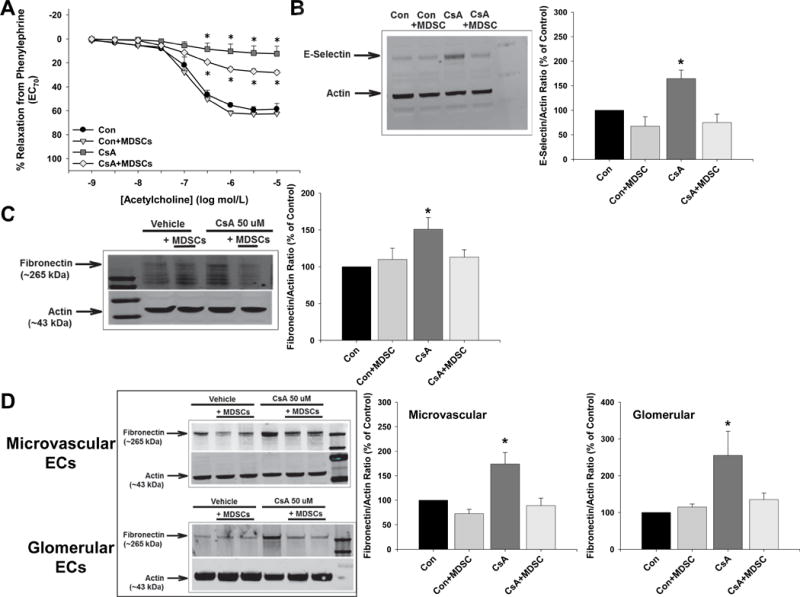
MDSCs prevent the direct vascular and renal effects of cyclosporine A. A, Aortas were isolated from control mice and incubated with DMSO-saline vehicle (Control) or cyclosporine A (CsA) for 24 hours in the absence and presence of 75,000 MDSCs isolated from control mice and then relaxation responses to acetylcholine were measured following contraction with phenylephrine. B, The rest of the aortic tissue from above was used to measure e-selectin levels by immunoblotting and normalized to the loading control actin. C, Right kidneys were isolated from control mice and incubated with DMSO-saline vehicle (Control) or CsA for 24 hours in the absence and presence of 75,000 MDSCs isolated from control mice. Fibronectin levels were then measured by immunoblotting and normalized to the loading control actin. D, Cultured microvascular and glomerular endothelial cells were treated with DMSO-saline vehicle (Control) or CsA in the absence and presence of 75,000 MDSCs isolated from control mice and fibronectin levels were measured by immunoblotting and normalized to the loading control actin. There were no significant group × treatment interactions as determined by 2-way ANOVA.
Inhibition of Calcineurin Decreases MDSCs
To first demonstrate that MDSCs may be directly affected by the calcineurin inhibitor CsA, we isolated MDSCs from the spleens and bone marrow from different control mice and measured calcineurin protein levels by immunoblotting. MDSCs do contain calcineurin (Figure 4A). Next, we treated isolated MDSCs with increasing concentrations of CsA for 24 hours and found that the number of live MDSC numbers were decreased significantly in a dose-dependent manner (Figure 4B). The same experiments were also performed with varying concentrations of the calcineurin autoinhibitory peptide (CAIP) and the selective NFAT inhibitor VIVIT. Both CAIP and VIVIT dose-dependently decreased the numbers of live MDSCs when compared to DMSO vehicle-treated MDSCs (Figure 4C and 4D).
Figure 4.
