

SUMMARY
The pathogenesis of Human Ebola virus disease (EVD) is complex. EVD is characterized by high levels of virus replication and dissemination, dysregulated immune responses, extensive virus- and host-mediated tissue damage, and disordered coagulation. To clarify how host responses contribute to EVD pathophysiology, we performed multi-platform ‘omics analysis of peripheral blood mononuclear cells and plasma from EVD patients. Our results indicate that EVD molecular signatures overlap with those of sepsis, imply that pancreatic enzymes contribute to tissue damage in fatal EVD, and suggest that EBOV infection may induce aberrant neutrophils whose activity could explain hallmarks of fatal EVD. Moreover, integrated biomarker prediction identified putative biomarkers from different data platforms that differentiated survivors and fatalities early after infection. This work reveals insight into EVD pathogenesis, suggests an effective approach for biomarker identification, and provides an important community resource for further analysis of human EVD severity.
eTOC SUMMARY
Eisfeld et al. comprehensively evaluated changes in host molecules in plasma and peripheral immune cells of Ebola virus disease (EVD) patients. Their results suggest new mechanisms of EVD pathogenesis and putative biomarkers for predicting EVD outcomes. Moreover, datasets associated with this work are an important community resource for further research.
INTRODUCTION
The West African Ebola virus (EBOV) outbreak of 2013 to 2016 was the most devastating human EBOV epidemic to date, causing >23,000 laboratory-confirmed cases and >11,000 deaths (WHO, 2016). The likelihood of future outbreaks of similar or greater impact is unclear, but given the lack of any approved countermeasures for prevention or treatment of EBOV disease (EVD), it is critical to expand our knowledge of EVD pathogenicity in humans to support countermeasure development.
EVD pathogenesis is marked by extensive virus replication and systemic spread, dysregulated immune responses, extensive tissue damage and organ dysfunction, and disordered coagulation (reviewed in (Messaoudi et al., 2015)). Lymphopenia and elevated pro-inflammatory cytokines in plasma are typical, especially in fatal infections. Nonetheless, T lymphocytes are robustly activated and, in survivors, become more specific toward EBOV proteins over time (Ruibal et al., 2016). In contrast, T lymphocytes in fatal infections exhibit pronounced immunosuppressive marker (PD-1/PDCD1 and CTLA4) expression and low EBOV protein specificity (Ruibal et al., 2016). The role of antigen presenting cells (APCs) is not fully understood, but a recent report suggested that monocytes may be inefficiently activated (Ludtke et al., 2016). Notably, systemic inflammation and immune dysfunction, as well as other clinical EVD findings (i.e., coagulopathies, vascular leakage, and organ dysfunction), are characteristic of classical sepsis caused by other disease agents (Hellman, 2015).
The recent EBOV outbreak afforded a rare opportunity to perform multi-platform ‘omics analysis of blood samples collected from EVD patients with the goal of identifying host response mechanisms that contribute to EVD severity.
RESULTS
Study design and patient cohorts
At three different hospitals (see Table S1), blood samples were collected from EVD patients after initial diagnosis, and serial samples were collected from survivors over the course of EVD and recovery (Figure 1A; S1, S2, and S3 refer to the first, second and third samples collected from survivors). Patients with fatal EVD succumbed to the infection before additional samples could be collected. In total, we obtained 29 samples from 11 EVD survivors and 9 samples from 9 EVD fatalities. For comparison, we collected blood samples from 10 healthy volunteers. Samples were transported to our field laboratory and processed for ‘omics and other analyses (Fig. S1). Statistical analysis of clinical and demographic data revealed no significant differences between the survivor and fatality groups with regard to sex, clinical presentation at diagnosis (i.e., dry versus wet disease; the ‘dry’ stage is characterized by fever, malaise, and myalgia; the ‘wet’ stage is distinguished by gastrointestinal symptoms, suggesting more advanced disease), age, time from symptom onset to the first sample, or the Ebola treatment center location (Fig. 1B and C; clinical and demographic data are provided in Table S1). As such, these parameters were not used for correlation analysis with ‘omics data downstream. There were no significant differences between the ages or sexes of the EVD patients and the healthy controls (Fig. 1C).
Figure 1. Study design and patient demographics.
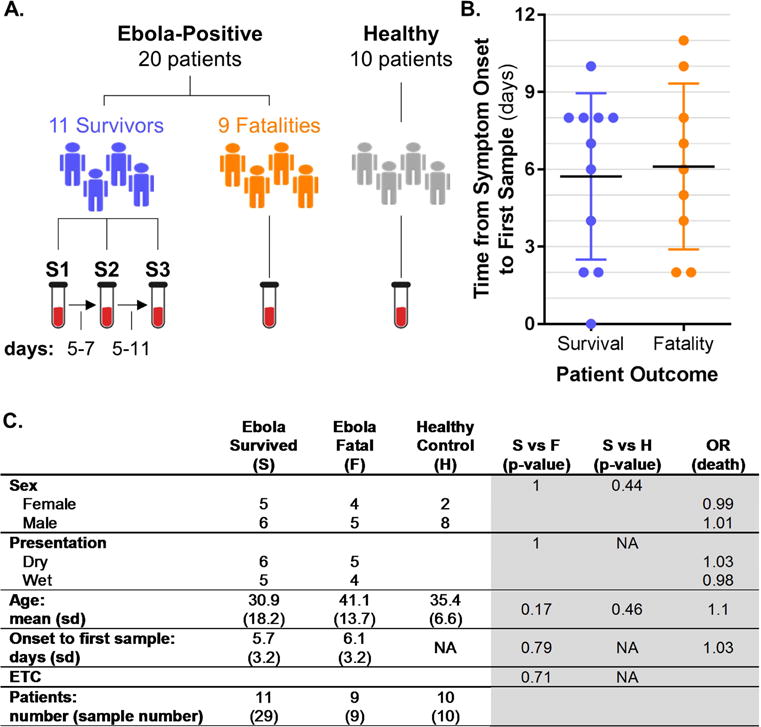
(A) Overview of blood sample collection from EVD patients and healthy controls. Serial samples from survivors are indicated by S1, S2, and S3. The number of days that elapsed between S1 and S2 or S2 and S3 collections is indicated at the bottom left. (B) The number of days between symptom onset and the first sample collection. (C) Statistical analysis of clinical and demographic data. Patient data are on the left (white background); and on the right (gray background), columns list chi-square (sex, disease presentation, and Ebola treatment center (ETC)) or t-test (age and time from symptom onset to first sample) results for comparisons between survivor (S) and fatality (F) or healthy control (H) patients. Odds ratios (OR) were estimated by logistic regression models with death as the outcome. For continuous parameters (age and time from symptom onset to first sample) the OR was estimated for a difference of 10 years (age) or 3 days (time from symptom onset to first sample). NA, not applicable. See also Figure S1 and Table S1.
Ebola fatalities can be differentiated from Ebola survivors by viral load and host responses
In recent and past EBOV outbreaks, higher viral load at the time of admission has been correlated with fatal outcome (Nanclares et al., 2016; Schieffelin et al., 2014), and patients with fatal EVD express higher plasma inflammatory cytokines compared with survivors (Ruibal et al., 2016; Wauquier et al., 2010). Consistent with these reports, EVD fatalities in our cohort exhibited significantly higher peripheral blood mononuclear cell (PBMC) viral loads on admission compared with EVD survivors (Fig. 2A; Student’s T-test, P = 0.0302). PBMC RNA-seq and plasma proteomics data corroborated this finding (Fig. S2A and B). Upon admission, EVD survivors exhibited significantly elevated levels of IL6, TNF, and IL10 (P = 0.0000008, P = 0.0005, and P = 0.01, respectively; measured by ELISA), which were diminished at later time points (Fig. 2B; also see Table S2). In fatal EVD cases, plasma IL6 and TNF were higher than in EVD survivors (fold change [FC] = 5, P = 0.002; and FC = 5, P = 0.001, respectively) (Fig. 2B), in line with previous observations (Ruibal et al., 2016; Wauquier et al., 2010). Thus, EVD fatalities can be differentiated from survivors by both viral loads and plasma cytokine levels upon admission.
Figure 2. EVD survivors and fatalities are differentiated by viral load and host responses.
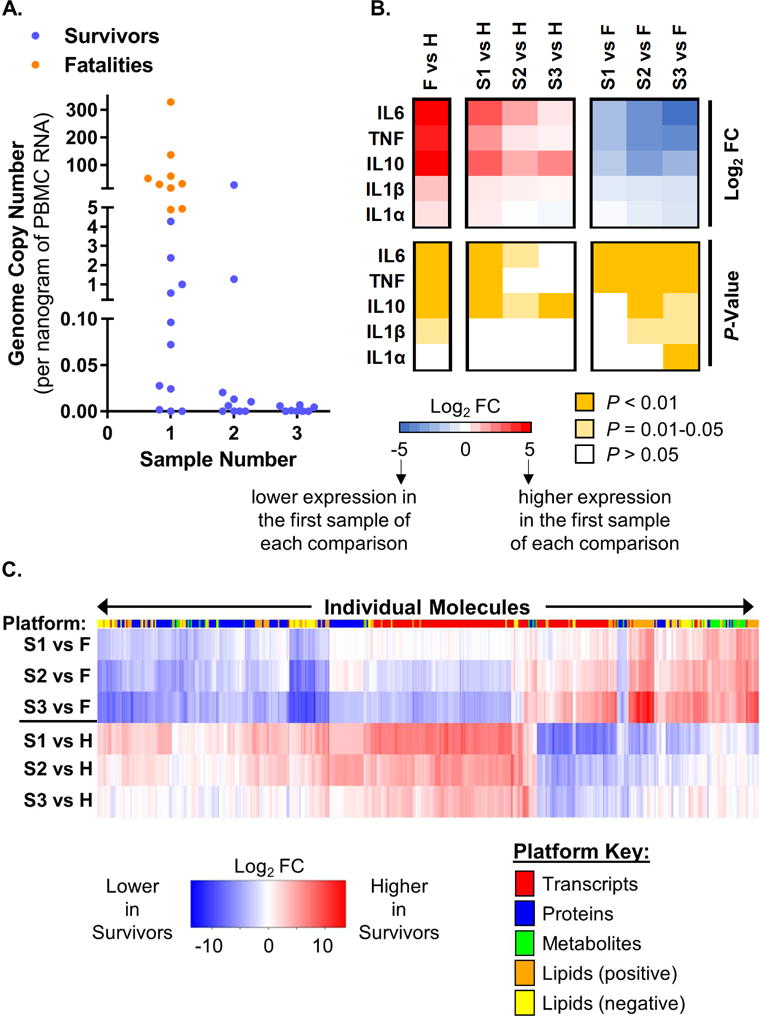
(A) EBOV load (copies per nanogram of input RNA) as determined by qRT-PCR of RNA from PBMCs. (B) Average cytokine levels (as determined by ELISAs) and associated P-values for EVD patients (fatalities, ‘F’; survivors’ first, second, and third samples, ‘S1’, ‘S2’, and ‘S3’) compared with healthy controls (H); or for S1/S2/S3 vs. F comparisons. Specific comparisons are represented in columns and assayed cytokines are represented in rows. For the expression heat map, F/S1/S2/S3 vs. H comparison values are displayed as the direction of expression in the EVD sample and S1/S2/S3 vs. F comparison values are displayed as the direction of expression in survivors. FC, fold change. (C) Expression trends for representative molecules from transcriptomics (P < 0.000001), proteomics (P < 0.001), metabolomics (P < 0.001), and lipidomics (P < 0.0001). Columns show log2 FC values for individual molecules (IDs not shown), and the color-coded bar above the heat map indicates the type of molecule in each column. Rows represent different comparison groups, and FC values are displayed as the direction of expression in the survivor samples versus the comparator. Lipid species identified by positive and negative ionization are shown separately. See also Figure S2 and Table S2.
Next, we clustered significantly altered factors from all platforms together to identify trends in the host response data (Fig. 2C). Comparing serial samples from EVD survivors to those from healthy controls, we observed the highest level of differential expression on admission and smaller differences for later samples as the infection resolved (Fig. 2C and Fig. S2C). An analogous comparison between EVD survivors and fatalities revealed host factors that were differentially expressed between the groups on admission (Fig. 2C and Fig. S2C). These differences were amplified when samples from fatalities were compared with samples from survivors taken at later time points (Fig. 2C), most likely reflecting recovery in the latter group.
