

Summary
Aims
Intracellular calcium plays an important role in neuronal hyperexcitability that leads to seizures. One calcium influx route of interest is the transient receptor potential vanilloid type 1 (TRPV1) channel. Here, we evaluated the effects of capsazepine (CPZ), a potent blocker of TRPV1 channels on acoustically evoked seizures (audiogenic seizures, AGS) in the genetically epilepsy‐prone rat (GEPR‐3), a model of inherited epilepsy.
Methods
Male and female GEPR‐3s were used. For the acute CPZ treatment study, GEPR‐3s were tested for AGS susceptibility before and after treatment with various doses of CPZ (0, 1, 3, and 10 mg/kg; ip). For semichronic CPZ treatment study, GEPR‐3s were tested for AGS susceptibility before and after 5‐day CPZ treatment at the dose of 1 mg/kg (ip). The prevalence, latency, and severity of AGS were recorded and analyzed.
Results
We found that acute CPZ pretreatment reduced the seizure severity in male GEPR‐3s; the effect was dose‐dependent. In female GEPR‐3s, however, CPZ treatment completely suppressed the seizure susceptibility. Furthermore, semichronic CPZ treatment suppressed seizure susceptibility in female GEPR‐3s, but only reduced the seizure severity in male GEPR‐3s.
Conclusions
These findings suggest that the TRPV1 channel is a promising molecular target for seizure suppression, with female GEPR‐3s exhibiting higher sensitivity than male GEPR‐3s.
Keywords: audiogenic seizures, capsazepine, generalized tonic‐clonic seizures, wild running seizures
1. INTRODUCTION
Epilepsies are disorders of neuronal excitability characterized by spontaneous recurrent seizures. Of an estimated 50 million individuals worldwide diagnosed with epilepsy, approximately 30% of these patients live with uncontrolled seizures.1, 2 Therefore, there is an urgent need for new antiepileptic treatments based on novel underlying mechanisms for neuronal hyperexcitability that leads to seizure generation. Ca2+ entry into neurons plays an important role in seizure generation, as intracellular [Ca2+] rises—and extracellular [Ca2+] decreases—during epileptiform activity.3, 4 Thus, inhibition of Ca2+ influx is a promising mechanistic approach for therapeutic interventions against seizures and epilepsies. One Ca2+ entry pathway of interest is the transient receptor potential vanilloid type 1 (TRPV1) channel, a nonselective cation channel with high Ca2+ permeability.5 Although TRPV1 channels have been implicated in the pathophysiology of seizures and epilepsies, conflicting reports have emerged. For instance, increased expression of TRPV1 channels was reported in the cortex and hippocampus from patients with mesial temporal lobe epilepsy.6 Similarly, the levels of expression of TRPV1 channels were increased in the dentate gyrus of mice exhibiting pilocarpine‐induced limbic status epilepticus.7 These findings suggest that preventing upregulation of TRPV1 channels should suppress limbic seizures. Surprisingly, activation of TRPV1 channels suppressed kainic acid‐induced limbic status epilepticus.8 In models of tonic‐clonic seizures, inhibition of TRPV1 channels prevented the development of clonic and tonic‐clonic seizures following amygdala kindling.9 In addition, inhibition of TRPV1 channels increased the seizure threshold for clonic seizures but had no signifi‐cant effect on pentylenetetrazole (PTZ)‐induced tonic seizures.10 The prevalence of PTZ‐induced generalized tonic‐clonic seizures was also reduced in adult mice lacking TRPV1 channels in the brain and previously subjected to a single febrile seizure episode at postnatal day 14.11 However, activation of TRPV1 channels suppressed tonic seizures and generalized tonic‐clonic seizures induced by maximal electroshock and PTZ, respectively.12
To further evaluate the role of TRPV1 channels in epilepsy, here, we report on the effects of capsazepine (CPZ), a potent TRPV1 channel antagonist, on seizure susceptibility in the moderate seizure severity substrain of the genetically epilepsy‐prone rat (GEPR‐3), an inherited model of epilepsy.13
2. MATERIALS AND METHODS
2.1. Animals
Seven‐week‐old male and female GEPR‐3 rats were used; these animals were obtained from our animal colony maintained at Georgetown University. The GEPR‐3s were housed in a temperature/humidity‐controlled room on a 12‐h/12‐h light/dark cycle with free access to food and water. All efforts were made to minimize the number of animals used in these experiments. All experimental procedures were approved by the Georgetown University Animal Care and Use Committee and were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals.14
2.2. Drug and treatment protocol
Capsazepine (CPZ) (N‐[2‐(4‐Chlorophenyl)ethyl]‐1,3,4,5‐tetrahydro‐7,8‐dihydroxy‐2H‐2‐benzazepine‐2‐carbothioamide) (Sigma‐Aldrich, St. Louis, USA) was freshly prepared (dissolution in a mixture of 0.9% saline and 0.2% dimethyl sulfoxide) prior to use. The GEPR‐3s were randomly separated into groups of n = 6‐8. CPZ treatment was performed in a blinded fashion.
2.2.1. Acute capsazepine treatment
The GEPR‐3s were first tested for AGS susceptibility prior to CPZ administration; GEPR‐3s that exhibited seizures were given an intraperitoneal (ip) CPZ injection in a volume of 0.2 mL/kg body weight at a dose of either 1, 3, or 10 mg/kg. Doses of the drug were selected from our preliminary experiments and previous studies.9, 10 Subsequent testing occurred 0.5, 1, 2, and 24 hours after CPZ administration.
2.2.2. Subchronic capsazepine treatment
This treatment involved fresh preparation of CPZ at the dose of 1 mg/kg. The drug was administered ip for 5 consecutive days during the week, with seizure testing being carried out on the 5th day. As with the acute treatment protocol, seizure testing was carried out at 0.5, 1, 2, and 24 hours after CPZ administration.
2.3. Acoustically evoked seizure testing
Seizure testing involved placing the animal into a Plexiglas chamber located in a sound attenuating cubic equipped with ventilation fan, light, sound generator, and video monitoring system (Med Associates, St Albans, VT, USA). To induce audiogenic seizures (AGS), an acoustic stimulus that consisted of pure tones at a 100‐105 decibels sound pressure levels intensity at 12 kHz was presented until either seizure was elicited or 60 seconds passed with no seizure. The GEPR‐3s were closely monitored following CPZ administration as well as during and following seizure testing. Convulsive seizure behavior was classified into stages: stage 0, no seizures in response to acoustic stimulus; stage 1, one episode of wild running seizures (WRS); stage 2, two or more episodes of WRS; stage 3, one episode of WRS followed by tonic‐clonic seizures characterized by tonic dorsiflexion of the neck, tonic flexion of shoulder and bouncing clonic seizures (or clonus, ie, tonic‐clonic seizures while the animal is lying on its belly); stage 4, two or more episodes of WRS followed by tonic‐clonic seizures; stage 5, one episode of WRS followed by tonic‐clonic seizures and forelimb extension; and stage 6, two episodes of WRS followed by tonic‐clonic seizures and forelimb extension.13, 15 The maximum response was recorded for each animal. In another set of experiments, GEPR‐3s were subjected to an assessment of general behavior (up to 24 hours) following CPZ administration. The following behavior abnormalities were recorded including lethargy, ataxia, tremor, Straub's tail, and spontaneous seizures. At the end of the experiment, animals were euthanized with a lethal dose of Nembutal (100 mg/kg, ip).