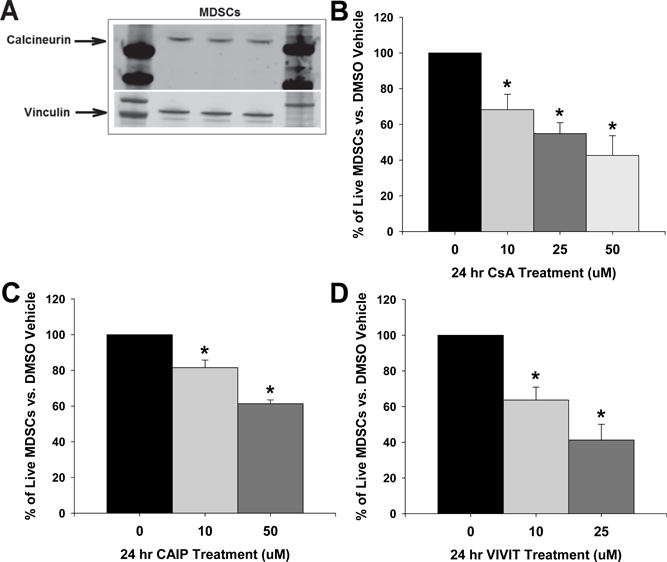
Inhibition of calcineurin in MDSCs reduces their numbers. A, Splenic MDSCs were isolated from control mice and calcineurin was measured by immunoblotting to confirm that MDSCs express calcineurin. Live MDSCs were measured by flow cytometry following 24 hour treatment with either DMSO-saline vehicle or (B) cyclosporine A (CsA), (C) the calcineurin autoinhibitory peptide (CAIP), or (D) the selective NFAT inhibitor VIVIT. Results are expressed as mean + SEM. *P<0.05 vs. 0 group and n=MDSCs from 4–6 control mice for each. There were no significant group × treatment interactions as determined by 2-way ANOVA.
DISCUSSION
CsA remains in clinical use despite the known detrimental cardiovascular and renal side effects as well as its negative effects on Tregs. CsA inhibits the calcium-dependent phosphatase calcineurin, which prevents de-phosphorylation and nuclear translocation of NFAT, and NFAT-mediated transcription of self-proliferation signals which is how it suppresses T cells. Whether this mechanism also occurs in MDSCs leading to their reduction in CsA-induced hypertension, or whether MDSCs increase in a compensatory manner like in other experimental models of hypertension,27 was unknown. Additionally, we wanted to determine whether augmenting MDSCs could ameliorate CsA-induced vascular and renal injury and hypertension. The main findings of the current study are that treatment with either IL-33 or MDSCs was able to eradicate the hypertension, endothelial dysfunction, and vascular and glomerular injury caused by CsA in mice and that direct calcineurin inhibition decreases MDSC levels.
MDSCs exert beneficial effects in a number of inflammatory and autoimmune diseases including atherosclerosis, experimental autoimmune encephalomyelitis, inflammatory bowel disease, alopecia, and Type 1 diabetes.17–19, 23, 24, 36–42 In the current study, MDSC augmentation through two different strategies, either IL-33 therapy or MDSC adoptive transfer, ameliorated the CsA-induced hypertension and vascular and renal injury similarly. One potential mechanism is through the induction of Tregs as both IL-33 and MDSCs directly have been reported to increase Treg levels.24, 25, 29 However, given the known negative effects of CsA on Treg levels, it was unknown how Treg levels would change in the setting of both MDSC augmentation and CsA. Here we demonstrate that increased MDSC levels by adoptive transfer exerted their beneficial effects independent of Tregs, as their levels remained decreased.