Plasma metabolome signatures correspond to clinical symptoms of EVD
We currently know little about the metabolic signatures of EVD. Therefore, we examined our plasma metabolomics dataset to identify metabolic signatures associated with outcome (Fig. S2C and Table S2). A prominent signature observed nearly exclusively among EVD fatalities was an acute reduction in plasma free amino acids (PFAAs), involving predominantly essential and conditionally essential amino acids (Fig. 3A). Since PFAAs (e.g., glutamine) are consumed in inflammatory states to fuel immune cell proliferation and phagocytosis (Soeters and Grecu, 2012), a strong reduction in PFAA levels among EVD fatalities suggests immune cell activation in these cases. EVD fatalities also exhibited significantly reduced plasma glucose and fructose, concomitant with substantial sucrose accumulation (Fig. 3A), suggesting perturbation of sucrose catabolism. Since sucrose is normally digested by enzymes in the duodenal microvilli, its abundance in EVD fatalities suggests the possibility that intestinal tissue damage (which could impair sucrose digestion) is increased in more severe cases of EVD. Other notable metabolic signatures included significantly increased plasma inositols (precursors to signal transduction molecules) in patients with fatal outcomes; and increased 2-hydroxybutyric acid in both EVD fatalities and survivors in the acute phase of illness (i.e., in the initial sample) (Fig. 3A). Accumulation of 2-hydroxybutyric acid (produced by the liver when high levels of glutathione synthesis deplete L-cysteine) suggests that oxidative stress – possibly mediated through production of reactive oxygen species by activated neutrophils or macrophages – is a characteristic of EVD regardless of outcome. Further, higher levels of 2-hydroxybutyric acid in EVD survivors may imply that the liver is more active in mitigating the effects of oxidative stress in less severe EVD.
Figure 3. EVD signatures in the plasma metabolome and lipidome.
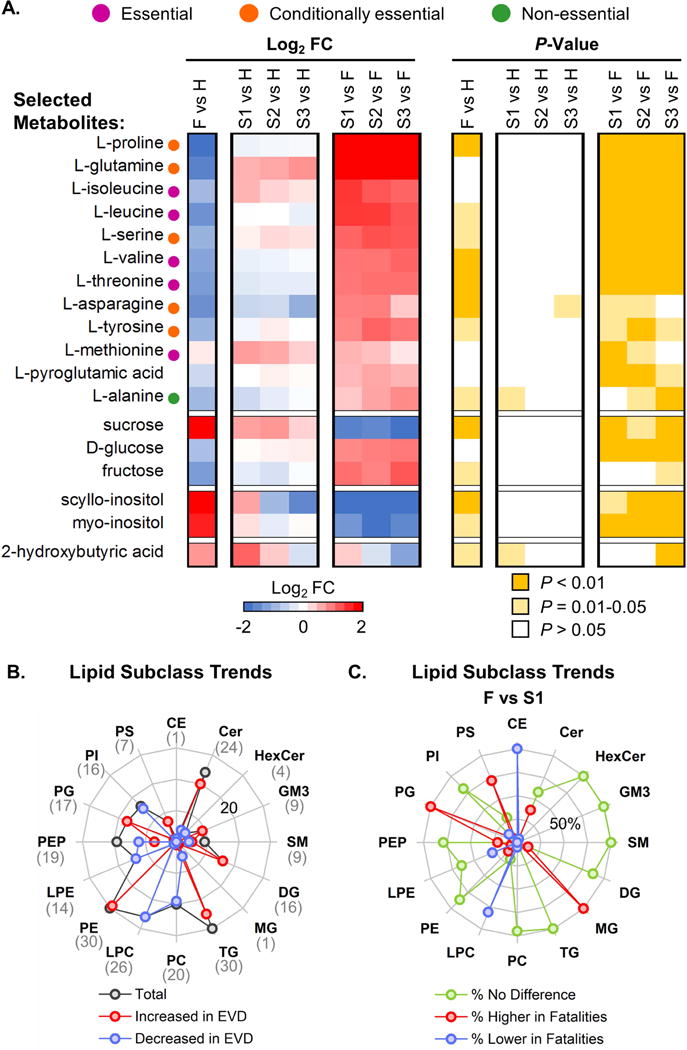
(A) Selected average plasma metabolite expression levels and associated P-values for EVD patients (data are represented in the same way as in Fig. 2B). FC, fold change. (B) The number of significantly changed (P < 0.01) plasma lipid species (from both positive and negative ionization analyses) that were increased (red line) or decreased (blue line) in EVD in at least one comparison (F vs. H, S1 vs. H, S2 vs. H, or S3 vs. H) for different lipid subclasses. The total number of significantly changed lipid species for each lipid subclass is shown by the dark gray line and is indicated in parentheses below each label at the edge of the radar map. The highest value depicted by the radar map is 30 (outermost concentric circle in light grey), and the line corresponding to 20 is labeled on the panel. Regardless of EVD outcome, lipid species in each subclass trended in the same direction. (C) The proportion of lipid species for each lipid subclass depicted in panel B that exhibit significantly higher (red line) or lower (blue line) expression in EVD fatalities relative to S1 (P < 0.01). The proportion of lipid species exhibiting no significant expression difference between EVD survivors (S1) and fatalities is also shown (green line). The highest value shown by the radar map is 100% (outermost concentric circle in light grey), and the line corresponding to 50% is labeled on the panel. CE, cholesterol ester; Cer, ceramide; HexCer, monohexosylceramide; GM3, ganglioside GM3; SM, sphingomyelin; DG, diacylglycerides; MG, monoacylglycerolipids; TG, triglycerides; PC, diacylglycerophosphocholine; LPC, monoacylglycerophosphocholine; PE, diacylglycerophosphoethanolamine; LPE, monoacylglycerophosphoethanolamine; PEP, PE plasmalogen; PG, diacylglycerophosphoglycerol; PI, diacylglycerophosphoinositol; PS, monoacylglycerophosphoserine. See also Table S2.
Lipid signatures differentiate fatal and non-fatal EVD
Alterations in various plasma lipid species are associated with inflammatory diseases including sepsis (Filippas-Ntekouan et al., 2017). Therefore, we next focused on our plasma lipidomics dataset to identify signatures of EVD. Plasma lipidomics analyses (Fig. S2C and Table S2) revealed alterations in lipid subclasses that define both EVD in general and fatal EVD (Fig. 3B and C). General signatures of EBOV infection include increases in plasma diacylglycerides (DG) and diacylglycerophosphoethanolamine (PE) species, and decreases in diacylglycerophosphocholine (PC), diacylglycerophosphoinositol (PI), monoacylglycerophosphoethanolamine (LPE), and PE plasmalogen (PEP) species (Fig. 3B and C). In fatalities, diacylglycerophosphoglycerol (PG) and a subset of monoacylglycerophosphoserine (PS) and ceramide (Cer) species were significantly increased compared with survivors, whereas monoacylglycerophosphocholine (LPC) species were significantly decreased (Fig. 3C). Cholesterol ester (CE) and monoacylglycerolipid (MG) were also significantly decreased and increased, respectively, in EVD fatalities (Fig. 3C); however, only a single significantly altered lipid was identified for each. Although substantial additional work will be required to dissect the implications of these lipid signatures for EVD pathogenesis, it is worth noting the following: (i) PS is a major component of procoagulant platelet microparticles (Lacroix and Dignat-George, 2012), which are secreted by activated platelets, and increased PS plasma levels in EVD fatalities suggests increased platelet activation in these patients; and (ii) reduced plasma LPC is a correlate of inflammatory state and septic shock (Park et al., 2014), and the significant reduction of LPC in fatalities is consistent with immune and septic shock-related pathophysiology.
Macrophages and neutrophils are strongly activated in EVD
We next explored our plasma proteomics dataset (Fig. S2C and Table S2) by using pathway enrichment analysis (Table S3). Enrichment of the ‘Cell Adhesion Molecules’ pathway (KEGG hsa04514) was a general characteristic of EBOV infection, since this pathway was highly enriched in both survivors and fatalities compared with healthy controls (Fig. 4A). Upregulated components included multiple class I major histocompatibility complex antigens (see Table S2) and adhesion molecules found on endothelial cells (e.g., VCAM1), macrophages (e.g., CD14, CD163, FCGR3A/CD16), and neutrophils (FCGR3B/CD16b) (Fig. 4B). Increases in soluble CD14 and CD163 are indicators of macrophage activation and immunological stress in diseases marked by immunopathology, including sepsis (Etzerodt and Moestrup, 2013; Sandquist and Wong, 2014). Moreover, ectodomain shedding of FCGR3A and FCGR3B is stimulated by activation and phagocytosis in macrophages and neutrophils, respectively (Wang et al., 2013; Webster et al., 2006). These data strongly implicate activated macrophages and neutrophils in the host response to EVD, suggest that activation may be more pronounced in fatalities, and are consistent with metabolomics and lipidomics signatures that predict immune cell activation and septic-like immune responses in EVD fatalities (Fig. 3). Interestingly, we also observed an increase in soluble VSIG4 – a co-stimulatory molecule expressed on macrophages – only in fatalities (Fig. 4B). Since VSIG4 is a potent inhibitor of T cell activation (Vogt et al., 2006), we suggest that VSIG4 shedding could be one mechanism by which T cell activation is impaired in fatal EVD (Ruibal et al., 2016).
Figure 4. EVD signatures in the plasma proteome.
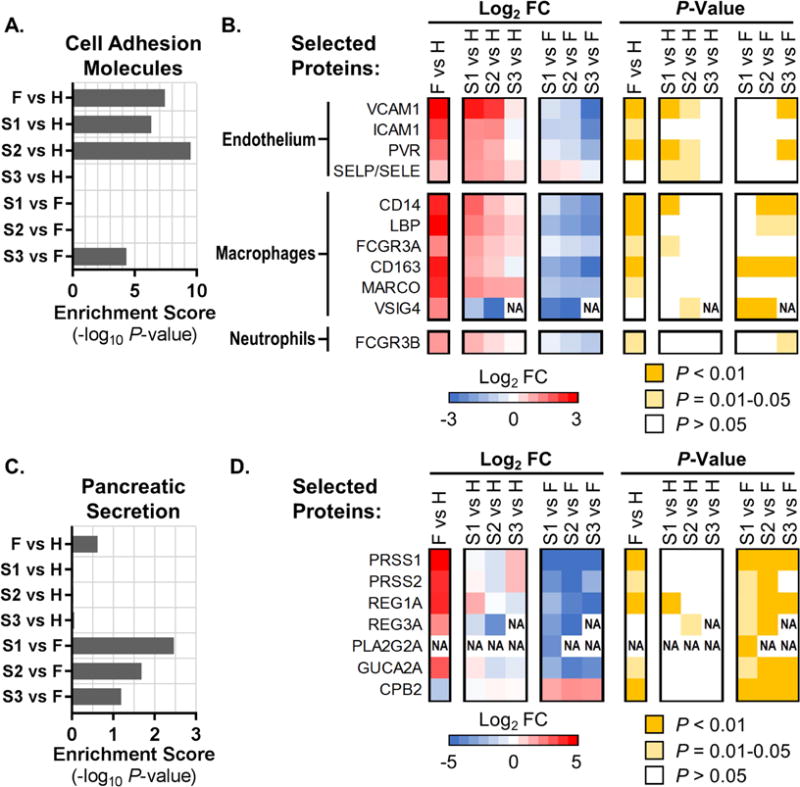
Pathway enrichments and heat maps showing average pathway protein expression levels (log2 fold change, FC) and associated P-values for ‘Cell Adhesion Molecules’ (KEGG pathway hsa04514; panels (A) and (B)), and ‘Pancreatic Secretion’ (KEGG pathway hsa04972; panels (C) and (D)). (A and C) KEGG pathway enrichment scores as the negative log10 of the enrichment P-value for EVD patients (fatalities, ‘F’; survivors’ first, second, and third samples, ‘S1’, ‘S2’, and ‘S3’) compared with healthy controls (H); or for S1/S2/S3 vs. F comparisons. (B and D) Expression levels and associated P-values for a selected set of plasma proteins (indicated by Entrez Gene Official Symbols) included in the respective KEGG pathways (data are represented in the same way as in Fig. 2B). ‘NA’ in heat maps indicates that FC and P-values were not calculated due to an insufficient number of values in one of the conditions. In panel (B), ‘SELP/SELE’ indicates a protein profile that cannot be assigned to one of these proteins due to their high homology. See also Table S2 and Table S3.
Pancreatic enzyme leakage is associated with fatal EVD
The ‘Pancreatic Secretion’ pathway (KEGG hsa04972) was also significantly enriched in the proteomics comparison between EVD survivors and fatalities (Fig. 4C). Indeed, the levels of several factors secreted by pancreatic acinar cells – including pancreatic trypsins (PRSS1 and PRSS2), lipase (PLA2G2A) and regenerating family proteins REG1A and REG3A – were strongly increased in the plasma of EVD fatalities (Fig. 4D). While some evidence suggests that REG1A is a biomarker of sepsis (Llewelyn et al., 2013) and/or intestinal damage mediated by inflammation (Vives-Pi et al., 2013), increased circulating levels of pancreatic trypsins and lipase are associated with acute pancreatitis (Malfertheiner and Kemmer, 1991), which can be caused by viral infection. Symptoms of acute pancreatitis include severe abdominal pain, and complications may include acute respiratory distress, disseminated intravascular coagulopathy and kidney failure (Agarwal and Pitchumoni, 1993), all of which have been observed in patients with severe EVD (Leligdowicz et al., 2016). These observations suggest a potential role for EBOV-induced pancreatic tissue damage and the consequential effects of systemic release of pancreatic enzymes in EVD pathogenesis.