2.4. Rectal temperature
To determine the effects of CPZ administration on core body temperatures, rectal temperatures were measured in freely moving male and female GEPR‐3s using a Physitemp TCAT‐2LV Controller (Physitemp Instrument INC, Clifton, NJ, USA). Core body temperatures were recorded 0.5 hour before CPZ administration (time zero corresponds to time before administering CPZ), and at 0.5, 1, 2, and 24 hours after CPZ administration. The effects of CPZ on rectal temperature were expressed as the difference between rectal temperatures measured at time zero (baseline) and at 0.5, 1, 2, and 24 hours after CPZ administration. The GEPR‐3s were euthanized at the end of the experiment.
2.5. Statistical analyses
Data were analyzed in a blinded fashion. Data for rectal temperatures were assessed using repeated measurements ANOVA (and Bonferroni correction, if necessary) with time for injection as the repeated factor and treatment as the between‐group factor. Following pharmacological pretreatment and seizure testing, GEPR‐3s that did not display seizures within the 60‐sec observation period were considered to be protected from seizure activity. Only data obtained in control conditions and on the first seizure testing following administration of CPZ were included in the analysis. For each group, the incidence of WRS and clonus components of AGS in the GEPR‐3s was recorded. The time interval from the start of acoustic stimulus to the appearance of the first episode of WRS was recorded as the seizure latency. The prevalence of WRS and clonus was analyzed using the McNemar's test that compares categorical data when subjects serve as their own controls. The seizure severity was analyzed using the Wilcoxon signed‐rank test that compares ordinal data in subjects before and after treatment. The seizure latency was analyzed using paired t test. The cutoff for statistical significance was P < 0.05, and no multiple comparisons and adjustment were performed. Data are presented as the mean ± SEM for seizure latency, median seizure score ± median average deviation (M.A.D.) for seizure severity, and percentages (%) ± standard error of proportion for the prevalence of WRS and clonus.
3. RESULTS
3.1. Effects of various doses of acute CPZ treatment on acoustically evoked seizure susceptibility in GEPR‐3s
Most of the male and female GEPR‐3s tested under control conditions exhibited WRS that progressed into clonus. First, we evaluated the effects of acute CPZ treatment at a dose of 1 mg/kg on the occurrence of WRS and clonus in both female and male GEPR‐3s. In controls (pre‐CPZ treatment), all female (n = 7, Figure 1A) and male GEPR‐3s (n = 8, Figure 1B) had WRS, while 86% of females (Figure 1C) and 100% of males (Figure 1D) had clonus. Quantification showed that CPZ significantly reduced the prevalence of WRS in both female (Figure 1A) and male (Figure 1B) GEPR‐3s. More precisely, in female GEPR‐3s, CPZ reduced the prevalence of WRS by 29% (χ2 = 27.03, df = 1, P < 0.001), 57% (χ2 = 55.02, df = 1, P < 0.001), 57% (χ2 = 55.02, df = 1, P < 0.001), and 14% (χ2 = 12.07, df = 1, P < 0.001) after 0.5, 1, 2, and 24 hours, respectively, when compared with controls (Figure 1A). Similarly, in male GEPR‐3s, CPZ reduced the prevalence of WRS by 25% (χ2 = 23.04, df = 1, P < 0.001), 25% (χ2 = 23.04, df = 1, P < 0.001), 37% (χ2 = 35.03, df = 1, P < 0.001), and 13% (χ2 = 11.08, df = 1, P < 0.001) at 0.5‐, 1‐, 2‐, and 24‐hour posttreatment time points, respectively, when compared with controls (Figure 1B). CPZ at 1 mg/kg also significantly reduced the prevalence of clonus in female GEPR‐3s by 29% (χ2 = 4.56, df = 1, P < 0.05), 43% (χ2 = 13.75, df = 1, P < 0.001), 73% (χ2 = 38.67, df = 1, P < 0.001), and 29% (χ2 = 19.24, df = 1, P < 0.001) after 0.5‐, 1‐, 2‐, and 24‐hour post‐CPZ treatment time points, respectively, when compared with controls (Figure 1C). In male GEPR‐3s, the prevalence of clonus was reduced by 37% (χ2 = 36.03, df = 1, P < 0.001), 50% (χ2 = 48.02, df = 1, P = 0.0001), 50% (χ2 = 48.02, df = 1, P < 0.001), and 37% (χ2 = 35.03, df = 1, P < 0.001) 0.5‐, 1‐, 2‐, and 24‐hour post‐CPZ treatment time points, respectively, when compared with controls (Figure 1D). We also evaluated the effects of CPZ treatment on the latency to develop seizures in the GEPR‐3s. In female GEPR‐3s (Figure 1E), CPZ nonsignificantly delayed the onset of seizures at 0.5‐hour (to 36.43 ± 6.5 seconds, n = 7, t = 2.15, df = 6, P < 0.07) and 24‐hour (to 32.43 ± 4.84 seconds, n = 7; t = 1.97, df = 2, P < 0.09) posttreatment time points, when compared with controls (22.7 ± 1.57 seconds, n = 7). CPZ significantly delayed the onset of seizures at 1 hour (to 46.57 ± 6.38 seconds, n = 7; t = 4.26, df = 6, P < 0.01) and 2 hours (to 48.57 ± 6.38 seconds, n = 7; t = 4.26, df = 6, P < 0.01), when compared with controls (22.7 ± 1.57 seconds, n = 7; Figure 1E). Similarly, CPZ significantly increased the seizure latency 2‐hour (to 36.75 ± 5.25 seconds, n = 8, t = 2.46, df = 7, P < 0.05) posttreatment time point in male GEPR‐3s, when compared with controls (23.83 ± 0.68 seconds, n = 8; Figure 1F). The seizure latency was nonsignificantly increased at 0.5‐hour, 1‐hour, and 24‐hour posttreatment time points when compared with controls (Figure 1F). Finally, we evaluated the extent to which CPZ treatment at a dose of 1 mg/kg affected the seizure severity in the GEPR‐3s. Quantification showed that CPZ significantly suppressed the occurrence of seizures in female GEPR‐3s at the 2‐hour (Z = 2.11, P < 0.05) posttreatment time point, when compared with controls (Figure 1G). At 1‐ and 24‐hour posttreatment time points, CPZ nonsignificantly reduced the seizure severity (Figure 1G). In male GEPR‐3s, CPZ nonsignificantly reduced the seizure severity 2‐hour posttreatment time point, when compared with controls (Figure 1H).
Figure 1.