Another new finding is that MDSCs exhibit direct organ and cellular protective effects. Aortas and kidneys isolated from control mice and then treated with CsA demonstrated vascular dysfunction, inflammation, and injury. It is noted that although there are increased aortic fibronectin levels in CsA-hypertensive mice, only endothelium-dependent relaxation responses are mitigated. This is likely due to the relatively short period of CsA-treatment for one week, whereas chronic administration for months would likely lead to major fibrosis and smooth muscle dysfunction. Additionally, microvascular and glomerular endothelial cells treated with CsA also exhibit injury as measured by increased fibronectin levels. The presence of MDSCs was able to completely block these CsA-induced effects in both organs and cells. Given the lack of immune cell infiltration during CsA treatment in these experiments, the mechanism likely includes the known ability of MDSCs to secrete the anti-inflammatory cytokines IL-4, IL-10, IL-13, and other factors.17, 25, 26
These anti-inflammatory cytokines have been reported to reduce vascular injury and blood pressure in various models.43–50 While measures of media levels of these cytokines in the current study may be helpful, we would not be able to identify the cellular source as endothelial cells can produce and secrete anti-inflammatory cytokines as well. Studies are underway to systematically determine which cytokine, or more likely cytokines, mediate the direct vascular and renal protective effects of MDSCs during CsA treatment. MDSCs lacking these anti-inflammatory cytokines either alone or in combination (i.e., via knockdown or isolated from cytokine knockout mice) will be determined whether they lose their ex vivo and in vitro protective effects. The cytokine(s) that are determined to play a major role in the beneficial MDSC effects will be tested in CsA-hypertensive mice. We would hypothesize that MDSCs lacking the crucial anti-inflammatory cytokine(s) would have no beneficial effects in CsA-treated mice.
In addition to the direct anti-inflammatory organ and cellular effects, in vivo, the known ability of MDSCs to reduce T cells likely also contributed to the blood pressure reduction as calcineurin inhibitors are known to increase pro-inflammatory T cells despite an overall reduction in total T cells.9, 32 The beneficial blood pressure effects of MDSCs align with previous results from Shah and colleagues who demonstrated that adoptive transfer of MDSCs from hypertensive mice into angiotensin II-treated hypertensive mice one day prior to starting the angiotensin II infusion and two days later prevented the development of hypertension.27 Additionally, weekly treatment with MDSCs after angiotensin II-induced hypertension was established was able to normalize blood pressure. Further demonstrating their beneficial effects during hypertension, they reported that depletion of MDSCs worsened the angiotensin II-induced hypertensive phenotype. Given the strong anti-hypertensive effects of MDSCs, it is promising that autologous cell therapy with MDSCs may be feasible soon as groups are generating MDSCs from various cells including mouse embryonic stem cells and hematopoietic stem cells as well as human peripheral blood mononuclear cells.42, 51
We demonstrate that CsA-induced hypertension is associated with a decrease in MDSCs, unlike other forms of hypertension that are associated with a compensatory increase in peripheral levels of MDSCs.27 Hypertension in mice induced by angiotensin II infusion, NOS inhibition, or high salt diet were all associated with increased levels of circulating MDSCs, which were reported to take on an immature and immunosuppressive phenotype. This increase in MDSCs was time-dependent as well as blood pressure-dependent. The authors determined that MDSCs in these forms of hypertension did not utilize iNOS or arginase to mediate their immunosuppressive activity but rather NADPH oxidase-derived hydrogen peroxide killing of T cells. It was not determined whether there were direct vascular or renal anti-inflammatory effects in these models independent of T cell reduction.
The reduction of MDSC levels caused by CsA is likely due to the inhibition of the calcineurin-nuclear factor of activated T cells (NFAT) pathway as two other inhibitors of this pathway, CAIP and VIVIT, dose-dependently decreased live MDSCs. The transcription factor NFAT has been reported to work together with the cell fate transcription factor family CCAAT-enhancer binding proteins (C/EBP) to form a composite enhancer complex.52 Shah and colleagues reported an increase in C/EBP-beta expression, which is crucial for MDSC formation,53 in MDSCs from angiotensin II-induced hypertensive mice.27 They also reported decreased expression of C/EBP-alpha, which induces myeloid differentiation, in MDSCs from hypertensive mice. A previous study demonstrated that NFAT and C/EBP can both bind a specific promoter in cells of the monocyte and macrophage lineage only and not in cells of a T-cell origin.54 Additionally, CsA-mediated inhibition of calcineurin/NFAT increased C/EBP-alpha binding, which in the case of MDSCs would cause their differentiation into monocytes and macrophages. This may explain why CsA decreases MDSC levels instead of MDSC levels increasing as in other forms of hypertension.