PBMC transcriptional signatures revealed by network analysis
Analysis of plasma ‘omics datasets suggested that strong innate immune cell activation and sepsis-like inflammatory responses are associated with EVD (Figs. 3 and 4). Next, we examined whether our PBMC transcriptome dataset (Fig. S2C and Table S4) would substantiate these findings. We employed the Multiscale Embedded Gene Co-Expression Network Analysis (MEGENA) approach (Song and Zhang, 2015), which identifies modules of co-expressed genes from large transcriptional datasets. MEGENA identified 27 first-level ‘parent’ modules comprising the EVD co-expression network (including both differentially expressed transcripts and transcripts that remain unchanged after infection; Fig. S3A and Table S5), of which 12 exhibited significant overlap (q < 0.05) with increased or decreased transcripts from one or more of the following comparisons: fatalities vs. healthy controls (F vs. H), survivors (sample 1) vs. healthy controls (S1 vs. H), or survivors (sample 2) vs. healthy controls (S2 vs. H) (Fig. S3B; module overlap calculations were determined for transcripts exhibiting a q-value < 0.01 for EVD vs. healthy control comparisons; and for comparisons in which fewer than 50 transcripts exhibited a q-value < 0.01 – that is, for S1/S2/S3 vs. F and S3 vs. H – module overlap scores are not shown). Overall, more modules were enriched in the F vs. H condition compared with the S1/S2 vs. H conditions, suggesting that coordinated transcriptional programs in PBMCs are more strongly activated in fatal EVD. Moreover, fewer enriched modules were observed in the S2 vs. H comparison relative to S1 vs. H, most likely indicating recovery.
Module 2 (which is related to mitosis and includes the MKI67/Ki-67 proliferation marker) was strongly up-regulated in all comparisons (Fig. S3B, Fig. S4A and Table S5), suggesting that a subset of PBMCs undergo expansion and/or activation during EVD. The Module 2 signature is not likely associated with lymphocytes, because lymphocyte-specific markers exhibited either no change in expression levels in any condition (e.g., CD8A, CD8B, CD19) or a mixed pattern between EVD survivors and fatalities (e.g., CD4) (Table S4). Rather, because Module 2 includes multiple mitotic regulators (Fig. S4B) that are known to be transcriptionally associated with monocyte-to-macrophage differentiation (Martinez et al., 2006), monocyte differentiation may be a common feature of the host response to EVD, regardless of outcome.
Myeloid cells express antiviral genes and may be sensitized to necroptosis in fatal EVD
Three PBMC transcriptional modules (Modules 3, 18, and 27) exhibited unique enrichment for increased transcripts in fatalities compared with healthy controls. Module 3 included 197 transcripts that were significantly increased (q < 0.01) and 20 transcripts that were significantly decreased in the F vs. H comparison (Fig. 5A). In contrast, fewer transcripts were significantly altered and were changed to a lesser degree in EVD survivor (S1/S2/S3 vs. H) comparisons (Fig. 5A). This module is enriched for genes involved in innate immune responses (Table S5). In addition, we identified significant changes in transcripts for myeloid cell chemokine receptors (CCR1 and CCRL2) and other macrophage-specific receptors (FCGR3A/CD16, FCGR1A, and MS4A7), suggesting a strong contribution of cells of myeloid origin (see Tables S4 and S5).
Figure 5. EBOV infection strongly induces antiviral and anti-apoptotic gene expression in PBMCs.
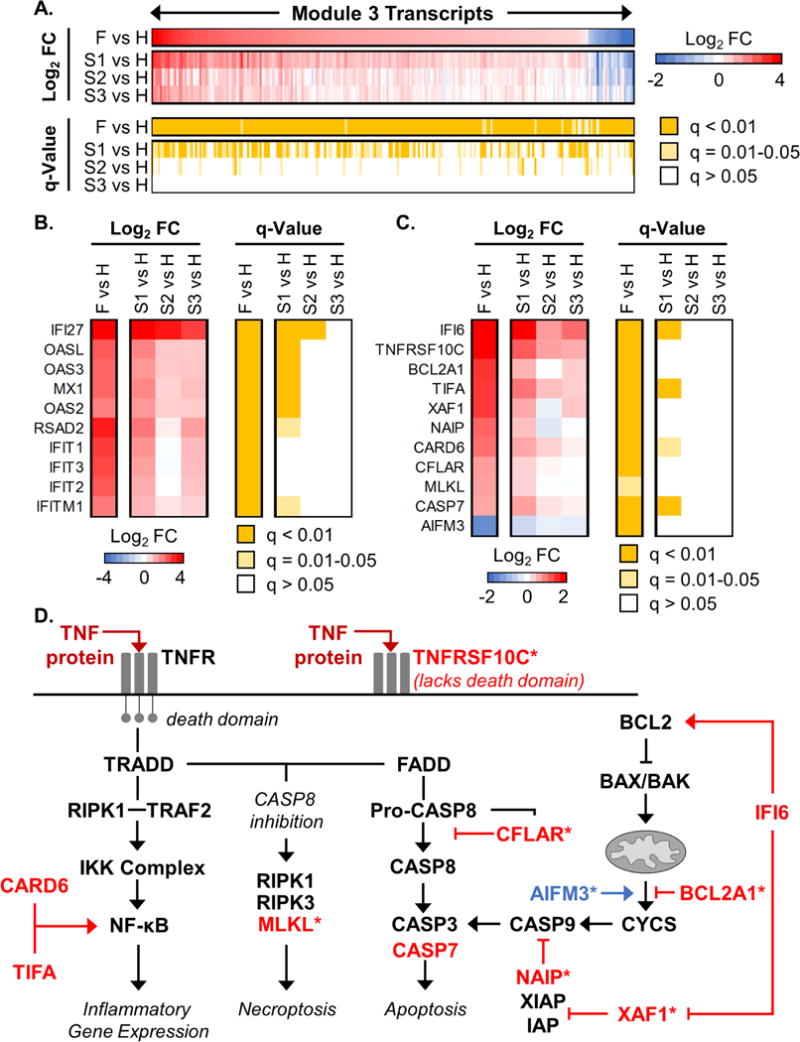
Shown are data associated with MEGENA Module 3, derived from PBMC transcriptome data. (A) Average PBMC expression levels and associated q-values for all transcripts exhibiting significantly altered expression (q-value < 0.01) in at least one condition when comparing EVD patients (fatalities, ‘F’; survivors’ first, second, and third samples, ‘S1’, ‘S2’, and ‘S3’) to healthy controls (H). For the transcriptome heat map, values are displayed as the direction of expression in the EVD patient. Columns show expression and q-values for individual transcripts (IDs not shown) and rows represent different comparison groups. FC, fold change. (B and C) Transcript level and q-value heat maps for a subset of Module 3 interferon-stimulated gene transcripts (B) and apoptosis-associated transcripts (C); data are represented in the same way as in Fig. 2B. Individual transcripts are represented as Entrez Gene Official Symbols. (D) Molecules involved in TNF receptor signaling leading to activation of inflammatory gene expression, apoptosis, and necroptosis, as well as those contributing to apoptosis though the mitochondrial pathway. Transcripts that are significantly altered in Module 3 (q-value < 0.05) are shown in red or blue text, indicating increased or decreased transcript expression in EVD, respectively. The TNF protein, which is elevated in the plasma of both EVD survivors and fatalities, is indicated by dark red text. Asterisks (*) indicate transcripts that were significantly changed only in EVD fatalities. See also Figure S3, Figure S4, Table S4, and Table S5.
Although previous reports have indicated that EBOV infection impairs antiviral signaling in APCs (Messaoudi et al., 2015), we observed up-regulation of both type I and type II IFN-stimulated genes in Module 3 (Fig. 5B), suggesting that antiviral signaling is at least partially intact in myeloid cells during human EBOV infection in vivo. This observation is consistent with the notion that myeloid cells are strongly activated in EVD, as suggested by shedding of macrophage-specific cell adhesion molecules (Fig. 4) and metabolic signatures of immune cell activation and systemic oxidative damage (Fig. 3).
Interestingly, Module 3 also included multiple regulators of apoptosis whose expression may alter apoptotic activity through both extrinsic (TNF-mediated) and intrinsic (mitochondria-mediated) mechanisms (i.e., CFLAR, BCL2A1, AIFM3, NAIP, XAF1, and IFI6; Fig. 5C and D). When extrinsic apoptotic signaling is impaired during TNF receptor activation, cells may be sensitized to necroptosis-dependent cell death regulated by the MLKL protein (Silke et al., 2015). Accordingly, we suggest that myeloid cells may be sensitized to necroptosis in fatal EVD, due to abundant levels of TNF in circulation (Fig. 2B), increased expression of the pro-caspase 8 cleavage inhibitor CFLAR (Fig. 5C and D), and increased expression of the necroptosis regulator MLKL (Fig. 5C and D). The effects of necroptosis on disease pathology are context-specific (Silke et al., 2015), and how necroptosis contributes to EVD pathogenesis remains to be explored.
EBOV infection may induce abnormal neutrophils
Module 18 exhibited the strongest unique enrichment for the F vs. H comparison group (Fig. S3B). Of the 101 Module 18 transcripts that were differentially expressed in EVD fatalities (FC > 1.5, q-value < 0.01), only 16 were also differentially expressed in the first sample from EVD survivors (S1 vs. H), but exhibited reduced fold changes (Fig. 6A; Fig. S5A). Moreover, few differentially expressed transcripts were observed in samples from EVD survivors at later time points, indicating that the Module 18 transcriptional response was resolved during recovery (Fig. 6A). Module 27 exhibited expression dynamics similar to Module 18 (Fig. S5A and B). Thus, Modules 18 and 27 are more strongly represented among EVD fatalities, suggesting they contribute to EVD pathogenicity.
Figure 6. Neutrophils may play a key role in EBOV pathogenicity.
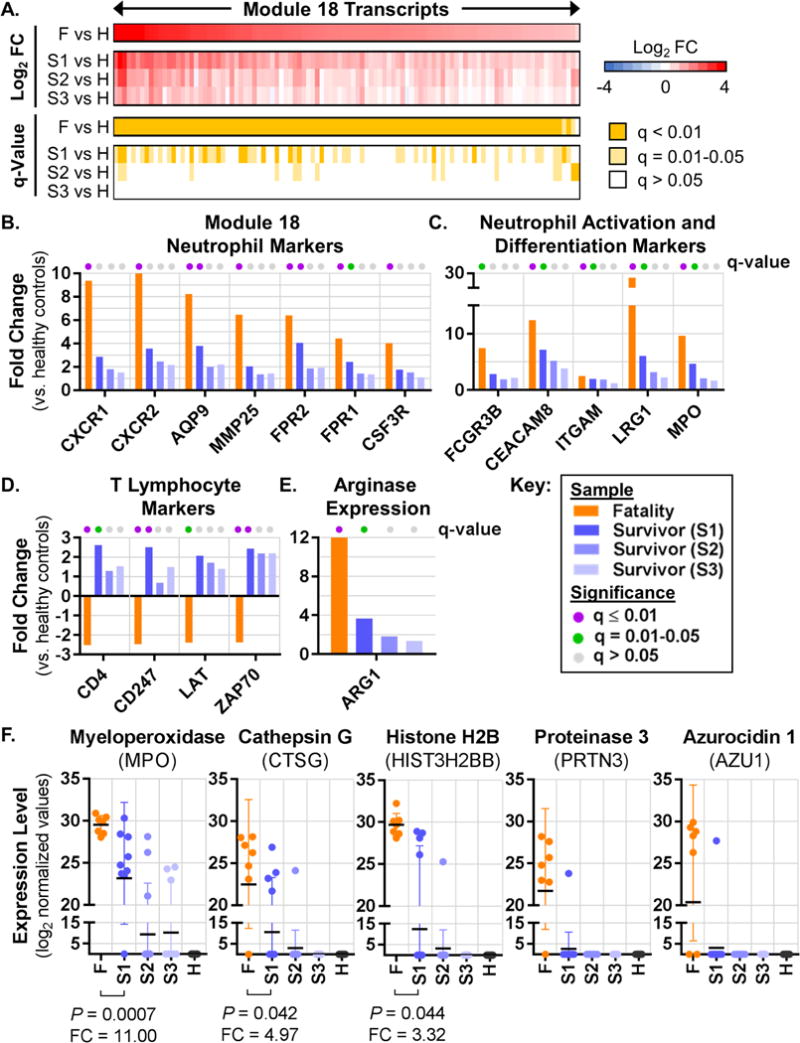
(A) Average PBMC transcript expression levels and associated q-values for all Module 18 transcripts that were significantly altered (q < 0.01) in at least one condition when comparing EVD patients with healthy controls (data are represented in the same way as in Fig. 5A). FC, fold change. (B–E) Average expression levels for individual transcripts in EVD vs. healthy control comparisons, with q-values indicated by the colored dots at the top of each bar. Panel (B) shows neutrophil markers from Module 18, panel (C) shows neutrophil activation and differentiation markers from Modules 18 and 27, panel (D) shows T lymphocyte markers from other modules, and panel (E) shows ARG1 expression from Module 27. (F) Normalized protein expression values for each sample from individual EVD patients or healthy controls. FC and P-values for the S1 vs. F comparison for MPO, CTSG and HIST3H2BB are shown below each plot; FC and P-values were not calculated for PRTN3 or AZU1 due to an insufficient number of samples in the S1 group. All transcript and protein IDs are given as Entrez Gene Official Symbols. See also Figure S3, Figure S5, Table S4, and Table S5.