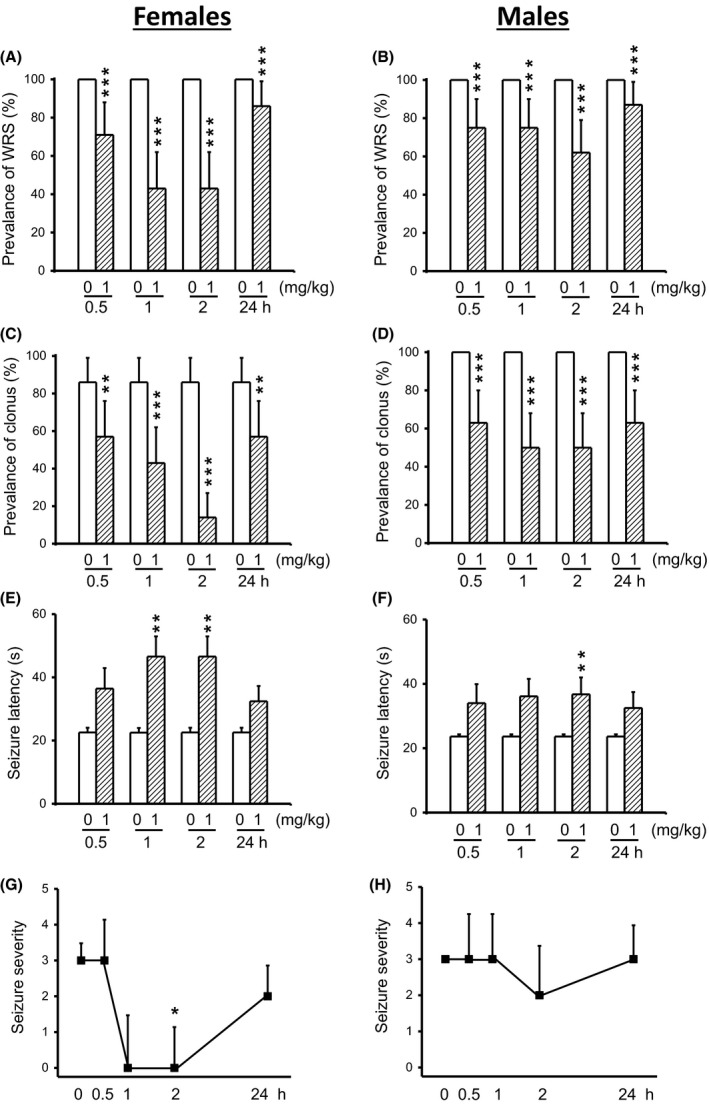
Effects of acute capsazepine (CPZ) treatment at a dose of 1 mg/kg on occurrence of acoustically evoked seizures in female and male GEPR‐3s. The anticonvulsant effects of CPZ were evaluated at different posttreatment time points of 0.5, 1, 2, and 24 h. CPZ at a dose of 1 mg/kg (ip) markedly reduced the incidence of WRS in both female (n = 7, panel A) and male (n = 8, panel B) GEPR‐3s. CPZ also reduced the prevalence of clonus in both female (panel C) and male (panel D) GEPR‐3s. CPZ also delayed the onset of seizure in either female (panel E) or male (panel F) GEPR‐3s. Time course of the effects of CPZ showed a transient, but complete seizure suppression by the 1st hour posttreatment in female GEPR‐3s (panel G), while no anticonvulsant effect was observed in male GEPR‐3s (panel H). For the prevalence of WRS and clonus, data represent the percentage (%) ± SE of proportion, and McNemar's test was used for statistical analysis. For latency, data represent mean ± SEM, and paired t test was used for analysis. For seizure severity, data represent the median score ± median average deviation, and Wilcoxon signed‐rank test was used for statistical analysis. Opened and filled bar graphs represent controls (pre‐CPZ) and CPZ‐treated GEPR‐3s, respectively. *P < 0.05, **P < 0.01, ***P < 0.001
Next, we evaluated the effects of acute CPZ treatment at a dose of 3 mg/kg on the expression of acoustically evoked seizures in both female and male GEPR‐3s. In control conditions, WRS occurred in all female (n = 8) and male (n = 8) GEPR‐3s (Figure 2A,B), while 75% of females and 100% of males had clonus (Figure 2C,D). Quantification showed that CPZ altered the occurrence of WRS and clonus in the GEPR‐3s. Thus, the prevalence of WRS was significantly reduced in females by 63% (χ2 = 51.02, df = 1, P < 0.001), 75% (χ2 = 73.01, df = 1, P < 0.001), 63% (χ2 = 51.02, df = 1, P < 0.001), and 37% (χ2 = 35.03, df = 1, P < 0.001) at 0.5‐, 1‐, 2‐, and 24‐hour posttreatment time points, respectively, when compared with controls (Figure 2A). In male GEPR‐3s, the prevalence of WRS was reduced by 25% (χ2 = 23.04, df = 1, P < 0.0001), 25% (χ2 = 23.04, df = 1, P < 0.001), 37% (χ2 = 35.03, df = 1, P < 0.001), and 50% (χ2 = 48.02 df = 1, P < 0.001) at 0.5‐, 1‐, 2‐, and 24‐hour post‐CPZ treatment time points, respectively, when compared with controls (Figure 2B). At 3 mg/kg, the prevalence of clonus was also significantly reduced in female GEPR‐3s by 50% (χ2 = 15.23, df = 1, P < 0.001), 67% (χ2 = 24.01, df = 1, P < 0.001), 50% (χ2 = 15.23, df = 1, P < 0.001), and 33% (χ2 = 7.67, df = 1, P < 0.01) at 0.5‐, 1‐, 2‐, and 24‐hour, post‐CPZ treatment time points, respectively, when compared with controls (Figure 2C). In males, CPZ treatment also reduced the prevalence of clonus by 37% (χ2 = 35.03, df = 1, P < 0.001), 50% (χ2 = 48.02, df = 1, P < 0.001), 75% (χ2 = 73.01, df = 1, P < 0.001), and 75% (χ2 = 73.01, df = 1, P < 0.001) at 0.5‐, 1‐, 2‐, and 24‐hour post‐CPZ treatment time points, respectively, when compared with controls (Figure 2D). The anticonvulsant effects of CPZ were associated with changes in seizure onset. Thus, in female GEPR‐3s (Figure 2E), CPZ significantly delayed the onset of seizures at 0.5‐hour (to 47.75 ± 6.03 seconds, n = 8; t = 4.56, df = 7, P < 0.001), 1‐hour (to 52.37 ± 5.01 seconds, n = 8; t = 5.18, df = 7, P < 0.001), 2‐hour (to 48.37 ± 5.7 seconds, n = 8; t = 4.95, df = 7, P < 0.001), and 24‐hour (to 41.75 ± 5.6 seconds, n = 8; t = 2.62, df = 7, P < 0.01) posttreatment time points, when compared with controls (25.25 ± 2.1 seconds, n = 8). In male GEPR‐3s (Figure 2F), CPZ significantly increased the seizure latency at 1‐hour (to 36.5 ± 5.19 seconds, n = 8, t = 2.87, df = 7, P < 0.01), 2‐hour (to 40.12 ± 5.95 seconds, n = 8; t = 3.35, df = 7, P < 0.01), and 24‐hour (to 43.25 ± 6.4 seconds, n = 8; t = 3.6, df = 7, P < 0.01), but not at 0.5‐hour (to 33 ± 6.07 seconds, n = 8, t = 2.22, df = 7, P < 0.06) posttreatment time points, when compared with controls (22.63 ± 2.03 seconds, n = 8). CPZ treatment also altered the seizure severity in the GEPR‐3s. Quantification showed that at 3 mg/kg, CPZ suppressed seizures in female GEPR‐3s (n = 8) at 0.5 hour (Z = 2.12, P < 0.05) and 1 hour (Z = 2.22, P < 0.05), when compared with controls (Figure 2G). CPZ nonsignificantly reduced the seizure severity at 0.5 hour (Z = 1.98, P < 0.06) and 2‐hour (Z = 1.98, P < 0.06) posttreatment time points, when compared with controls (Figure 2G). In males GEPR‐3s, the seizure severity significantly decreased 2‐hour (Z = 2.13, P < 0.05) and 24‐hour (Z = 2.16, P < 0.05) posttreatment time point when compared with controls (Figure 2H).