Limitations of the current study include the blood pressure measures, lack of MDSC subset characterization and surface markers, and indirect IL-33 effects. We acknowledge that tail-cuff blood pressure measures are not as accurate as telemetry and every effort was made to acclimate the mice to the procedure which was performed at the same time of day. Tail-cuff measures were utilized to confirm that CsA induced hypertension and to test whether IL-33 or MDSC adoptive transfer attenuated the hypertension. Future studies using direct measures of blood pressure can confirm these effects as well as examine diurnal changes. Shah and colleagues reported that MDSCs that increase during hypertension in mice are both granulocytic and monocytic, but that granulocytic MDSCs are more prevalent. Additionally, IL-33 affects many different cell types.55 A recent paper reported that IL-33 treatment in mice expanded anti-inflammatory type 2 innate lymphoid cells in the kidneys and this protected against adriamycin-induced glomerulosclerosis. Also, IL-33 has been reported to increase Tregs in addition to MDSCs,28, 29 but the role of Tregs in the beneficial effects of IL-33 treatment in CsA-treated mice was not assessed although they did not increase following MDSC adoptive transfer. Lastly, we did not assess where the injected MDSCs localized to but Shah and colleagues reported that injected MDSCs traffic to the kidney and spleen in hypertensive mice. Studies are underway to determine whether they infiltrate blood vessels and kidneys or remain in the circulation in the setting of CsA-induced hypertension. Nonetheless, augmenting MDSCs had direct protective cardiovascular and renal effects against CsA-induced injury.
In conclusion, the direct inhibition of calcineurin/NFAT in MDSCs reduces their numbers and this loss of MDSCs and their anti-inflammatory effects contribute to the development of CsA-induced toxicity and hypertension. Augmenting MDSCs reduces the detrimental cardiovascular and renal effects of CsA in mice and may be a therapeutic strategy for CsA-treated patients.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
1) What Is New?
CsA decreases MDSCs by inhibiting calcineurin and this contributes to the development of organ injury and hypertension.
IL-33 or MDSC adoptive transfer ameliorates the vascular and renal toxicity and hypertension caused by CsA.
2) What Is Relevant?
CsA is a commonly used immunosuppressive drug, but it promotes hypertension and cardiovascular and renal damage.
Preventing these negative side effects would improve the health of CsA-treated patients.
3) Summary
Increasing levels of beneficial MDSCs prevents the hypertension and vascular and renal injury induced by CsA.
Acknowledgments
SOURCES OF FUNDING
This work was supported by NIH grant HL084299 (BMM). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health.
Footnotes
PERSPECTIVES
CsA inhibition of calcineurin in MDSCs decreases their numbers and the reduction of MDSC anti-inflammatory effects contributes to the development of vascular and renal toxicity and hypertension. Increasing MDSC levels reduces the organ injury and hypertension caused by CsA.
DISCLOSURE
There are no conflicts of interest to disclose.
References
- 1.Ponticelli C. Cyclosporine: From renal transplantation to autoimmune diseases. Ann N Y Acad Sci. 2005;1051:551–558. doi: 10.1196/annals.1361.099. [DOI] [PubMed] [Google Scholar]
- 2.Hamawy MM. Molecular actions of calcineurin inhibitors. Drug News Perspect. 2003;16:277–282. doi: 10.1358/dnp.2003.16.5.829315. [DOI] [PubMed] [Google Scholar]