Module 18 is functionally enriched for plasma membrane receptors and G-protein coupled receptor signaling (Table S5). Intriguingly, many genes in this module are neutrophil-associated, including CSF3R (which controls neutrophil production, differentiation, and function), receptors that regulate neutrophil chemotaxis (CXCR1, CXCR2, FPR1, FPR2, and AQP9), and a neutrophil-specific matrix metalloprotease (MMP25) (Fig. 6B). Module 27 contains neutrophil subset-specific markers (CD177 and OLFM4), neutrophil collagenase (MMP8), and other transcriptional markers of granulocytes in blood (IL1RN and GCA) (Palmer et al., 2006) (Fig. S5C), as well as multiple transcripts encoding neutrophil granule proteins (e.g., CHIT, LTF, and BPI; see Tables S4 and S5). Together, these observations suggest that two transcriptional modules that are uniquely enriched in EVD fatalities are associated with transcriptional programs expressed in neutrophils, implying a potential role for neutrophils in regulating the outcome of EVD in humans.
In normal physiological states, neutrophils do not fractionate with PBMCs in density gradient preparations of blood, due to their higher density relative to mononuclear cells. However, low density neutrophils (LDNs; also known as monocyte-derived suppressor cells [MDSCs]) exhibiting pathologic functions have been identified in PBMC fractions from patients with systemic lupus erythematosus (SLE) (Smith and Kaplan, 2015) and sepsis (Darcy et al., 2014), among other conditions. A general characteristic of LDNs is the concomitant expression of neutrophil activation markers (suggesting a mature phenotype) and neutrophil differentiation markers (which are normally expressed in immature granulocytes) (Carmona-Rivera and Kaplan, 2013). In our neutrophil transcriptional modules, we observed strongly increased expression of neutrophil activation markers (FCGR3B/CD16b, CEACAM8/CD66b, and ITGAM/CD11b) and neutrophil azurophilic granule proteins that are typically expressed during granulopoiesis (MPO and LRG1) (Fig. 6C). These observations are consistent with the mixed maturity phenotype observed for LDNs in other systems, and support the notion that LDNs could develop during EVD.
A potential role for abnormal neutrophils in T lymphocyte dysregulation in EVD
In sepsis, T lymphocytes exhibit impaired proliferation and increased apoptosis (Ward and Bosmann, 2012), similar to EVD patients. Sepsis-derived LDNs (i.e., ‘neutrophil MDSCs’) that express arginase (ARG1) suppress T lymphocyte proliferation by catalyzing breakdown of arginine (an essential metabolite for cell cycle progression) and reducing T cell receptor zeta chain (TCRζ/CD247) expression (Darcy et al., 2014). While a recent study has implicated increased expression of immunosuppressive molecules (CTLA4 and PD-1/PDCD1) in the dysregulation of T lymphocyte function during EVD (Ruibal et al., 2016), the mechanisms that control T lymphocyte survival and proliferation have not been fully elucidated. We speculate that LDNs that emerge during EVD could contribute to reductions in T lymphocyte numbers in a manner similar to what has been described in sepsis, on the basis of the following observations: (i) CD4 transcript expression (a proxy for CD4 T lymphocyte levels) exhibited significantly reduced expression in EVD fatalities, but was significantly increased in Sample 1 of EVD survivors (Fig. 6D); (ii) TCRζ/CD247 and two other T lymphocyte signaling molecule transcripts (LAT and ZAP70) exhibited expression profiles that mirrored that of CD4 (Fig. 6D); (iii) the ARG1 transcript is a component of Module 27, one of the neutrophil-associated modules, and is highly induced in fatal EVD (Fig. 6E); and (iv) levels of urea, a metabolic by-product of arginine breakdown by ARG1, were significantly increased in the plasma of EVD fatalities relative to those in the first sample from EVD survivors (log2 FC = 2.5, P = 0.056; Table S2). These observations are consistent with reduced circulating CD4 T lymphocytes in fatal EVD that occur as a result of neutrophil-mediated, ARG1-dependent arginase consumption and down-regulation of TCRζ/CD247 expression.
Neutrophils may enhance EVD-associated tissue damage
Another well-established feature of LDNs is their increased capacity to release neutrophil extracellular traps (NETs) (Kaplan and Radic, 2012), which are composed of cellular DNA, core histones, and azurophilic granule-derived antimicrobial proteins. NETs contribute to host defense by trapping microorganisms and limiting their spread, and by serving as a scaffold for thrombus formation on endothelium. However, excess NET formation (or impaired NET clearance) can lead to tissue damage and coagulopathies, and has been implicated in the pathogenesis of highly inflammatory conditions (e.g., sepsis) and other viral hemorrhagic fevers (Kaplan and Radic, 2012; Raftery et al., 2014). Consistent with the possibility of NET formation in EVD, core histone proteins (e.g., HIST3H2BB), and azurophilic granule proteins (MPO, CTSG, PRTN3, and AZU1) are present in the plasma of EVD patients but not that of healthy individuals (Fig. 6F). All five proteins were detected in higher proportions of patients in the fatality group; moreover, MPO, CTSG, and HISTH2BB protein levels were significantly increased in samples from fatalities (Fig. 6F; PRTN3 and AZU1 ratios were not calculated since each was detected only in a single survivor sample). These observations suggest that mechanisms promoting the release of NET-associated proteins, which have significant potential to cause systemic tissue damage when present at excess levels, are activated by EVD and up-regulated in fatal EVD. It is important to note that mechanisms other than NET formation could be responsible for increasing plasma levels of histones and neutrophil granule proteins in EVD patients (e.g., necrotic cell death). However, regardless of the mechanism, neutrophils are likely to be involved, given that MPO, CTSG, PRTN3 and AZU1 are predominantly expressed in this cell type.
Altogether, these observations suggest that neutrophils – specifically, LDN – contribute to EVD pathogenesis by dysregulating adaptive immunity and promoting tissue damage. However, definitive identification of LDN populations in EVD patients will require PBMC surface staining with LDN-specific markers to establish an increase in the absolute proportion of this cell type compared to healthy controls. This confirmation, in combination with the results presented here, would strongly point to LDN activity as a unifying explanation for critical correlates of fatal EVD.
Comparison with other human transcriptome datasets
We directly compared our PBMC transcriptome dataset to that of humans with sepsis (Severino et al., 2014), and we identified a common T-lymphocyte-associated signature between EVD fatalities and patients with septic shock (i.e., PD-1 signaling; Fig. S6A and Table S5). We also compared our PBMC transcriptomics data to that of a longitudinal study of a single EVD survivor (Kash et al., 2017), and identified many common signatures, as well as signatures that were unique to each dataset (for examples, see Fig. S6B; also see Table S5). These findings further emphasize similarities between EVD and sepsis-related disease, demonstrate that our data faithfully represent host responses in humans with EVD, and highlight EVD host response signatures that may vary depending on host genetics or environment.
Biomarkers that predict EVD fatality
Finally, we used our ‘omics, cytokine, and clinical datasets to perform biomarker discovery and assessment. We employed a nested, leave-one-out cross validation (LOOCV) design on each dataset individually and for all datasets combined (Fig. S7A), focusing on markers that predict survival early in EVD. Lipids, metabolites, cytokines (ELISA-based), and clinical variables (primarily driven by viral load) appear to provide the strongest predictive power of survival at day 8 from symptom onset (Fig. S7B). The metabolite L-threonine and the vitamin-D binding protein (VTDB) perfectly stratify patients by outcome, exhibit better predictive ability than viral load, and are striking not only for their stratification of outcome, but also because their expression is relatively stable in survivors over time (Fig. 7A). Because L-threonine and VTDB plasma levels can predict outcomes several days prior to death (death occurs ~11–12 days from symptom onset, on average) (Velasquez et al., 2015) (Fig. 7A), these markers may be informative about discrete characteristics of infections leading to death, in contrast to events that return to baseline over time. Other individual markers are nearly as distinctive. While our results are promising, follow-up studies will be necessary to better estimate the true precision of the putative biomarkers identified here.
Figure 7. Biomarkers that may predict outcomes of EVD.
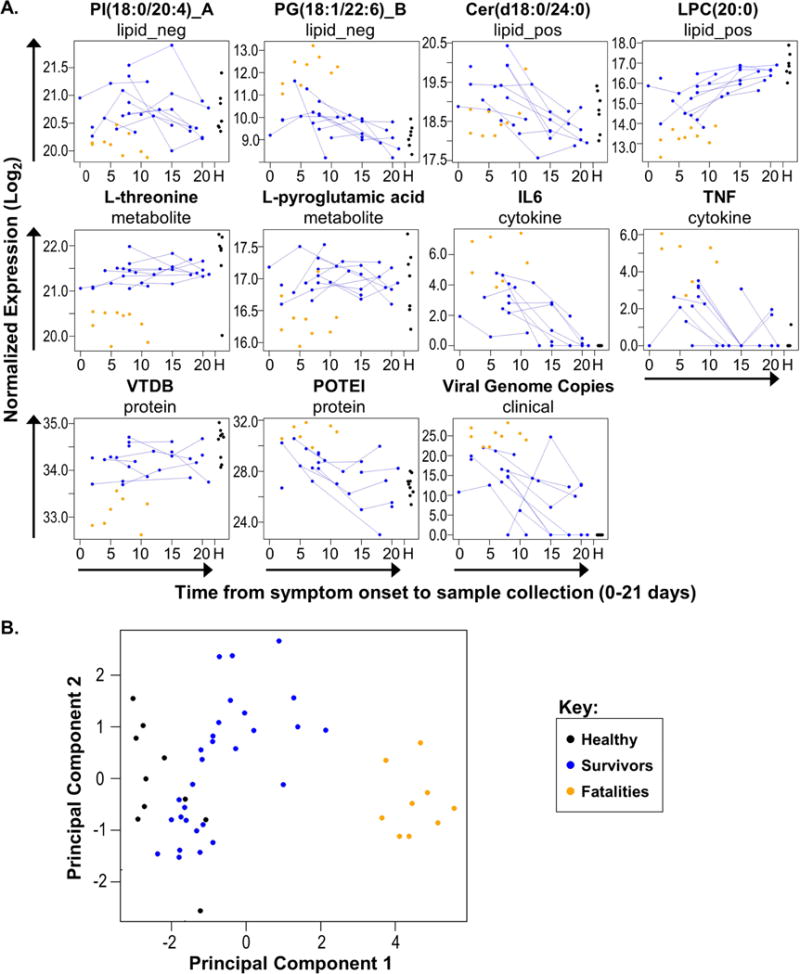
(A) Plots for 11 candidate biomarkers. Log2 normalized expression for each feature is plotted against the days from onset on which the corresponding sample was collected. Within each plot, each dot represents a single patient sample, colored by study group. Longitudinal samples from the same patient are connected with lines. Samples from healthy patients are shown together at the far right of each plot (indicated by an ‘H’ on the X-axis). Above each panel, the name of the candidate biomarker is given, along with the dataset from which it was derived. ‘lipid_neg’ and ‘lipid_pos’ indicate lipids identified by negative ionization or positive ionization LC-MS/MS, respectively. ‘_A’ or ‘_B’ designations indicate that the lipid has an isomer that differs only in structural arrangement. Cytokine (IL6 and TNF) data are derived from plasma ELISA analysis. (B) Patient sample scores from probabilistic principal components analysis using the 11 candidate biomarkers shown in panel (A). Samples assessed in this analysis were not included in the biomarker prediction analysis pipeline due to missing data. Each dot represents an individual patient sample. See also Figure S7 and Table S6.