Figure 2.

Effects of acute capsazepine (CPZ) treatment at a dose of 3 mg/kg on occurrence of acoustically evoked seizures in GEPR‐3s. The anticonvulsant effects of CPZ were evaluated on the prevalence and severity of seizures in both female (n = 8) and male (n = 8) GEPR‐3s at different posttreatment time points of 0.5, 1, 2, and 24 h. CPZ at a dose of 3 mg/kg (ip) markedly reduced the incidence of WRS in both female (panel A) and male (panel B) GEPR‐3s. CPZ also reduced the prevalence of clonus in both female (panel C) and male (panel D) GEPR‐3s. CPZ delayed the onset of seizure onset in either female (panel E) or male (panel F) GEPR‐3s. Time course of the effects of CPZ showed a transient, but complete seizure suppression in female GEPR‐3s by first half‐hour posttreatment (panel G). In male GEPR‐3s, CPZ significantly reduced the seizure severity by the 2nd hour posttreatment (panel H). The prevalence of WRS and clonus, seizure latency, and seizure severity was analyzed as described in Figure 1. Opened and filled bar graphs represent controls (pre‐CPZ) and CPZ‐treated GEPR‐3s, respectively. *P < 0.05, **P < 0.01, ***P < 0.001
In another set of experiments, we evaluated the extent to which acute CPZ treatment at a dose of 10 mg/kg alters the expression of acoustically evoked seizures in both female (n = 8) and male (n = 8) GEPR‐3s. CPZ treatment reduced the prevalence of WRS in both female and male GEPR‐3s (Figure 3). Quantification showed that the prevalence of WRS was reduced by 63% (χ2 = 61.02, df = 1, P < 0.001), 75% (χ2 = 73.01, df = 1, P < 0.001), 100% (χ2 = 98.01, df = 1, P < 0.001), and 87% (χ2 = 85.01, df = 1, P < 0.001) at 0.5‐, 1‐, 2‐, and 24‐hour post‐CPZ treatment time points in female GEPR‐3s, respectively, when compared with controls (Figure 3A). In male GEPR‐3s, the prevalence of WRS was reduced by 25% (χ2 = 23.40, df = 1, P < 0.001), 37% (χ2 = 35.03, df = 1, P < 0.001), 50% (χ2 = 48.02, df = 1, P < 0.001), and 63% (χ2 = 61.02, df = 1, P < 0.001) at 0.5‐, 1‐, 2‐, and 24‐hour post‐CPZ treatment time points, respectively, when compared with controls (Figure 3B). CPZ treatment also altered the expression of clonus in both female and male GEPR‐3s. At 10 mg/kg, CPZ reduced the prevalence of clonus in females by 67% (χ2 = 24.01, df = 1, P < 0.01) and 83% (χ2 = 33.62, df = 1, P < 0.001) at 0.5‐ and 1‐hour posttreatment time points, respectively, when compared with controls (Figure 3C). Interestingly, clonus seizures were completely suppressed at 2‐ and 24‐hour post‐CPZ treatment (χ2 = 43.81, df = 1, P < 0.001), when compared with controls (Figure 3C). In male GEPR‐3s, CPZ pretreatment also reduced the prevalence of clonus by 37% (χ2 = 35.53, df = 1, P < 0.001), 63% (χ2 = 60.52, df = 1, P < 0.0001), 87% (χ2 = 85.51, df = 1, P < 0.001), and 87% (χ2 = 85.51, df = 1, P < 0.001) at 0.5‐, 1‐, 2‐, and 24‐hour posttreatment time points, respectively, when compared with controls (Figure 3D). In female GEPR‐3s (Figure 3E), the anticonvulsant effect of CPZ was associated with a significant delay in the onset of seizures at 0.5‐hour (CPZ: 48.87 ± 5.44 seconds, n = 8; t = 3.96, df = 7, P < 0.01), 1‐hour (CPZ: 52.5 ± 4.9 seconds, n = 8; t = 5.2, df = 7, P < 0.001), 2‐hour (CPZ: 60 ± 0 seconds, n = 8; t = 27.14, df = 7, P < 0.001), and 24‐hour (CPZ: 56.5 ± 3.5 seconds, n = 8; t = 8.89, df = 7, P < 0.001) posttreatment time points, when compared with controls (22.75 ± 1.37, n = 8). In male GEPR‐3s (Figure 3F), CPZ significantly increased the seizure latency at 1‐hour (to 41 ± 5.72 seconds, n = 8, t = 2.97, df = 7, P < 0.01), 2‐hour (to 44.37 ± 5.94 seconds, n = 8; t = 3.95, df = 7, P < 0.01), and 24‐hour (to 48 ± 5.87 seconds, n = 8; t = 4.49, df = 7, P < 0.001), but not at 0.5‐hour (to 33.75 ± 5.03 seconds, n = 8) posttreatment time points, when compared with controls (23.38 ± 2.03 seconds, n = 8). At a dose of 10 mg/kg, CPZ completely suppressed seizure susceptibility 0.5‐hour (z = 2.12, P < 0.05), 1‐hour (Z = 2.34, P < 0.01), 2‐hour (Z = 2.57, P < 0.01), and 24‐hour (Z = 2.53, P < 0.01) post‐CPZ treatment time points in females GEPR‐3s when compared with controls (Figure 3G). In male GEPR‐3s, however, CPZ treatment significantly reduced the seizure severity 2‐hour (Z = 2.34, P < 0.01) and 24‐hour (Z = 2.37, P < 0.01) time points, but not at 1‐hour time point (Z = 1.93, P < 0.06), when compared with controls (Figure 3F).
Figure 3.