- 3.Justo P, Lorz C, Sanz A, Egido J, Ortiz A. Intracellular mechanisms of cyclosporin a-induced tubular cell apoptosis. J Am Soc Nephrol. 2003;14:3072–3080. doi: 10.1097/01.asn.0000099383.57934.0e. [DOI] [PubMed] [Google Scholar]
- 4.Lindenfeld J, Miller GG, Shakar SF, Zolty R, Lowes BD, Wolfel EE, Mestroni L, Page RL, 2nd, Kobashigawa J. Drug therapy in the heart transplant recipient: Part II: Immunosuppressive drugs. Circulation. 2004;110:3858–3865. doi: 10.1161/01.CIR.0000150332.42276.69. [DOI] [PubMed] [Google Scholar]
- 5.Luke RG. Mechanism of cyclosporine-induced hypertension. Am J Hypertens. 1991;4:468–471. doi: 10.1093/ajh/4.5.468. [DOI] [PubMed] [Google Scholar]
- 6.Nankivell BJ, Borrows RJ, Fung CL, O’Connell PJ, Chapman JR, Allen RD. Calcineurin inhibitor nephrotoxicity: Longitudinal assessment by protocol histology. Transplantation. 2004;78:557–565. doi: 10.1097/01.tp.0000128636.70499.6e. [DOI] [PubMed] [Google Scholar]
- 7.Roullet JB, Xue H, McCarron DA, Holcomb S, Bennett WM. Vascular mechanisms of cyclosporin-induced hypertension in the rat. J Clin Invest. 1994;93:2244–2250. doi: 10.1172/JCI117222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bocian K, Borysowski J, Wierzbicki P, Wyzgal J, Klosowska D, Bialoszewska A, Paczek L, Gorski A, Korczak-Kowalska G. Rapamycin, unlike cyclosporine a, enhances suppressive functions of in vitro-induced CD4+CD25+ Tregs. Nephrol Dial Transplant. 2010;25:710–717. doi: 10.1093/ndt/gfp586. [DOI] [PubMed] [Google Scholar]
- 9.Chiasson VL, Pakanati AR, Hernandez M, Young KJ, Bounds KR, Mitchell BM. Regulatory T-cell augmentation or interleukin-17 inhibition prevents calcineurin inhibitor-induced hypertension in mice. Hypertension. 2017;70:183–191. doi: 10.1161/HYPERTENSIONAHA.117.09374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fourtounas C, Dousdampanis P, Sakellaraki P, Rodi M, Georgakopoulos T, Vlachojannis JG, Mouzaki A. Different immunosuppressive combinations on T-cell regulation in renal transplant recipients. Am J Nephrol. 2010;32:1–9. doi: 10.1159/000313940. [DOI] [PubMed] [Google Scholar]
- 11.Hijnen D, Haeck I, van Kraats AA, Nijhuis E, de Bruin-Weller MS, Bruijnzeel-Koomen CA, Knol EF. Cyclosporin a reduces CD4(+)CD25(+) regulatory T-cell numbers in patients with atopic dermatitis. J Allergy Clin Immunol. 2009;124:856–858. doi: 10.1016/j.jaci.2009.07.056. [DOI] [PubMed] [Google Scholar]
- 12.Lim DG, Koo SK, Park YH, Kim Y, Kim HM, Park CS, Kim SC, Han DJ. Impact of immunosuppressants on the therapeutic efficacy of in vitro-expanded CD4+CD25+FoxP3+ regulatory T cells in allotransplantation. Transplantation. 2010;89:928–936. doi: 10.1097/TP.0b013e3181d3c9d4. [DOI] [PubMed] [Google Scholar]
- 13.Miroux C, Morales O, Carpentier A, Dharancy S, Conti F, Boleslowski E, Podevin P, Auriault C, Pancre V, Delhem N. Inhibitory effects of cyclosporine on human regulatory T cells in vitro. Transplant Proc. 2009;41:3371–3374. doi: 10.1016/j.transproceed.2009.08.043. [DOI] [PubMed] [Google Scholar]
- 14.Pino-Lagos K, Michea P, Sauma D, Alba A, Morales J, Bono MR, Fierro A, Rosemblatt M. Cyclosporin a-treated dendritic cells may affect the outcome of organ transplantation by decreasing CD4+CD25+ regulatory T cell proliferation. Biol Res. 2010;43:333–337. [PubMed] [Google Scholar]
- 15.San Segundo D, Fabrega E, Lopez-Hoyos M, Pons F. Reduced numbers of blood natural regulatory T cells in stable liver transplant recipients with high levels of calcineurin inhibitors. Transplant Proc. 2007;39:2290–2292. doi: 10.1016/j.transproceed.2007.07.076. [DOI] [PubMed] [Google Scholar]