During virus outbreaks, adequate blood volumes are often difficult to obtain, assays for some biomolecules may fail, and clinical data may be incomplete. Using 11 distinct biomarkers (predicted by our LCOOV analysis), we predicted outcomes for samples that were not included in our biomarker prediction analysis pipeline due to lack of data from one or more analysis platform (Fig. 7B; also see Table S6). These results suggest that a combination of redundant biomarkers may be useful to ensure accurate outcome prediction in real-life scenarios, which in turn, may improve outcomes for the most at-risk patients.
DISCUSSION
The overarching goals of this work were to use multi-platform ‘omics analysis to develop hypotheses of EVD pathogenesis, generate a high-quality resource of datasets for the research community, and provide a standard for comparison with datasets generated in animal models of EVD pathogenesis or other human disease systems. Our analysis of our ‘omics datasets established their quality and revealed insights into EVD pathogenesis that need to be further explored: (i) the role of pancreas-associated enzymes in EVD-induced tissue damage; (ii) the role of necroptosis signaling in myeloid cell functions during EBOV infection; and (iii) the contribution of neutrophils to EVD-induced dysregulation of adaptive immunity and tissue damage. In addition, we demonstrated that our datasets are a valuable resource for identifying biomarkers that predict EVD outcomes, which may be useful for developing diagnostic assays. We further uncovered commonalities between molecular signatures of EVD and sepsis, supporting the concept that overlapping mechanisms regulate outcomes related to these conditions, and suggesting that sepsis-related research could be harnessed to develop new hypotheses regarding mechanisms of EVD pathogenesis and/or potential treatment options.
It is important to emphasize that PBMC samples used to generate our transcriptomics dataset consisted of a mixture of immune cells, and that infection-induced changes in transcript expression levels could reflect a combination of changes in immune cell populations and differences in immune cell activation. In our high containment field laboratory, we could not isolate individual immune cell populations for transcriptomics profiling or quantify immune cell populations to correlate with transcript expression profiles. Nonetheless, our network analysis approach separated PBMC transcriptomics data into modules of co-expressed transcripts that included well-established markers of different immune cell types, which enabled us to infer immune cell population-specific contributions to EVD pathogenicity. Additional studies will be required to precisely gauge the impact of EVD-driven changes in peripheral immune cell populations on PBMC transcript expression, particularly for neutrophils, which are markedly increased in the periphery of some EVD patients (Hunt et al., 2015) but probably accumulate at similar levels in survivors and fatalities (Ludtke et al., 2016).
Another challenge that needs to be addressed in future efforts is how to best integrate PBMC transcriptomics data, which reflects systemic alterations in immune cell function during EVD; and plasma proteomics, lipidomics, and metabolomics data, which reflect the aggregate responses of various cell types and organ systems that are affected by EVD. Such integration will require increased knowledge of host responses in specific infected cells/organs and factors that are secreted due to activation or induction of cell death pathways by EBOV infection.
In summary, we have demonstrated the value of a multi-platform ‘omics approach, which provides a more complete context for understanding host response contributions to disease pathology relative to individual platforms. Ultimately, this work expands our knowledge of human EVD pathogenesis and may facilitate future development of prophylactic or therapeutic countermeasures. Moreover, we suggest that multi-platform ‘omics analysis should be applied to samples from other human disease systems to facilitate a clearer understanding of disease pathophysiology and to accelerate identification of disease biomarkers.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be addressed by the Lead Contact, Yoshihiro Kawaoka (yoshihiro.kawaoka@wisc.edu).
Experimental Models and Subject Details
Ethics and human subjects
All work performed in this study was approved by the Sierra Leone Ethics and Scientific Review Committee, the Research Ethics Review Committee of the Institute of Medical Science at the University of Tokyo, and the University of Wisconsin (UW)-Madison Health Sciences Institutional Review Board (IRB). IRB approval was also obtained at Icahn School of Medicine at Mount Sinai and at Pacific Northwest National Laboratory prior to any sample shipments to these institutions. EBOV-positive subjects – as determined by quantitative real-time reverse transcriptase polymerase chain reaction (qRT-PCR) of whole blood specimens (performed by a World Health Organization Collaborating Center or Chinese or British laboratories in Freetown, Sierra Leone) – were recruited from three Ebola Treatment Centers (ETCs) in Freetown, Sierra Leone (located at the Joint Military Unit [JMU] 34th Regimental Military Hospital at Wilberforce; the Hastings Police Training School 1; or the Police Training School 2) from February through May of 2015. Healthy subjects were recruited from healthcare workers and laboratory technicians at JMU during the same time period, none of whom had previously experienced EBOV disease. Three of the healthy subjects provided two samples each; samples from these patients were not treated as longitudinal because (i) the time between the first and second sample collection was approximately 4 months, and (ii) samples were correlated no more within the same patient than between patients. Consent was obtained from all subjects prior to enrollment, and for children under the age of 18, consent was provided by the child’s parent or guardian. Basic demographic data (i.e., sex, age, time from symptom onset to admission to an ETC, and disease type [i.e., wet vs. dry] upon admission) was collected from each patient at the time of enrollment (Table S1). None of the patients involved in the study described herein received experimental treatments for EBOV infection (e.g., whole blood, plasma or serum from survivors, antibody cocktails, or antiviral drugs). All patients received supportive care.
Patient samples
EBOV-positive patients were enrolled in the study after diagnosis and admission to an ETC, and an initial blood sample (≤ 3 ml) was collected. Additional (serial) blood samples were collected from most EBOV survivors over the course of their disease at intervals of ≥ 5 days. Because patients that succumbed to EBOV infection typically died within 3–4 days from admission to an ETC, we were not able to collect serial samples from these patients. Single samples were collected from healthy patients recruited from healthcare workers and laboratory personnel at one of the ETC sites. All samples were collected using potassium-EDTA blood collection tubes. All samples used in the current study are described in Table S1.
Virus strains
Patients enrolled in this study naturally acquired infection by the West African Zaire EBOV, Makona strain. To validate EBOV inactivation by chemical extraction procedures, we used EbolaΔVP30 virus (based on the Zaire EBOV Mayinga strain), which encodes a reporter gene in the place of an essential transcription factor (VP30) and can only grow in cells expressing the VP30 protein (Halfmann et al., 2008).
Commercially obtained human blood samples
For EBOV inactivation studies, human (male, African American) blood samples collected in potassium EDTA-coated tubes were obtained from a commercial vendor (Zen-Bio, Inc.).
Cell lines
We used the Vero-VP30 cell line – a derivative of the Vero cell line (female African green monkey [Cercopithecus aethiops] kidney epithelial cells) that constitutively expresses the Zaire EBOV (Mayinga strain) VP30 protein (Halfmann et al., 2008) – to generate EbolaΔVP30 virus stocks, and to evaluate chemically-treated samples for the presence of live EBOV. Vero-VP30 cells were maintained in Minimum Essential Medium (MEM; Gibco) containing 10% fetal bovine serum (FBS; Sigma Aldrich) and a 1X penicillin/streptomycin (P/S) cocktail (Gibco) at 37°C in a humidified atmosphere of 5% CO2. Vero-VP30 cells have not been authenticated through DNA analysis or karyotyping. Liquid nitrogen stocks are mycoplasma-free (as determined by a polymerase chain reaction [PCR]-based assay), and growing cultures were regularly tested for mycoplasma contamination and discarded immediately if contamination was identified.
Method Details
Biosafety
Blood sample collection was performed only by trained technical personnel, and samples were immediately transported to a research laboratory at the JMU site for processing. At the JMU research laboratory, samples containing EBOV were processed using Rapid Containment Kits (RCKs, Germfree); that is, portable, battery-operated, double HEPA-filtered, negative-pressure field laboratory containment units. Only personnel with extensive training and experience with biocontainment procedures engaged in the processing of EBOV-positive blood samples. Personnel wore personal protective equipment while handling EBOV-positive samples in RCKs, including designated scrubs and shoes, full-body gowns, gloves, hairnets, N95 masks, and eye protection. Standard operating procedures (SOPs) for processing EBOV-positive blood samples were reviewed and approved by the UW-Madison Environmental Health and Safety Department.
Following extraction and inactivation by use of various protocols (additional information below), samples were shipped from Freetown, Sierra Leone, to the UW-Madison under export permits provided by the Pharmacy Board of Sierra Leone and with the approval of the UW-Madison Environmental Health and Safety Department. Some samples were subsequently shipped to Pacific Northwest National Laboratory (PNNL) or to the Icahn School of Medicine at Mount Sinai (ISMMS) for further analyses. Prior to any shipments within the United States, all extraction/inactivation SOPs and supporting data were reviewed and approved by the United States Centers for Disease Control and the UW-Madison Environmental Health and Safety Department, and relevant SOPs and supporting data were further reviewed and approved by receiver institutions.
Blood processing and PBMC isolation
Within 3 hours of collection, blood samples were processed to separate PBMCs and plasma using Ficoll-Paque PLUS (GE Healthcare). Briefly, blood was mixed 1:1 with sterile 1× phosphate buffered saline (PBS), layered over 3 ml of Ficoll-Paque in a 15-ml SepMate tube (STEMCELL Technologies), and centrifuged for 10 minutes at 1,200 × g. Following centrifugation, plasma was harvested in 2–4 aliquots of 0.5 to 1 ml and immediately frozen at −80°C, and PBMCs were collected and washed one time with sterile 1× PBS. After washing, the PBMC pellet was suspended to homogeneity in 1 ml of TRIzol reagent (Life Technologies), allowed to incubate for 10 minutes at room temperature, and subsequently frozen at −80°C. Frozen TRIzol ext racts were shipped to the UW-Madison.
Total RNA extraction from PBMC
After shipment to UW-Madison, inactivated TRIzol PBMC samples were thawed and passed through a QiaShredder column (Qiagen). QiaShredder eluates were mixed with 200 μl of chloroform, vortexed vigorously, and incubated on ice for 10 minutes. Subsequently, phases were separated by centrifugation (12,000 × g for 15 minutes at 4°C), and the upper phase was collect ed and used for total RNA extraction with miRNEasy columns (Qiagen) according to the manufacturer’s instructions. An aliquot of the extracted total RNA was reserved for qRT-PCR analysis of viral RNA and the remainder was shipped to ISMMS for RNA sequencing (RNA-seq) analysis.
PBMC RNA-seq analysis
Total RNA from PBMC samples was DNase treated with 1 U of Baseline Zero DNase (Epicentre) at 37°C for 30 minu tes, cleaned with 1.8× volume of AMPureXP beads (Beckman-Coulter), and eluted in nuclease-free water. RNA quality was assessed using an Agilent Bioanalyzer (all samples exhibited RNA integrity numbers > 9) and quantified using the Qubit RNA Broad Range Assay kit (Thermo Fisher). Up to 500 ng of each DNase-treated sample was used for library preparation. Briefly, globin and ribosomal RNAs were depleted using the Globin-Zero Gold rRNA Removal Kit (Illumina) according to the manufacturer’s instructions, and then purified with 1.6× volume of AMPureXP beads. Ribosomal RNA depletion was confirmed by using Agilent Bioanalyzer analysis and noting the absence of ribosomal peaks. Next, 8.5 μl of Elute, Prime, and Fragment Mix from the TruSeq Stranded Total RNA Library Prep Kit (Illumina) were added to each sample, followed by fragmentation at 94°C for 8 minutes to yield a median fragment size distribution of 155 nt and a final library of 309 nt. Libraries were prepared according to the manufacturer’s instructions by using the TruSeq Stranded Total RNA Library Prep Kit, incorporating different barcoded adaptors for each sample and amplifying libraries for 15 cycles. Following final library quality control on the Agilent Bioanalyzer to confirm the expected size distributions, libraries were pooled and sequenced on the Illumina HiSeq 4000 platform in a 100-bp paired-end read run format.
Following de-multiplexing, read sequences were trimmed at the 3′-end after reaching a base with a PHRED quality score lower than 10, or after encountering 15 bases with a PHRED score lower than 28, using custom scripts. Next, 3′ Illumina adapter sequences were removed using cutadapt with a minimal overlap of 6 bp, allowing for an adapter error rate of 15%. Reads less than 50 nt (for paired-end 100 nt reads) in length after quality and adapter trimming were removed from further analysis. Full-length adapter-trimmed reads were mapped to the human (hg38) and viral (EBOV/G3683/KM034562.1) reference genomes using STAR v2.5 with the corresponding gene annotations (Gencode GRCh37/V23 for the human genome), using detection of chimeric alignments with a minimum mapped length and a minimum chimeric overhang junction of 15 nt, and default settings for all other arguments. Total mapped read counts per gene were determined using featureCounts with default settings.
Quantification of EBOV genomic RNA in PBMCs
EBOV was quantified in PBMC RNA by using the Ebola 2014 outbreak genesig qRT-PCR kit (which detects the EBOV Makona strain nucleoprotein gene) and oasig™ one-step qRT-PCR master mix (Primerdesign) according to the manufacturer’s instructions. Genome copy numbers were calculated based on a standard curve generated by using a positive control template (provided in the kit) and were normalized to the amount of input RNA. Cycle threshold (ct) values, total copy number, and copy number per nanogram for each sample are provided in Table S1.