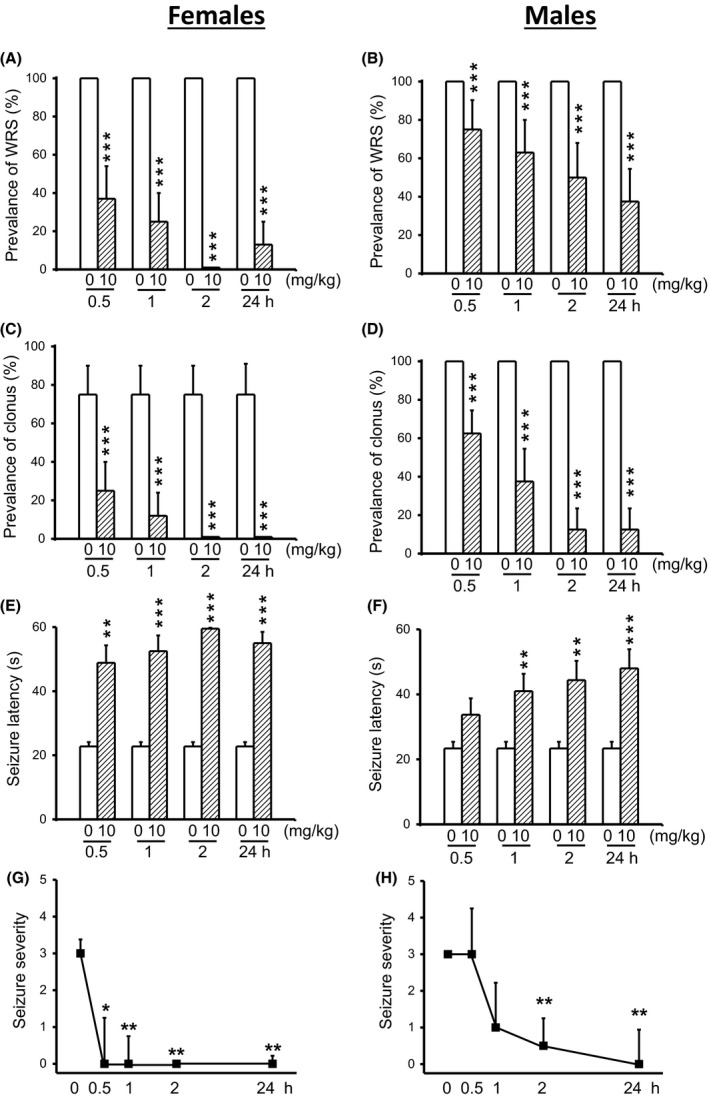
Effects of acute capsazepine (CPZ) treatment at a dose of 10 mg/kg on occurrence of acoustically evoked seizures in GEPR‐3s. The anticonvulsant effects of CPZ were evaluated on the prevalence and severity of seizures in both female and male GEPR‐3s at different posttreatment time points of 0.5, 1, 2, and 24 h. CPZ at a dose of 10 mg/kg (ip) markedly suppressed or reduced the incidence of WRS in female (n = 8, panel A) and male (n = 8, panel B) GEPR‐3s, respectively. CPZ also suppressed or reduced the prevalence of clonus in female (panel C) and male (panel D) GEPR‐3s, respectively. CPZ delayed the onset of seizure onset in either female (panel E) or male (panel F) GEPR‐3s. Note that seizures were completely suppressed 2‐hour posttreatment, and only one animal exhibited seizure 24‐hour following CPZ treatment. Time course of the effects of CPZ showed a long‐lasting seizure suppression in female GEPR‐3s by the first half‐hour posttreatment (panel G). In male GEPR‐3, however, a long‐lasting attenuation of seizure severity was observed by the 2nd hour posttreatment time point (panel H). The prevalence of WRS and clonus, seizure latency, and seizure severity was analyzed as described in Figure 1. Opened and filled bar graphs represent controls (pre‐CPZ) and CPZ‐treated GEPR‐3s, respectively. *P < 0.05, **P < 0.01, ***P < 0.001
3.2. Effects of semichronic CPZ treatment on acoustically evoked seizure susceptibility in GEPR‐3s
We evaluated whether a five consecutive daily (semichronic) CPZ treatment at a dose of 1 mg/kg enhanced anticonvulsant effects seen following acute CPZ treatment (see Figure 1). In control conditions (pre‐CPZ treatment), all female (n = 6) and male (n = 6) GEPR‐3s exhibited WRS and clonus (Figure 4). Quantification showed that semichronic CPZ treatment at a dose of 1 mg/kg significantly reduced the prevalence of WRS by 14% (χ2 = 12.07, df = 1, P < 0.001), 71% (χ2 = 69.01, df = 1, P < 0.001), 86% (χ2 = 84.01, df = 1, P < 0.001), and 57% (χ2 = 55.01, df = 1, P < 0.001) at 0.5‐ 1‐, 2‐, and 24‐hour posttreatment time points in female GEPR‐3s, respectively, when compared with controls (Figure 4A). The prevalence of WRS was also reduced in male GEPR‐3s by 17% (χ2 = 15.06, df = 1, P < 0.001), 33% (χ2 = 31.03, df = 1, P < 0.001), 50% (χ2 = 48.02, df = 1, P < 0.001), and 17% (χ2 = 15.06, df = 1, P < 0.001) at 0.5‐, 1‐, 2‐, and 24‐hour posttreatment time points, respectively, when compared with controls (Figure 4B). Next, we evaluated the effects of CPZ semichronic treatment on the occurrence of clonus in the GEPR‐3s. Quantification also showed that CPZ reduced the prevalence of clonus in female GEPR‐3s by 29% (χ2 = 27.03, df = 1, P < 0.001), 86% (χ2 = 84.01, df = 1, P < 0.001), 86% (χ2 = 84.01, df = 1, P < 0.001), and 71% (χ2 = 69.01 df = 1, P < 0.001) at 0.5‐, 1‐, 2‐, and 24‐hour posttreatment time points, respectively, when compared with controls (Figure 4C). In male GEPR‐3s, CPZ reduced the prevalence of clonus by 17% (χ2=15.06, df = 1, P < 0.001), 50% (χ2 = 48.02, df = 1, P < 0.001), 83% (χ2 = 81.02, df = 1, P < 0.001), and 83% (χ2 = 81.02, df = 1, P < 0.001) at 0.5‐, 1‐, 2‐, and 24‐hour post‐CPZ treatment time points, respectively, when compared with controls (Figure 4D). In female GEPR‐3s (Figure 4E), the anticonvulsant effect was associated with a significant delay in the onset seizure only at the 24‐hour (controls: 22 ± 2.82 seconds, n = 2; CPZ: 23.38 ± 2.03 seconds, n = 6, t = 3.91, df = 5, P < 0.01) post‐CPZ treatment time point. The latency was nonsignificantly increased at 1 hour (31.67 ± 6.5 seconds, n = 6, t = 2.3, df = 5, P < 0.06) and 2 hours (35.5 ± 8.01 seconds, n = 6, t = 2.42, df = 5, P < 0.06) following CPZ treatment, when compared with controls (Figure 4E). In male GEPR‐3s (Figure 4F), the occurrence of seizure was also delayed at 24‐hour (controls: 21.5 ± 1.47 seconds, n = 6; CPZ: 37 ± 4.88 seconds, n = 6; t = 2.94, df = 5, P < 0.01), but not at 0.5‐hour posttreatment time points. CPZ treatment nonsignificantly delayed the seizure latency at 1‐hour (to 42 ± 8.07 seconds, n = 6; t = 2.31, df = 5, P < 0.07) and 2‐hour (to 34.67 ± 5.33 seconds, n = 6; t = 2.39, df = 5, P < 0.06) posttreatment time points, when compared with controls. We also evaluated the extent to which semichronic CPZ treatment affects the seizure severity in the GEPR‐3s. The time course of seizure severity showed that semichronic CPZ treatment suppressed acoustically evoked seizures at 1‐hour (Z = 2.22, P < 0.05) and 2‐hour (Z = 2.33, P < 0.05) posttreatment time points in female GEPR‐3s, when compared with controls. CPZ has no effect on seizure severity at 0.5 hour following semichronic treatment, when compared with controls (Figure 4G). In male GEPR‐3s, CPZ significantly reduced the seizure severity at 2 hours (Z = 1.92, P < 0.06) and 24 hours (Z = 1.92, P < 0.06) but not at 0.5 and 1 hour following a semichronic treatment, when compared with controls (Figure 4H).