- 16.van de Wetering J, Koumoutsakos P, Peeters A, van der Mast BJ, de Kuiper P, JN IJ, Weimar W, Baan CC. Discontinuation of calcineurin inhibitors treatment allows the development of FoxP3+ regulatory T-cells in patients after kidney transplantation. Clin Transplant. 2011;25:40–46. doi: 10.1111/j.1399-0012.2010.01311.x. [DOI] [PubMed] [Google Scholar]
- 17.Dilek N, Vuillefroy de Silly R, Blancho G, Vanhove B. Myeloid-derived suppressor cells: Mechanisms of action and recent advances in their role in transplant tolerance. Front Immunol. 2012;3:208. doi: 10.3389/fimmu.2012.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greten TF, Manns MP, Korangy F. Myeloid derived suppressor cells in human diseases. Int Immunopharmacol. 2011;11:802–807. doi: 10.1016/j.intimp.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh UP, Singh NP, Singh B, Hofseth LJ, Taub DD, Price RL, Nagarkatti M, Nagarkatti PS. Role of resveratrol-induced CD11b(+) Gr-1(+) myeloid derived suppressor cells (mdscs) in the reduction of CXCR3(+) T cells and amelioration of chronic colitis in IL-10(−/−) mice. Brain Behav Immun. 2012;26:72–82. doi: 10.1016/j.bbi.2011.07.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Solito S, Bronte V, Mandruzzato S. Antigen specificity of immune suppression by myeloid-derived suppressor cells. J Leukoc Biol. 2011;90:31–36. doi: 10.1189/jlb.0111021. [DOI] [PubMed] [Google Scholar]
- 22.Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010;70:68–77. doi: 10.1158/0008-5472.CAN-09-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Gu X, Xiang J, Chen Z. Myeloid-derived suppressor cells participate in preventing graft rejection. Clin Dev Immunol. 2012;2012:731486. doi: 10.1155/2012/731486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chou HS, Hsieh CC, Charles R, Wang L, Wagner T, Fung JJ, Qian S, Lu LL. Myeloid-derived suppressor cells protect islet transplants by B7-H1 mediated enhancement of T regulatory cells. Transplantation. 2012;93:272–282. doi: 10.1097/TP.0b013e31823ffd39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dilek N, Poirier N, Usal C, Martinet B, Blancho G, Vanhove B. Control of transplant tolerance and intragraft regulatory T cell localization by myeloid-derived suppressor cells and CCL5. J Immunol. 2012;188:4209–4216. doi: 10.4049/jimmunol.1101512. [DOI] [PubMed] [Google Scholar]
- 26.Pan PY, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, Divino CM, Chen SH. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010;70:99–108. doi: 10.1158/0008-5472.CAN-09-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shah KH, Shi P, Giani JF, Janjulia T, Bernstein EA, Li Y, Zhao T, Harrison DG, Bernstein KE, Shen XZ. Myeloid suppressor cells accumulate and regulate blood pressure in hypertension. Circ Res. 2015;117:858–869. doi: 10.1161/CIRCRESAHA.115.306539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gajardo T, Morales RA, Campos-Mora M, Campos-Acuna J, Pino-Lagos K. Exogenous interleukin-33 targets myeloid-derived suppressor cells and generates periphery-induced FoxP3(+) regulatory T cells in skin-transplanted mice. Immunology. 2015;146:81–88. doi: 10.1111/imm.12483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turnquist HR, Zhao Z, Rosborough BR, Liu Q, Castellaneta A, Isse K, Wang Z, Lang M, Stolz DB, Zheng XX, Demetris AJ, Liew FY, Wood KJ, Thomson AW. IL-33 expands suppressive CD11b+ Gr-1(int) and regulatory T cells, including ST2L+ FoxP3+ cells, and mediates regulatory T cell-dependent promotion of cardiac allograft survival. J Immunol. 