Protein extraction from plasma
The 14 most abundant plasma proteins (albumin, α1-antitrypsin, transferrin, haptoglobin, α2-macroglobulin, α1-acid glycoprotein [orosomucoid], fibrinogen, complement C3, IgG, IgA, IgM, high density lipoprotein [HDL; apolipoproteins A-I and A-II], and low density lipoprotein [LDL; mainly apolipoprotein B]) were simultaneously immunodepleted by using Seppro IgY14 spin columns (Sigma-Aldrich) according to the manufacturer’s instructions. Briefly, 10 μl of plasma (from Ficoll-Paque PLUS preparations, thawed from −80°C) were diluted with 500 μl of 1× d ilution buffer – in duplicate for each sample – and filtered using 0.45-μm Spin-X centrifuge tube filters (Costar). Pre-filtered samples were then mixed with IgY14 beads and rotated end-over-end for 15 minutes. Immunodepleted eluates from IgY14 columns were collected by centrifugation and duplicate samples were combined. IgY14 beads were then washed with 500 μl of 1× dilution buffer, and eluates from this wash step were collected and combined with the original eluates. Immunodepleted eluates (~2 ml per sample) were transferred to Amicon Ultra 4 Centrifugal Filter Units (Millipore) and centrifuged at 3,260 × g for ≥ 36 minutes to concentrate eluates to 150 μl total volume. Concentrated eluates were subsequently mixed with urea to a final concentration of 8 M, incubated at room temperature for 15 minutes, and then frozen at −80°C. Frozen 8 M urea extracts were shipped to the UW-Madison.
Plasma proteomics analysis
Plasma extracted with 8 M urea was shipped to PNNL for proteomics analysis. Dithiothreitol was added to a final concentration of 5 μM, and samples were denatured and reduced by incubating for 1 h at 37°C with 800 rpm constant shaking in Thermomixer R (Eppendorf). Then, iodoacetamide was added to a final concentration of 20 mM, and sample alkylation was carried out for 1 h at 37°C with 800 rpm constant shaking in Thermomixer R. Subsequently, samples were diluted 5-fold with a solution of 50 mM ammonium bicarbonate and 1 mM calcium chloride prior to the addition of sequence-grade trypsin (Promega) at a 1:50 enzyme-to-protein ratio. Enzymatic digestion was carried out overnight (~16 h) at 37°C with 800 rpm constant shaking in Th ermomixer R. Following digestion, peptides were desalted using a 96-well plate C18 SPE (Strata C18-E, Phenomenex), eluted in 600 μl of 80% acetonitrile/0.1% trifluoroacetic acid and lyophilized, and rehydrated in 25 mM ammonium bicarbonate. Concentrations were determined by BCA assay, and samples were normalized to a final peptide concentration of 0.3 μg/μl.
Peptide samples were analyzed by LC-MS/MS. The LC component was a Waters nano-Acquity M-Class dual pumping UPLC system (Milford, MA) configured for on-line trapping of a 5-μL injection at 3 μL/min with reverse direction elution onto the analytical column at 300 nL/min. Columns were packed in-house using 360 μm o.d. fused silica (Polymicro Technologies Inc.) with 1-cm sol-gel frits for media retention (Maiolica et al., 2005) and contained Jupiter C18 media (Phenomenex) in 5 μm particle size for the trapping column (100 μm i.d. × 4cm long) and 3 μm particle size for the analytical column (75 μm i.d. × 70 cm long). Mobile phases consisted of (i) 0.1% formic acid in water; and (ii) 0.1% formic acid in acetonitrile with the following gradient profile (min, %ii): 0, 1; 2, 8; 20, 12; 75, 30; 97, 45; 100, 95; 110, 95; 115, 1; 150, 1.
MS analysis was performed using a Q-Exactive HF mass spectrometer (Thermo Scientific, San Jose, CA) outfitted with a home-made nano-electrospray ionization interface. Electrospray emitters were home-made using 150 μm o.d. × 20 μm i.d. chemically etched fused silica (Kelly et al., 2006). The ion transfer tube temperature and spray voltage were 325° C and 2.2 kV, respectively. Data were collected for 100 min following a 15 min delay from sample injection. FT-MS survey spectra were acquired from 400–2000 m/z at a resolution of 30k (AGC target 3e6), and data-dependent FT-HCD-MS/MS spectra were acquired for the top 12 most abundant ions in each survey spectrum with an isolation window of 2.0 m/z and at a resolution of 15k (AGC target 1e5) using a normalized collision energy of 30 and a 60 sec exclusion time.
LC–MS/MS raw data were converted into dta files using Bioworks Cluster 3.2 (Thermo Fisher Scientific), and the MSGF+ algorithm was used to search MS/MS spectra against the Human Uniprot 2016-04-13 database with 20154 entries, plus Zaire_Ebola virus 2014-07-10 sequence containing 7 viral protein entries. The key search parameters used were ±20 ppm tolerance for precursor ion masses, +2.5 Da and −1.5 Da window on fragment ion mass tolerances, MSGF+ high resolution HCD scoring model, no limit on missed cleavages but a maximum peptide length of 50 residues, partial or fully tryptic search, variable oxidation of methionine (15.9949 Da), and fixed alkylation of cysteine (carbamidomethyl, 57.0215 Da). The decoy database searching methodology was used to control the false discovery rate (FDR) at the unique peptide level to <1% and subsequent protein level to <0.5% (% FDR = ((reverse identifications*2)/total identifications)*100). Identification and quantification of the detected peptide peaks were performed by using the label-free Accurate Mass and Time (AMT) tag approach (Zimmer et al., 2006). Briefly, an AMT tag database was created from the MS/MS results, and in-house developed informatics tools (including algorithms for peak-picking and determining isotopic distributions and charge states, which are publicly available at ncrr.pnnl.gov/software) were used to process the LC-MS data and correlate the resulting LC-MS features to an AMT tag database. Further downstream data analysis incorporated all possible detected peptides into a visualization program, VIPER, to automatically correlate LC-MS features to the peptide identifications in the AMT tag database. The resulting post-VIPER matching proteomics data were filtered to achieve an absolute average mass error of 1.08 ppm and an absolute average net elution time error of 0.16%.
Lipid and metabolite extraction from plasma
Lipids and metabolites were extracted from the same plasma sample by using an established chloroform/methanol extraction procedure. Briefly, 150 μl of plasma (from Ficoll-Paque PLUS preparations, thawed from −80°C) were mixed with 600 μl of a 2:1 chloroform:methanol solution in siliconized 2-ml tubes, vortexed vigorously, and incubated at room temperature for 20 minutes. Following incubation, the samples were vortexed again and centrifuged at 12,000 × g for 10 minutes to separate phases. After centrifugation, the upper (aqueous/methanol) phase (containing metabolites) and the lower (organic/chloroform) phase (containing lipids) were transferred to fresh siliconized tubes (the protein interlayer was discarded), evaporated to dryness using a speedvac and frozen at −80°C. Frozen, dried metabolite and lipid extracts were shipped to the UW-Madison.
Plasma metabolomics analysis
Dried metabolite extracts of plasma were shipped to PNNL for metabolomics analysis. Extracts were chemically derivatized using a modified version of the protocol used to create FiehnLib. Briefly, extracts were dried again to remove any residual moisture accrued as a result of storage at −80°C. T o protect carbonyl groups and reduce the number of tautomeric isomers, 20 μL of methoxyamine in pyridine (30 mg/mL) were added to each sample, followed by vortexing for 30 s and incubation at 37°C with generous shaking (1,000 rpm) for 90 min. At this point, the sample vials were inverted one time to capture any condensation of solvent at the cap surface, followed by a brief centrifugation at 1,000 × g for 1 min. To derivatize hydroxyl and amine groups to trimethylsilyated (TMS) forms, 80 μL of N-methyl-N-(trimethylsilyl) trifluoroacetamide (MSTFA) with 1% trimethylchlorosilane (TMCS) were then added to each vial, followed by vortexing for 10 s and incubation at 37°C with shaking (1,000 rpm) for 30 min. Again, the sample vials were inverted one time, followed by centrifugation at 1,000 × g for 5 min. The samples were allowed to cool to room temperature and were analyzed on the same day. Samples were analyzed according to the method used to create FiehnLib. An Agilent GC 7890A coupled with a single quadrupole MSD 5975C (Agilent Technologies) was used, and the samples were blocked and analyzed in random order. An HP-5MS column (30 m × 0.25 mm × 0.25 μm; Agilent Technologies) was used for untargeted metabolomics analyses. The sample injection mode was splitless, and 1 μL of each sample was injected. The injection port temperature was held at 250°C throughout the analysis. The GC oven was held at 60°C for 1 min after injection, and the temperature was then increased to 325°C by 10°C/min, followed by a 10-min hold at 325 °C. The helium gas flow rate was determined by the Agilent Retention Time Locking function based on analysis of deuterated myristic acid and was in the range of 0.45–0.5 ml/min. Data were collected over the mass range 50 – 550 m/z. A mixture of FAMEs (C8–C28) was analyzed once per day together with the samples for retention index alignment purposes during subsequent data analysis.
GC-MS raw data files were processed using the Metabolite Detector software, version 2.5.2 beta. Briefly, Agilent .D files were converted to netCDF format using Agilent Chemstation, followed by conversion to binary files using Metabolite Detector. Retention indices of detected metabolites were calculated based on the analysis of the FAMEs mixture, followed by their chromatographic alignment across all analyses after deconvolution. Metabolites were initially identified by matching experimental spectra to a PNNL-augmented version of FiehnLib, containing spectra and validated retention indices for over 850 metabolites, using a Metabolite Detector match probability threshold of 0.6 (combined retention index and spectral probability). All metabolite identifications were manually validated to reduce deconvolution errors during automated data-processing and to eliminate false identifications.
The NIST 14 GC-MS library was also used to cross validate the spectral matching scores obtained using the Agilent library and to provide identifications of unmatched metabolites. The three most abundant fragment ions in the spectra of each identified metabolite were automatically determined by Metabolite Detector, and their summed abundances were integrated across the GC elution profile; fragment ions due to trimethylsilylation (i.e. m/z 73 and 147) were excluded from the determination of metabolite abundance. A matrix of identified metabolites, unidentified metabolite features (characterized by mass spectra and retention indices and assigned as ‘unknown’), and their abundances was created for statistical analysis. Features resulting from GC column bleeding were removed from the data matrices prior to further data processing and analysis.
Data were imported into MatLab® R2014a and log2 transformed. Outliers were assessed by Pearson correlation and robust Mahalanobis distance, and then the log2 values were median centered.
Plasma lipidomics analysis
Dried lipid extracts of plasma were shipped to PNNL for lipidomics analysis. Extracted lipids were analyzed by LC-MS/MS using a Waters NanoAcquity UPLC system interfaced with a Velos Orbitrap mass spectrometer (Thermo Scientific, San Jose, CA). The electrospray ionization emitter and MS inlet capillary potentials were 2.2 kV and 12 V, respectively. Lipid extracts were reconstituted in 200 μl of methanol, and 7 μl of each sample was injected and separated over a 90-min gradient elution (mobile phase A: ACN/H2O (40:60) containing 10 mM ammonium acetate; mobile phase B: ACN/IPA (10:90) containing 10 mM ammonium acetate) at a flow rate of 30 μl/min (Table S7). Samples were analyzed in both positive and negative ionization (full scan range of 200–2,000 m/z) using HCD (higher-energy collision dissociation) and CID (collision-induced dissociation) on the top 6 most abundant ions to obtain high coverage of the lipidome. A normalized collision energy of 30 and 35 arbitrary units for HCD and CID were used, respectively. Both CID and HCD were set with a maximum charge state of 2 and an isolation width of 2 m/z units. An activation Q value of 0.18 was used for CID.
Confident lipid identifications were made by using LIQUID, which enables the examination of the tandem mass spectra for diagnostic ion fragments along with associated hydrocarbon chain fragment information. In addition, the isotopic profile, extracted ion chromatogram, and mass measurement error of precursor ions were examined for each lipid species. To facilitate quantification of lipids, a reference database for lipids identified from the MS/MS data was created, containing the lipid name, observed m/z, and retention time. Lipid features from each analysis were then aligned to the reference database based on their m/z and retention time using MZmine 2. Aligned features were manually verified and peak apex intensity values were exported for subsequent statistical analysis. Positive and negative ionization data were analyzed separately at all stages. Normalization and outlier detection were performed as described for proteomics.