Figure 4.
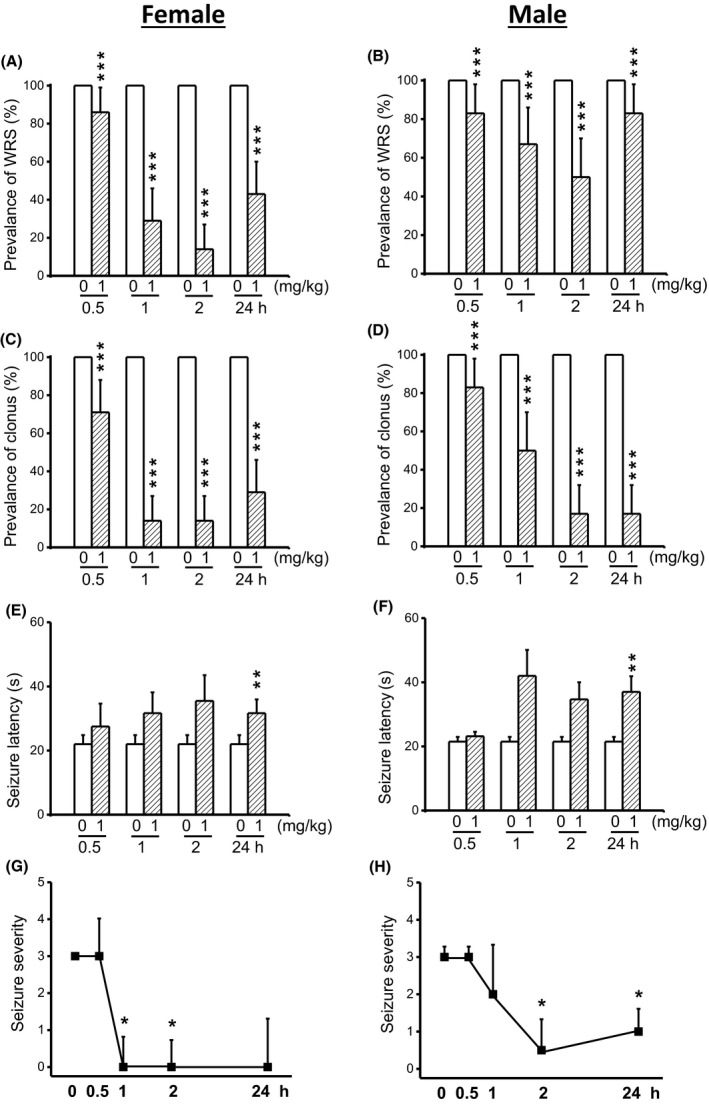
Effects of semichronic CPZ treatment on the expression of acoustically seizure susceptibility in GEPR‐3s. The effects of CPZ at a dose of 1 mg/kg (ip) were evaluated on the prevalence and severity of seizures in both female (n = 6) and male (n = 6) GEPR‐3s at different time points of 0.5, 1, 2, and 24 h. CPZ at a dose of 1 mg/kg (ip) markedly reduced the prevalence of WRS in female (panel A) and male (panel B) GEPR‐3s. CPZ treatment also reduced the prevalence of clonus in female (panel C) and male (panel D) GEPR‐3s. The anticonvulsant effect was associated with increased seizures latency in female (panel E) and male (panel F) GEPR‐3s. Time course of the effects of CPZ treatment showed a long‐lasting complete seizure suppression by the 1st hour posttreatment in female GEPR‐3s (panel G). In male GEPR‐3s, CPZ reduced the seizure severity by the 2nd hour posttreatment (panel H). The prevalence of WRS and clonus, seizure latency, and seizure severity was analyzed as described in Figure 1. Opened and filled bar graphs represent controls (pre‐CPZ) and CPZ‐treated GEPR‐3s, respectively. *P < 0.05, **P < 0.01, ***P < 0.001
3.3. Effects of acute and CPZ treatment on core body rectal temperatures in GEPR‐3s
Administration of CPZ at the tested doses did not induce abnormal behaviors (ie, reduced exploratory behavior, lethargy, ataxia, Straub's tail, and spontaneous seizures) in both female (n = 6‐8) and male (n = 6‐8). Similarly, test between‐subject effect revealed that acute (0, 1, 3, or 10 mg/kg, ip; Figure 5A,B) or semichronic (0, 1 mg/kg, ip, Figure 5C,D) CPZ treatment did not alter core body temperatures of both female (n = 6) and male (n = 6) GEPR‐3s.
Figure 5.