2011;187:4598–4610. doi: 10.4049/jimmunol.1100519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chatterjee P, Weaver LE, Doersch KM, Kopriva SE, Chiasson VL, Allen SJ, Narayanan AM, Young KJ, Jones KA, Kuehl TJ, Mitchell BM. Placental toll-like receptor 3 and toll-like receptor 7/8 activation contributes to preeclampsia in humans and mice. PLoS One. 2012;7:e41884. doi: 10.1371/journal.pone.0041884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiasson VL, Munshi N, Chatterjee P, Young KJ, Mitchell BM. Pin1 deficiency causes endothelial dysfunction and hypertension. Hypertension. 2011;58:431–438. doi: 10.1161/HYPERTENSIONAHA.111.172338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiasson VL, Talreja D, Young KJ, Chatterjee P, Banes-Berceli AK, Mitchell BM. FK506 binding protein 12 deficiency in endothelial and hematopoietic cells decreases regulatory T cells and causes hypertension. Hypertension. 2011;57:1167–1175. doi: 10.1161/HYPERTENSIONAHA.110.162917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cook LG, Chiasson VL, Long C, Wu GY, Mitchell BM. Tacrolimus reduces nitric oxide synthase function by binding to FKBP rather than by its calcineurin effect. Kidney Int. 2009;75:719–726. doi: 10.1038/ki.2008.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM. Interleukin-17 causes rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc Res. 2013;97:696–704. doi: 10.1093/cvr/cvs422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qiu LQ, Sinniah R, Hsu SI. Role of differential and cell type-specific expression of cell cycle regulatory proteins in mediating progressive glomerular injury in human IgA nephropathy. Lab Invest. 2004;84:1112–1125. doi: 10.1038/labinvest.3700144. [DOI] [PubMed] [Google Scholar]
- 36.Boros P, Ochando JC, Chen SH, Bromberg JS. Myeloid-derived suppressor cells: Natural regulators for transplant tolerance. Hum Immunol. 2010;71:1061–1066. doi: 10.1016/j.humimm.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brockhausen A, Varga G, Ehrchen J, Nippe N, Tsianakas A, Ross M, Lugering A, Roth J, Sunderkotter A. Glucocorticoid-stimulated monocytes (GCSM) control T cell-induced colitis in vivo. Ann Paediatr Rheumatol. 2012;1:25. [Google Scholar]
- 38.Chen S, Akbar SM, Abe M, Hiasa Y, Onji M. Immunosuppressive functions of hepatic myeloid-derived suppressor cells of normal mice and in a murine model of chronic hepatitis B virus. Clin Exp Immunol. 2011;166:134–142. doi: 10.1111/j.1365-2249.2011.04445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cripps JG, Gorham JD. MDSC in autoimmunity. Int Immunopharmacol. 2011;11:789–793. doi: 10.1016/j.intimp.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, Taylor PA, Panoskaltsis-Mortari A, Serody JS, Munn DH, Tolar J, Ochoa AC, Blazar BR. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood. 2010;116:5738–5747. doi: 10.1182/blood-2010-06-287839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Rompaey N, Le Moine A. Myeloid-derived suppressor cells: Characterization and expansion in models of endotoxemia and transplantation. Methods Mol Biol. 2011;677:169–180. doi: 10.1007/978-1-60761-869-0_12. [DOI] [PubMed] [Google Scholar]