Enzyme-linked immunosorbent assays
Plasma (from Ficoll-Paque PLUS preparations, thawed from −80°C) cytokine (IL6, TNF, IL10, IL1 α, and IL1β) levels were assessed using commercially available sandwich ELISAs (enzyme-linked immunosorbent assays) for human cytokines (Millipore or Thermo Fisher Scientific). Assays were performed according to the manufacturer’s instructions, and all samples were assessed in duplicate. Absorbance readings were performed by using an Infinite F50 (Tecan) plate reader, and cytokine levels were quantified (for the average of duplicate readings per sample) based on a standard curve using Microplate Manager Software 6 version 6.0 (Bio-Rad).
Biomarker prediction
To prioritize markers that are predictive early in infection, training sets were filtered using four time-from-disease-onset thresholds (8, 9, 10, and 11 days from symptom onset). Leave-one-out cross validation (LOOCV) was performed on each data source separately, and also for all data sources combined (transcriptomics, proteomics, metabolomics, lipidomics, cytokine ELISA, and clinical parameters). For analysis using combined data sources, data were centered and scaled prior to combination using standard methods. LOOCV was employed in iterations in which a single sample was first set aside as a test set and the remaining samples were considered for the training set (top-level cross-validation). Every sample was used in one iteration as a test set. To avoid potential training bias due to the longitudinal design of this study, the test and training sets were never permitted to overlap by patient. Once the test and training samples were defined for a given iteration, a second level of LOOCV was employed to determine the optimal model complexity (i.e., the number of features to include) for balancing variance and bias. Therefore, at each top level cross-validation iteration and each nested level of cross-validation, a logistic regression model was fit and tested against the nested level holdout (not the top level holdout) using the glmnet package in R. Parameter coefficients were constrained and selected using the least absolute shrinkage and selection operator (LASSO) on scaled training data. These estimates were combined to generate receiver-operator characteristic (ROC) curves for each data source at each time-from-onset filter. A limitation of this method is that only features with complete data are considered; therefore, if a feature (e.g., a protein) was not detected in every sample, then its potential use as a biomarker was not examined. To assess significance within this study, bootstrap permutations decoupling the response and predictors were applied to this same pipeline. For downstream principal components analysis and visualizations, a maximum of two features from each data-source were selected by prioritizing those with large coefficient estimates in low complexity models.
EBOV inactivation
A previous report described EBOV inactivation by using TRIzol extraction (Blow et al., 2004). To verify that 8 M urea treatment inactivates EBOV, 20 replicates of commercially obtained human plasma (140 μl each) were spiked with 10 μl of EbolaΔVP30 virus (Halfmann et al., 2008) (~9.5e6 infectious virus particles), followed by addition of urea to a final concentration of 8 M. Following a 10-minute incubation, urea-treated samples were diluted with 150 μl of MEM (Gibco) containing 10% FBS (Sigma Aldrich) and a 1× P/S cocktail (Gibco) and vortexed; the full volume of each replicate sample was then used to perform a focus forming unit (FFU) assay (employing a monoclonal mouse anti-EBOV VP40 protein antibody to detect replicating virus). In addition, 20 replicates of similarly treated samples were used to infect Vero-VP30 cells (Halfmann et al., 2008) for blind passaging (5 passages were performed, with a 7-day incubation for each). At the conclusion of the fifth passage, culture supernatants were assessed for the presence of EBOV by using the FFU assay. To verify that chloroform/methanol treatment inactivates EBOV, 20 replicates of commercially obtained human plasma were spiked with EbolaΔVP30 as described above, followed by addition of 600 μl of chloroform/methanol (2:1 ratio). Samples were vortexed and centrifuged to separate phases, and the metabolite and lipid fractions were dried in a speedvac. Dried samples (20 replicates for each fraction) were resuspended in MEM containing 10% FBS and 1× P/S and were used for FFU assays. Additionally, 20 replicates of similarly treated samples were used for blind passaging as described above. Both 8 M urea and chloroform/methanol treatments completely inactivated EBOV.
Quantification and Statistical Analysis
Clinical and demographic data
Several statistical methods were used to test for associations of clinical and demographic parameters with outcome. Odds ratios were estimated by logistic regression models with survival as the outcome. Continuous variables (age and time from symptom onset to first sample) were compared among outcome groups by t-test, and a chi-square test was used to compare sex, disease presentation (i.e., wet vs. dry) and Ebola treatment centers. For the continuous variables, odds ratios were estimated for a difference of 10 years (age) or 3 days (time from symptom onset to first sample). These data are shown in Figure 1C and are also described in the text of the Results.
Plasma ‘omics and ELISA data
All statistical analyses of ‘omics data were performed using the R framework. Normalized log2 transformed values were used for statistical analyses of cytokines, proteins, metabolites, and lipids. For differential expression (DE) discovery, models were fit independently for each feature (i.e., for each individual protein, metabolite, and lipid). To accommodate the correlation structure in longitudinal comparisons, generalized linear mixed models (GLMM) were fit using the nlme and lme4 packages, allowing for random slopes and intercepts within subjects, and fixed effects to contrast treatment group slopes. Comparisons between groups without repeated measures were performed using the limma package with an empirical Bayes shrinkage estimate for variance. Assessment of confounding variables was performed using three approaches: (i) outcome was directly modeled against demographic covariates, (ii) heat maps of candidate DE features were overlaid with demographic data for visual assessment, and (iii) residual variance from DE modeling was explored for unexplained correlation structure (e.g., batch effects). Surrogate latent variables were estimated using the sva package, and those from the first 2 principal components were used to assess potential bias from latent correlation structure. For plasma ‘omics datasets, fold-changes, P-values and Benjamin-Hochberg-adjusted P-values (i.e., q-values) for various group comparisons are reported in Table S2, and specific observations are highlighted in the figures, and in the Results and Figure Legends. Multiple hypothesis correction was not carried out for plasma ELISA data.
Pathway enrichment for proteomics
Pathway (or feature set) enrichment statistics were calculated in R using a log-rank test to compare the ranks of features falling within each pathway to those that do not. Ranks were determined by the statistical significance of the linear models used to contrast patient groups. The pathway and null curves were visualized by plotting Kaplan-Meier survival curves using the Survival package in R. Pathway enrichment results are reported in Table S3, and specific observations are highlighted in figures and in the Results and Figure Legends.
RNA-seq data
Raw fragment (i.e., paired-end read) counts were combined into a numeric matrix, with genes in rows and experiments in columns, and used as input for gene expression differential analysis with the Bioconductor Limma package after multiple filtering steps to remove low-expressed genes. First, gene counts were converted to FPKM (fragments per kb per million reads) using the RSEM package with default settings in strand-specific mode, and only genes with expression levels above 1 FPKM in at least 50% of samples were retained for further analysis. Additional filtering removed genes with less than 50 total reads across all samples or of less than 200 nucleotides in length. Normalization factors were computed on the filtered data matrix using the weighted trimmed mean of M-values (TMM) method, followed by voom mean-variance transformation in preparation for Limma linear modeling. Data were fitted to a design matrix containing all sample groups and pairwise comparisons were performed between sample groups (survivors’ samples 1–3, fatalities, and healthy controls). eBayes adjusted P-values were corrected for multiple testing using the Benjamin-Hochberg (BH) method and used to select genes with significant expression differences (q < 0.01). Fold-changes and q-values for various group comparisons are reported comprehensively in Table S4, and specific observations are highlighted in figures and in the Results and Figure Legends.
Network construction and enrichment analysis
The same filtered and normalized gene expression data matrix used in the differential gene expression analysis was also used as input for Multiscale Embedded Gene Co-expression network analysis (MEGENA) with the MEGENA v1.3.4 R package (Song and Zhang, 2015). The Pearson method was used to calculate co-expression correlation between gene pairs, with false discovery rate (FDR) calculation by permutation analysis (100 iterations) and an FDR cutoff of q < 0.05 to identify significant correlations. Following Planar Filtered Network construction, Multi Clustering Analysis and Multi-scale Hub Analysis were performed using 100 permutations. Significant modules and hubs were identified at FDR q < 0.05 and a resolution parameter cut-off (i.e., alpha) value of 1. Finally, each module was tested for significant gene set enrichment (padj<0.05) against the Molecular Signatures Database (MSigDB) using a Fisher’s exact test and Bonferroni correction to account for multiple testing, and module diagrams showing the co-expression relationships between modules and genes within each module were plotted. MEGENA module data are reported in Table S5, and specific observations are highlighted in the figures and in the Results and Figure Legends.
Comparisons with other published datasets
Publicly available human PBMC transcriptomics datasets (GSE48080 and GSE93861) (Kash et al., 2017; Severino et al., 2014) were obtained from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/). Datasets were quantile normalized and then an empirical Bayes-adjusted t-test was used to estimate differential expression between groups (i.e., severe sepsis or septic shock compared to healthy controls [GSE48080] or EVD survivor compared to healthy controls [GSE93861]). Pathway enrichment was performed as described for plasma proteomics (results are reported in Table S5). Heat maps comparing these datasets to our data were generated by using the ComplexHeatmap R package. Heat map values represent the log2 effect size (i.e., the fold-change standardized by the standard deviation). Specific observations are highlighted in figures and in the Results and Figure Legends.
Data and Software Availability
Demographic and clinical datasets
Anonymized patient demographic data, sample information and viral genome quantification data (qRT-PCR) are provided in Table S1.
Statistically processed datasets
Plasma ELISA, proteomics, metabolomics and lipidomics datasets (including fold-change, P-values, and FDR-adjusted q-values for various group comparisons) are provided in Table S2. Plasma protein pathway enrichment statistics are provided in Table S3. PBMC RNA-seq data (including fold-change and FDR-adjusted q-values for various group comparisons) are provided in Table S4. MEGENA module data are provided in Table S5, including module summaries, membership lists, overlaps with differentially expressed transcripts derived from various group comparisons, and pathway and process enrichment statistics. Enrichment statistics for other sepsis and EVD PBMC transcriptomics datasets are provided in Table S5.
Raw data
All raw GC-MS data (metabolites), LC-MS/MS data (lipids), and proteomics raw mass spectrometry data corresponding to instrument files, mzML, and MSGF+ MS/MS search for peptide identifications used to populate AMT tag databases have been deposited at the Mass Spectrometry Interactive Virtual Environment (MassIVE) at the University of California at San Diego, (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp), under the ID code MSV000080129.
Supplementary Material
▪ Table S1. Related to Figure 1, Figure 2, and Figure 7. Patient demographic and sample information.
▪ Table S2. Related to Figure 2, Figure 3, and Figure 4. Plasma cytokine and ‘omics data.
▪ Table S3. Related to Figure 4. Plasma protein pathway enrichment.
▪ Table S4. Related to Figure 5 and Figure 6. PBMC transcriptome data.
▪ Table S5. Related to Figure 5 and Figure 6. MEGENA data and comparisons with publicly available transcriptomics datasets.
HIGHLIGHTS.
Multi-omics analysis of peripheral blood from Ebola disease (EVD) patients was performed.
Pancreatic enzymes may contribute to host-mediated tissue damage in fatal EVD.
Neutrophils may dysregulate adaptive immunity and cause tissue damage in fatal EVD.
A redundant panel of diverse biomarkers can predict EVD outcomes.
Acknowledgments
This study was funded by a Health and Labor Sciences Research Grant (Japan); by grants for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Sports, Science and Technology of Japan (No. 16H06429, 16K21723, and 16H06434); by Emerging/Re-emerging Infectious Diseases Project of Japan; and by an administrative supplement to grant U19AI106772, provided by the National Institute of Allergy and Infectious Diseases, National Institutes of Health (USA). Next generation sequencing was carried out by the Mount Sinai Genomics Core Facility, and was supported in part by the Department of Scientific Computing at the ISMMS. Proteomics and metabolomics analyses were performed in the Environmental Molecular Sciences Laboratory, a national scientific user facility sponsored by the U.S. Department of Energy (DOE) Office of Biological and Environmental Research, and located at PNNL. Additional support was provided by National Institute of General Medical Sciences grant P41 GM103493. PNNL is a multi-program national laboratory operated by Battelle for the DOE under Contract DE-AC05-76RLO 1830. The authors would like to thank Zachary Najacht and Daniel Beechler (UW-Madison) and Thomas Korfeh, Francis Khoryama, Alex Bockarie, and Mohamed Nyallay (34th Regimental Military Hospital at Wilberforce, Freetown, Sierra Leone) for technical assistance; Amy E. S. Kuehn and Alexander Karasin (UW-Madison) and Ishamil Barrie (Project 1808, Freetown, Sierra Leone) for excellent administrative support; and Susan Watson for editing the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Conceptualization, Y.K., P.J.H., A.N., F.S., A.J.E., and G.N.; Methodology, A.J.E., P.J.H., J.P.W., J.M.J., H.v.B., and T.O.M.; Formal Analysis, J.P.W., J.E.K., K.E.B.-J., Z.P., K.G.S., B.-J.M.W.-R., and Y.-M.K.; Investigation, A.J.E., P.J.H., J.E.K., K.E.B.-J., Z.P., T.M., K.B.W., T.W., S.F., M.Y., J.M.J., Y.-M.K., C.P.C., M.A.G., M.E.M., K.K.W., and A.K.S.; Resources, Y.K., F.S., H.v.B., T.O.M., R.D.S., K.M.W., J.L.R., and A.N.; Data Curation, A.J.E., J.P.W., Z.P., J.E.K., K.E.B.-J., Z.P., K.G.S., B.-J.M.W.-R., and Y.-M.K.; Writing – Original Draft, A.J.E., J.P.W., J.E.K., K.E.B.-J., H.v.B., and Y.K.; Writing – Review and Editing, A.J.E., J.P.W., Y.K., G.N., P.J.H., J.E.K., K.E.B.-J., Z.P., T.M., K.B.W., T.W., S.F., M.Y., M.T., J.L.R., H.v.B., T.O.M., and K.M.W.; Visualization, A.J.E., J.P.W., Z.P., and H.v.B.; Supervision, Y.K., F.S., H.v.B., T.O.M., R.D.S., K.M.W., and J.L.R.; Project Administration, A.J.E. and P.J.H.; Funding Acquisition, Y.K., H.v.B., T.O.M., R.D.S., K.M.W., and J.L.R.