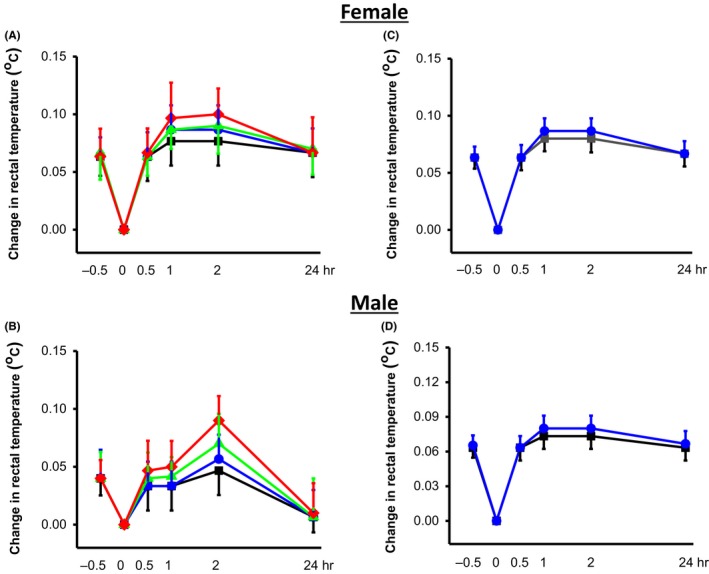
Effects of CPZ acute and semichronic treatments on core body temperature in GEPR‐3s. Acute CPZ treatment at all tested doses (0 mg/kg: black lines, 0.5 mg/kg: blue lines, 1 mg/kg: green lines and 10 mg/kg: red lines) did not significantly alter rectal temperature in both female (A, n = 6) and male (B, n = 6) GEPR‐3s. Similarly, semichronic treatment at either the dose of 0 (black lines) or 1 mg/kg (ip, blue lines) did not alter core body temperatures in both female (n = 6) and male (n = 6) GEPR‐3s. Repeated measures ANOVA was used to compare changes in rectal temperature
4. DISCUSSION
The main finding of this present study is that acute CPZ treatment suppressed AGS susceptibility in female GEPR‐3s or reduced the seizure severity in male GEPR‐3s. The seizure suppression was observed in female GEPR‐3s at all tested doses, whereas only higher doses were effective in attenuating the seizure severity in male GEPR‐3s following acute CPZ treatment. Semichronic CPZ treatment using the least effective acute CPZ tested dose suppressed seizure susceptibility in female GEPR‐3s, but only reduced the seizure severity in male GEPR‐3s. These data indicate that AGS in female GEPR‐3s is more sensitive to the effect of CPZ treatment compared to male GEPR‐3s; the underlying biological mechanism for this differential sex effect is yet unknown. As CPZ inhibits the activity of TRPV1 channels, these findings suggest that blockade of these channels may have a potent anticonvulsant activity in female GEPR‐3s. In the GEPR, seizure activity is initiated in the inferior colliculus (IC).16, 17 Thus, the suppression of AGS following CPZ treatment suggests that remodeling of TRPV1 channels, at least in the IC, may play an important role in AGS susceptibility in the GEPR‐3s.
TRPV1 channels are mainly located in nociceptive neurons of the peripheral nervous system suggesting low expression levels of these channels in the central nervous system.18, 19, 20 The presence of TRPV1 channels in the central nervous system suggests their broader role than in pain perception. Accordingly, increased expression of TRPV1 channels has been reported in the cortex and hippocampus obtained from patients with temporal lobe epilepsy.6 Similarly, increased expression of TRPV1 channels was found in the dentate gyrus of mice exhibiting pilocarpine‐induced status seizures.7 Thus, it is tempting to speculate that AGS susceptibility in the GEPR‐3s is associated with increased expression of TRPV1 channels in the network for AGS, including the IC.
The role of TRPV1 channels in the pathophysiology of epilepsy is complex. For instance, TRPV1 antagonist CPZ suppressed PTZ‐induced seizures, while the TRPV1 agonist capsaicin reduced the prevalence of maximal electroshock‐induced tonic seizures and PTZ‐induced tonic‐clonic seizures.12, 21 However, inhibition of TRPV1 channels had no effect on the severity of PTZ‐induced seizures.10, 22 Furthermore, the severity of PTZ‐induced tonic‐clonic seizures was decreased in TRPV1‐deficient mice.11 Surprisingly, decreased severity of PTZ‐induced tonic‐clonic seizures was also reported in mice with an overexpression of TRPV1 channels in the hippocampus.22 The severity of PTZ‐induced seizures was also reduced after intrahippocampal administration of either TRPV1 channel agonist or antagonist.22 Thus, it has been suggested that in mice with TRPV1 channel overexpression, PTZ administration was not able to further increase TRPV1 channel function and neuronal excitability in the hippocampus, leading to decreased seizure susceptibility22. Inhibition of TRPV1 channels also prevented the development of clonic and tonic‐clonic seizures following amygdala kindling, a model of limbic epileptogenesis.9 Consistent with these studies, we found that inhibition of TRPV1 channels suppressed AGS susceptibility in the GEPR‐3s. Tonic seizures and generalized tonic‐clonic seizures induced by maximal electroshock and PTZ in naive rodents or acoustic stimuli in the GEPR‐3s are mediated by the brainstem. Nevertheless, maximal electroshock‐induced tonic seizures and PTZ‐induced tonic seizures and generalized tonic‐clonic seizures are models of acute seizures, but not models of epilepsy per se. Thus, activation of TRVP1 channels suppresses seizures in models of acute seizures, while inhibition of these channels suppresses seizures in models of acquired and inherited epileptogenesis.
The underlying mechanisms of how blockade of TRPV1 channels suppresses AGS susceptibility in the GEPR‐3s are not yet fully understood. TRPV1 channel is a nonselective cation channel with high Ca2+ permeability, and its activation results in increased intracellular Ca2+ and neuronal excitability, leading to enhanced glutamate release.23, 24, 25, 26 Interestingly, altered intracellular Ca2+ and glutamate signaling in the IC are known to play important roles in the pathophysiology of seizures in GEPRs.27, 28 Whether Ca2+ signaling via TRPV1 channels interacting with glutamate signaling is critical in AGS susceptibility in GEPR‐3s remains unknown. Another putative mechanism underlying epileptogenesis is long‐term potentiation (LTP), where repeated synaptic stimulation lead to enhancement of synaptic efficiency.29 Interestingly, TRPV1 channels contributed to synaptic plasticity as their activation facilitated LTP and suppressed long‐term depression (LTD) in the hippocampus and amygdala.24, 30, 31 Such effects of TRPV1 channels on LTP and LTD may also occur in the IC and contribute to neuronal hyperexcitability associated with enhanced susceptibility to acoustically evoked seizures.
In conclusion, inhibition of TRPV1 channels suppressed acoustically evoked seizures in the GEPR‐3s, suggesting that these channels may provide a potential molecular target for the suppression of generalized tonic‐clonic seizures.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
This publication was made possible by the Public Health Service Grants R01 AA020073 (P.N.) from the National Institutes of Health (NIH), and its contents are the responsibility of the authors and do not necessarily represent the official views of NIH.
Cho SJ, Vaca MA, Miranda CJ, N'Gouemo P. Inhibition of transient potential receptor vanilloid type 1 suppresses seizure susceptibility in the genetically epilepsy‐prone rat. CNS Neurosci Ther. 2018;24:18–28. 10.1111/cns.12770
REFERENCES
- 1. WHO . World Health Organization: epilepsy: epidemiology, aetiology and prognosis. WHO Factsheet 2001.