- 42.Zhou Z, French DL, Ma G, Eisenstein S, Chen Y, Divino CM, Keller G, Chen SH, Pan PY. Development and function of myeloid-derived suppressor cells generated from mouse embryonic and hematopoietic stem cells. Stem Cells. 2010;28:620–632. doi: 10.1002/stem.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chatterjee P, Chiasson VL, Kopriva SE, Young KJ, Chatterjee V, Jones KA, Mitchell BM. Interleukin 10 deficiency exacerbates toll-like receptor 3-induced preeclampsia-like symptoms in mice. Hypertension. 2011;58:489–496. doi: 10.1161/HYPERTENSIONAHA.111.172114. [DOI] [PubMed] [Google Scholar]
- 44.Didion SP, Kinzenbaw DA, Schrader LI, Chu Y, Faraci FM. Endogenous interleukin-10 inhibits angiotensin II-induced vascular dysfunction. Hypertension. 2009;54:619–624. doi: 10.1161/HYPERTENSIONAHA.109.137158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kinzenbaw DA, Chu Y, Pena Silva RA, Didion SP, Faraci FM. Interleukin-10 protects against aging-induced endothelial dysfunction. Physiol Rep. 2013;1:e00149. doi: 10.1002/phy2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tinsley JH, South S, Chiasson VL, Mitchell BM. Interleukin-10 reduces inflammation, endothelial dysfunction, and blood pressure in hypertensive pregnant rats. Am J Physiol Regul Integr Comp Physiol. 2010;298:R713–719. doi: 10.1152/ajpregu.00712.2009. [DOI] [PubMed] [Google Scholar]
- 47.Chatterjee P, Chiasson VL, Seerangan G, Tobin RP, Kopriva SE, Newell-Rogers MK, Mitchell BM. Cotreatment with interleukin 4 and interleukin 10 modulates immune cells and prevents hypertension in pregnant mice. Am J Hypertens. 2015;28:135–142. doi: 10.1093/ajh/hpu100. [DOI] [PubMed] [Google Scholar]
- 48.Chatterjee P, Kopriva SE, Chiasson VL, Young KJ, Tobin RP, Newell-Rogers K, Mitchell BM. Interleukin-4 deficiency induces mild preeclampsia in mice. J Hypertens. 2013;31:1414–1423. doi: 10.1097/HJH.0b013e328360ae6c. discussion 1423. [DOI] [PubMed] [Google Scholar]
- 49.Ghoreschi K, Thomas P, Breit S, Dugas M, Mailhammer R, van Eden W, van der Zee R, Biedermann T, Prinz J, Mack M, Mrowietz U, Christophers E, Schlondorff D, Plewig G, Sander CA, Rocken M. Interleukin-4 therapy of psoriasis induces Th2 responses and improves human autoimmune disease. Nat Med. 2003;9:40–46. doi: 10.1038/nm804. [DOI] [PubMed] [Google Scholar]
- 50.Woods JM, Katschke KJ, Volin MV, Ruth JH, Woodruff DC, Amin MA, Connors MA, Kurata H, Arai K, Haines GK, Kumar P, Koch AE. IL-4 adenoviral gene therapy reduces inflammation, proinflammatory cytokines, vascularization, and bony destruction in rat adjuvant-induced arthritis. J Immunol. 2001;166:1214–1222. doi: 10.4049/jimmunol.166.2.1214. [DOI] [PubMed] [Google Scholar]
- 51.Lechner MG, Liebertz DJ, Epstein AL. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol. 2010;185:2273–2284. doi: 10.4049/jimmunol.1000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang TT, Chow CW. Transcription cooperation by NFAT.C/EBP composite enhancer complex. J Biol Chem. 2003;278:15874–15885. doi: 10.1074/jbc.M211560200. [DOI] [PubMed] [Google Scholar]
- 53.Hirai H, Zhang P, Dayaram T, Hetherington CJ, Mizuno S, Imanishi J, Akashi K, Tenen DG. C/EBPbeta is required for ‘emergency’ granulopoiesis. Nat Immunol. 2006;7:732–739. doi: 10.1038/ni1354. [DOI] [PubMed] [Google Scholar]
- 54.Dahiya S, Liu Y, Nonnemacher MR, Dampier W, Wigdahl B. CCAAT enhancer binding protein and nuclear factor of activated T cells regulate HIV-1 LTR via a novel conserved downstream site in cells of the monocyte-macrophage lineage. PLoS One. 2014;9:e88116. doi: 10.1371/journal.pone.0088116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miller AM. Role of IL-33 in inflammation and disease. J Inflamm. 2011;8:22. doi: 10.1186/1476-9255-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.