The authors do not have any conflicts of interest to declare.
References
- Agarwal N, Pitchumoni CS. Acute pancreatitis: a multisystem disease. Gastroenterologist. 1993;1:115–128. [PubMed] [Google Scholar]
- Blow JA, Dohm DJ, Negley DL, Mores CN. Virus inactivation by nucleic acid extraction reagents. J Virol Methods. 2004;119:195–198. doi: 10.1016/j.jviromet.2004.03.015. [DOI] [PubMed] [Google Scholar]
- Carmona-Rivera C, Kaplan MJ. Low-density granulocytes: a distinct class of neutrophils in systemic autoimmunity. Semin Immunopathol. 2013;35:455–463. doi: 10.1007/s00281-013-0375-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darcy CJ, Minigo G, Piera KA, Davis JS, McNeil YR, Chen Y, Volkheimer AD, Weinberg JB, Anstey NM, Woodberry T. Neutrophils with myeloid derived suppressor function deplete arginine and constrain T cell function in septic shock patients. Crit Care. 2014;18:R163. doi: 10.1186/cc14003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etzerodt A, Moestrup SK. CD163 and inflammation: biological, diagnostic, and therapeutic aspects. Antioxid Redox Signal. 2013;18:2352–2363. doi: 10.1089/ars.2012.4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippas-Ntekouan S, Liberopoulos E, Elisaf M. Lipid testing in infectious diseases: possible role in diagnosis and prognosis. Infection. 2017 doi: 10.1007/s15010-017-1022-3. Epub: May 8. [DOI] [PubMed] [Google Scholar]
- Halfmann P, Kim JH, Ebihara H, Noda T, Neumann G, Feldmann H, Kawaoka Y. Generation of biologically contained Ebola viruses. Proc Natl Acad Sci U S A. 2008;105:1129–1133. doi: 10.1073/pnas.0708057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellman J. Addressing the Complications of Ebola and Other Viral Hemorrhagic Fever Infections: Using Insights from Bacterial and Fungal Sepsis. PLoS Pathog. 2015;11:e1005088. doi: 10.1371/journal.ppat.1005088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt L, Gupta-Wright A, Simms V, Tamba F, Knott V, Tamba K, Heisenberg-Mansaray S, Tamba E, Sheriff A, Conteh S, et al. Clinical presentation, biochemical, and haematological parameters and their association with outcome in patients with Ebola virus disease: an observational cohort study. Lancet Infect Dis. 2015;15:1292–1299. doi: 10.1016/S1473-3099(15)00144-9. [DOI] [PubMed] [Google Scholar]
- Kaplan MJ, Radic M. Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol. 2012;189:2689–2695. doi: 10.4049/jimmunol.1201719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kash JC, Walters KA, Kindrachuk J, Baxter D, Scherler K, Janosko KB, Adams RD, Herbert AS, James RM, Stonier SW, et al. Longitudinal peripheral blood transcriptional analysis of a patient with severe Ebola virus disease. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aai9321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly RT, Page JS, Luo Q, Moore RJ, Orton DJ, Tang K, Smith RD. Chemically etched open tubular and monolithic emitters for nanoelectrospray ionization mass spectrometry. Anal Chem. 2006;78:7796–7801. doi: 10.1021/ac061133r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix R, Dignat-George F. Microparticles as a circulating source of procoagulant and fibrinolytic activities in the circulation. Thromb Res. 2012;129(Suppl 2):S27–29. doi: 10.1016/j.thromres.2012.02.025. [DOI] [PubMed] [Google Scholar]
- Leligdowicz A, Fischer WA, 2nd, Uyeki TM, Fletcher TE, Adhikari NK, Portella G, Lamontagne F, Clement C, Jacob ST, Rubinson L, et al. Ebola virus disease and critical illness. Crit Care. 2016;20:217. doi: 10.1186/s13054-016-1325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llewelyn MJ, Berger M, Gregory M, Ramaiah R, Taylor AL, Curdt I, Lajaunias F, Graf R, Blincko SJ, Drage S, et al. Sepsis biomarkers in unselected patients on admission to intensive or high-dependency care. Crit Care. 2013;17:R60. doi: 10.1186/cc12588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtke A, Ruibal P, Becker-Ziaja B, Rottstegge M, Wozniak DM, Cabeza-Cabrerizo M, Thorenz A, Weller R, Kerber R, Idoyaga J, et al. Ebola Virus Disease Is Characterized by Poor Activation and Reduced Levels of Circulating CD16+ Monocytes. J Infect Dis. 2016 doi: 10.1093/infdis/jiw260. [DOI] [PubMed] [Google Scholar]
- Maiolica A, Borsotti D, Rappsilber J. Self-made frits for nanoscale columns in proteomics. Proteomics. 2005;5:3847–3850. doi: 10.1002/pmic.200402010. [DOI] [PubMed] [Google Scholar]
- Malfertheiner P, Kemmer TP. Clinical picture and diagnosis of acute pancreatitis. Hepatogastroenterology. 1991;38:97–100. [PubMed] [Google Scholar]
- Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2006;177:7303–7311. doi: 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- Messaoudi I, Amarasinghe GK, Basler CF. Filovirus pathogenesis and immune evasion: insights from Ebola virus and Marburg virus. Nat Rev Microbiol. 2015;13:663–676. doi: 10.1038/nrmicro3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanclares C, Kapetshi J, Lionetto F, de la Rosa O, Tamfun JJ, Alia M, Kobinger G, Bernasconi A. Ebola Virus Disease, Democratic Republic of the Congo, 2014. Emerg Infect Dis. 2016;22:1579–1586. doi: 10.3201/eid2209.160354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer C, Diehn M, Alizadeh AA, Brown PO. Cell-type specific gene expression profiles of leukocytes in human peripheral blood. BMC Genomics. 2006;7:115. doi: 10.1186/1471-2164-7-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DW, Kwak DS, Park YY, Chang Y, Huh JW, Lim CM, Koh Y, Song DK, Hong SB. Impact of serial measurements of lysophosphatidylcholine on 28-day mortality prediction in patients admitted to the intensive care unit with severe sepsis or septic shock. J Crit Care. 2014;29:882, e885–811. doi: 10.1016/j.jcrc.2014.05.003. [DOI] [PubMed] [Google Scholar]
- Raftery MJ, Lalwani P, Krautkrmer E, Peters T, Scharffetter-Kochanek K, Kruger R, Hofmann J, Seeger K, Kruger DH, Schonrich G. beta2 integrin mediates hantavirus-induced release of neutrophil extracellular traps. J Exp Med. 2014;211:1485–1497. doi: 10.1084/jem.20131092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruibal P, Oestereich L, Ludtke A, Becker-Ziaja B, Wozniak DM, Kerber R, Korva M, Cabeza-Cabrerizo M, Bore JA, Koundouno FR, et al. Unique human immune signature of Ebola virus disease in Guinea. Nature. 2016;533:100–104. doi: 10.1038/nature17949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandquist M, Wong HR. Biomarkers of sepsis and their potential value in diagnosis, prognosis and treatment. Expert Rev Clin Immunol. 2014;10:1349–1356. doi: 10.1586/1744666X.2014.949675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieffelin JS, Shaffer JG, Goba A, Gbakie M, Gire SK, Colubri A, Sealfon RS, Kanneh L, Moigboi A, Momoh M, et al. Clinical illness and outcomes in patients with Ebola in Sierra Leone. N Engl J Med. 2014;371:2092–2100. doi: 10.1056/NEJMoa1411680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severino P, Silva E, Baggio-Zappia GL, Brunialti MK, Nucci LA, Rigato O, Jr, da Silva ID, Machado FR, Salomao R. Patterns of gene expression in peripheral blood mononuclear cells and outcomes from patients with sepsis secondary to community acquired pneumonia. PLoS One. 2014;9:e91886. doi: 10.1371/journal.pone.0091886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silke J, Rickard JA, Gerlic M. The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol. 2015;16:689–697. doi: 10.1038/ni.3206. [DOI] [PubMed] [Google Scholar]
- Smith CK, Kaplan MJ. The role of neutrophils in the pathogenesis of systemic lupus erythematosus. Curr Opin Rheumatol. 2015;27:448–453. doi: 10.1097/BOR.0000000000000197. [DOI] [PubMed] [Google Scholar]
- Soeters PB, Grecu I. Have we enough glutamine and how does it work? A clinician’s view. Ann Nutr Metab. 2012;60:17–26. doi: 10.1159/000334880. [DOI] [PubMed] [Google Scholar]
- Song WM, Zhang B. Multiscale Embedded Gene Co-expression Network Analysis. PLoS Comput Biol. 2015;11:e1004574. doi: 10.1371/journal.pcbi.1004574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasquez GE, Aibana O, Ling EJ, Diakite I, Mooring EQ, Murray MB. Time From Infection to Disease and Infectiousness for Ebola Virus Disease, a Systematic Review. Clin Infect Dis. 2015;61:1135–1140. doi: 10.1093/cid/civ531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives-Pi M, Takasawa S, Pujol-Autonell I, Planas R, Cabre E, Ojanguren I, Montraveta M, Santos AL, Ruiz-Ortiz E. Biomarkers for diagnosis and monitoring of celiac disease. J Clin Gastroenterol. 2013;47:308–313. doi: 10.1097/MCG.0b013e31827874e3. [DOI] [PubMed] [Google Scholar]
- Vogt L, Schmitz N, Kurrer MO, Bauer M, Hinton HI, Behnke S, Gatto D, Sebbel P, Beerli RR, Sonderegger I, et al. VSIG4, a B7 family-related protein, is a negative regulator of T cell activation. J Clin Invest. 2006;116:2817–2826. doi: 10.1172/JCI25673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wu J, Newton R, Bahaie NS, Long C, Walcheck B. ADAM17 cleaves CD16b (FcgammaRIIIb) in human neutrophils. Biochim Biophys Acta. 2013;1833:680–685. doi: 10.1016/j.bbamcr.2012.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PA, Bosmann M. A historical perspective on sepsis. Am J Pathol. 2012;181:2–7. doi: 10.1016/j.ajpath.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauquier N, Becquart P, Padilla C, Baize S, Leroy EM. Human fatal zaire ebola virus infection is associated with an aberrant innate immunity and with massive lymphocyte apoptosis. PLoS Negl Trop Dis. 2010;4 doi: 10.1371/journal.pntd.0000837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster NL, Kedzierska K, Azzam R, Paukovics G, Wilson J, Crowe SM, Jaworowski A. Phagocytosis stimulates mobilization and shedding of intracellular CD16A in human monocytes and macrophages: inhibition by HIV-1 infection. J Leukoc Biol. 2006;79:294–302. doi: 10.1189/jlb.0705382. [DOI] [PubMed] [Google Scholar]
- WHO. Ebola Situation Report - 30 March 2016 (World Health Organization) 2016 http://apps.who.int/ebola/current-situation/ebola-situation-report-30-march-2016.
- Zimmer JS, Monroe ME, Qian WJ, Smith RD. Advances in proteomics data analysis and display using an accurate mass and time tag approach. Mass Spectrom Rev. 2006;25:450–482. doi: 10.1002/mas.20071. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
▪ Table S1. Related to Figure 1, Figure 2, and Figure 7. Patient demographic and sample information.
▪ Table S2. Related to Figure 2, Figure 3, and Figure 4. Plasma cytokine and ‘omics data.
▪ Table S3. Related to Figure 4. Plasma protein pathway enrichment.
▪ Table S4. Related to Figure 5 and Figure 6. PBMC transcriptome data.
▪ Table S5. Related to Figure 5 and Figure 6. MEGENA data and comparisons with publicly available transcriptomics datasets.