- 2. French JA. Refractory epilepsy: clinical overview. Epilepsia. 2007;48(Suppl. 1):3‐7. [DOI] [PubMed] [Google Scholar]
- 3. Albowitz B, König P, Kuhnt U. Spatiotemporal distribution of intracellular calcium transients during epileptiform activity in guinea pig hippocampal slices. J Neurophysiol. 1997;77:491‐501. [DOI] [PubMed] [Google Scholar]
- 4. Delorenzo RJ, Sun DA, Deshpande LS. Cellular mechanisms underlying acquired epilepsy: the calcium hypothesis of the induction and maintenance of epilepsy. Pharmacol Ther. 2005;105:229‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pedersen SF, Owsianik G, Nilius B. TRP channels: an overview. Cell Calcium. 2005;38:233‐252. [DOI] [PubMed] [Google Scholar]
- 6. Sun F‐J, Guo W, Zheng D‐H, et al. Increased expression of TRPV1 in the cortex and hippocampus from patients with mesial temporal lobe epilepsy. J Mol Neurosci. 2013;49:182‐193. [DOI] [PubMed] [Google Scholar]
- 7. Bhaskara MD, Smith BN. Effects of TRPV1 activation on synaptic excitation in the dentate gyrus of a mouse model of temporal lobe epilepsy. Exp Neurol 2010;223:529‐536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee TH, Lee JG, Yon JM, et al. Capsaicin prevents kainic acid‐induced epileptogenesis in mice. Neurochem Int. 2011;58:634‐640. [DOI] [PubMed] [Google Scholar]
- 9. Shirazi MI, Izadi M, Amin M, Rezvani ME, Roohbakhsh A, Shamsizadeh A.Involvement of central TRPV1 receptors in pentylenetetrazole and amygdala kindling in male rats. Neurol Sci. 2014;35:1235‐1241. [DOI] [PubMed] [Google Scholar]
- 10. Socala K, Nieoczym D, Pierog M, Wlaz P. α‐spinasterol, a TRPV1 receptor antagonist, elevates the seizure threshold in three acute seizure tests in mice. J Neural Transm (Vienna). 2015;122:1239‐1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kong W‐l, Min J‐W, Liu Y‐L, Li JX, He XH, Peng BW. Role of TRPV1 in susceptibility to PTZ‐induced seizure following repeated hyperthermia challenges in neonatal mice. Epilepsy Behav 2014;31:276‐280. [DOI] [PubMed] [Google Scholar]
- 12. Chen C‐Y, Li W, Qu K‐P, Chen CR. Piperine exerts anti‐seizure effects via the TRPV1 receptor in mice. Eur J Pharmacol. 2013;714:288‐294. [DOI] [PubMed] [Google Scholar]
- 13. Mishra PK, Dailey JW, Reigel CE, Jobe PC. Audiogenic convulsions in moderate seizure genetically epilepsy‐prone rats (GEPR‐3s). Epilepsy Res. 1989;3:191‐198. [DOI] [PubMed] [Google Scholar]
- 14. National Research Council (U.S.) , Institute for Laboratory Animal Research (U.S.) , National Academies Press (U.S.) . Guide for the Care and Use of Laboratory Animal, 8th edn Washington, DC: National Academies Press; 2011. [Google Scholar]
- 15. Jobe PC, Picchioni AL, Chin L. Role of brain norepinephrine in audiogenic seizure in the rat. J Pharmacol Exp Ther. 1973;184:1‐10. [PubMed] [Google Scholar]
- 16. Browning RA. Neuroanatomical localization of structures responsible for seizures in the GEPR: lesion studies. Life Sci. 1986;39:857‐867. [DOI] [PubMed] [Google Scholar]
- 17. Faingold CL, Anderson CA. Loss of intensity‐induced inhibition in inferior colliculus neurons leads to audiogenic seizure susceptibility in behaving genetically epilepsy‐prone rats. Exp Neurol. 1991;113:354‐363. [DOI] [PubMed] [Google Scholar]
- 18. Mezey E, Toth ZE, Cortright DN, et al. Distribution of m RNA for vanilloid receptor subtype 1(VR1), and VR1‐like immunoreactivity, in the central nervous system of rat and human. Proc Natl Acad Sci USA. 2000;97:3655‐3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Toth A, Boczan J, Kedei N, et al. Expression and distribution of vanilloid receptor 1 (TRPV1) in adult rat brain. Brain Res Mol Brain Res. 2005;135:162‐168. [DOI] [PubMed] [Google Scholar]
- 20. Cristino L, de Petrocellis L, Pryce G, Baker D, Guglielmotti V, DiMarzo V. Immunohistochemical localization of cannabinoid type 1 and vanilloid transient receptor potential type 1 receptors in the mouse brain. Neuroscience. 2006;139:1405‐1415. [DOI] [PubMed] [Google Scholar]
- 21. Manna SS, Umathe SN. Involvement of transient receptor potential vanilloid type 1 channels in the pro‐convulsant effect of anandamide in pentylenetetrazole‐induced seizures. Epilepsy Res. 2012;100:113‐124. [DOI] [PubMed] [Google Scholar]
- 22. Jia Y‐F, Li Y‐C, Tang Y‐P, et al. Interference of TRPV1 function altered the susceptibility of PTZ‐induced seizures. Front Cell Neurosci. 2015;9:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kauer JA, Gibson HE. Hot flash: TRV channels in the brain. Trends Neurosci. 2009;32:215‐224. [DOI] [PubMed] [Google Scholar]
- 24. Gibson HE, Edwards JG, Page RS, Van Hook MJ, Kauer JA. TRPV1 channels mediate long‐term depression at synapses on hippocampal interneurons. Neuron. 2008;57:746‐759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chavez AE, Chiu CQ, Castillo PE. TRPV1 activation by endogenous anandamide triggers postsynaptic long‐term depression in dentate gyrus. Nat Neurosci. 2010;13:1511‐1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zsombok A, Bhaskaran MD, Gao H, Derbenev AV, Smith BN. Functional plasticity of central TRPV1 receptors in brainstem dorsal vagal complex circuits of streptozotocin‐treated hyperglycemic mice. J Neurosci. 2011;31:14024‐14031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Faingold CL, Naritoku DK, Copley CA, Randall ME, Riaz A, Anderson CA. Arnerić SP Glutamate in the inferior colliculus plays a critical role in audiogenic seizure initiation. Epilepsy Res. 1992;13:95‐105. [DOI] [PubMed] [Google Scholar]
- 28. N'Gouemo P, Faingold CL, Morad M. Calcium channels dysfunction in inferior colliculus neurons of the genetically epilepsy‐prone rat. Neuropharmacology. 2009;56:665‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Leite JP, Neder L, Arisi GM, Carlotti CG Jr, Assirati J, Moreira J. Plasticity, synaptic strength, and epilepsy: what can we learn from ultrastructure data? Epilepsia. 2005;46(Suppl 5):134‐141. [DOI] [PubMed] [Google Scholar]
- 30. Li HB, Mao RR, Zhang JC, Yang Y, Cao J, Xu L. Anti‐stress effect of TRPV1 channel on synaptic plasticity and spatial memory. Biol Psychiatry. 2008;64:286‐292. [DOI] [PubMed] [Google Scholar]
- 31. Zschenderlein C, Gebhardt C, von Bohlen Und Halbach O, Kulisch C, Albrecht D. Capsaicin‐induced changes in LTP in the lateral amygdala are mediated by TRPV1. PLoS ONE 2011;6:e16116. [DOI] [PMC free article] [PubMed] [Google Scholar]