

Abstract
Direct targeting to the kidneys is a promising strategy to improve drug therapeutic index for the treatment of kidney diseases. We sought to investigate the renal selectivity and safety of kidney-targeted “mesoscale” nanoparticle technology. We found that direct intravenous administration of these particles resulted in 26-fold renal selectivity and localized negligibly in the liver or other organs. The nanoparticles targeted the renal proximal tubular epithelial cells, as evidenced by intravital microscopy and ex vivo imaging. Mice treated with the nanoparticles exhibited no negative systemic consequences, immune reaction, liver impairment, or renal impairment. The localization of material selectively to the renal tubules is uncommon, and this work portends the development of renal-targeted drugs for the treatment of kidney diseases.
Keywords: biomaterials, drug carriers, pharmacokinetics, nanoparticles, renal disease, drug development
INTRODUCTION
Acute kidney injury (AKI) and chronic kidney disease (CKD) are major health problems of increasing worldwide prevalence and severity1, 2. AKI accounts for 1% of hospital admissions in the US3 and approximately 25% of patients in the intensive care unit (ICU) develop AKI. Of these, up to 5% will require renal replacement therapy, for whom mortality rate ranges between 40 and 60%4–8. In addition, it is estimated that over 20 million US adults (~11%) have chronic kidney disease (CKD)9. This risk of death increases as CKD progresses, ranging from a 20 to 80 percent increase in mortality hazard among patients with early CKD, to over a 3-fold risk increase in patients with more advanced disease10. Despite their prevalence and associated morbidity and mortality, the treatment and prevention of both AKI and CKD is limited by the lack of novel therapeutic approaches that could ameliorate their progression or severity.
Multiple clinical trials have failed to demonstrate beneficial effects of a variety of pharmacologic approaches in the treatment and prevention of AKI in spite of promising pre-clinical data11, 12. The reasons for these failures are probably multifactorial but are likely due at least in part to poor pharmacokinetics of the different compounds tested in humans13, 14. In the case of CKD, few strategies have demonstrated an effect on the progression of CKD to end stage renal disease15, 16. The development of strategies that target specific renal compartments may overcome this problem and result in specific treatments for both acute and chronic renal disease by targeting therapeutics to the area of injury. The successful translation of such an approach to humans may have major impacts on the morbidity and mortality associated with acute and chronic kidney diseases, as well as many other kidney-related medical conditions.
Our group recently developed nanoparticle-based delivery systems that localize specifically to the kidneys17. We found that large (350–400 nm) diameter polymer-based “mesoscale nanoparticles” (MNPs) localized 5–7 times more efficiently to the kidneys than any other organ upon intravenous tail vein administration17. The localization appeared to be predominantly tubular in nature and persisted up to 7 days in the epithelial cells. These findings reveal a novel material in the nanotechnology literature with potentially profound applications in renal disease18.
Here, we sought to understand the clinical promise of this nanoparticle platform by characterizing in detail its renal localization and investigating the safety of selective renal targeting. To improve upon the renal selectivity of MNPs, we first hypothesized that altering the administration route may affect the renal-specific localization of MNPs. We next investigated the hypothesis that MNP dose may impact the relative specificity of the particles for the kidney. We then employed advanced intravital imaging techniques to assess the tubular localization of these particles. Finally, to ensure the safety of our nanoparticle platform, we performed longer-term biodistribution studies and found that enhanced renal localization has no negative systemic consequences or on kidney function. With this work, we have probed the parameters of nanoparticle renal targeting that are consequential for their use in the clinic, allowing the technology to advance to therapeutic efficacy studies.
METHODS
Particle Formulation and Characterization
Mesoscale nanoparticles (MNPs) were formed from poly(lactic-co-glycolic acid) conjugated to polyethylene glycol (PLGA-PEG). The block copolymer was conjugated as we previously described17. Briefly, 5 g (90–130 μmol) carboxylic acid-terminated PLGA (50:50; MW 28–54 kDa) (Aldrich; St. Louis, MO) was dissolved in methylene chloride with 1.2 mmol N-hydroxysuccinimide (NHS) and 1.2 mmol 1-ethyl-3-(3-(dimethylamino)propyl)-carbodiimide (EDC) and stirred for approximately 30 minutes. PLGA-NHS was then precipitated with ethyl ether and washed with 50:50 ethyl ether:methanol and dried under vacuum. PLGA-NHS (1 g, 18–26 μmol) was mixed with 50 μmol amine-PEG-carboxylic acid (MW 5 kDa) (Nanocs; New York, NY) in chloroform and 220 μmol N,N-diisopropylethylamine overnight. Conjugated PLGA-PEG was precipitated and washed with cold methanol then dried under vacuum. 1H NMR was used to confirm conjugation as previously described19.
Fluorescent MNPs were formed from PLGA-PEG and 3,3′-diethylthiadicarbocyanine iodide (DEDC) (Acros Organics; Geel, Belgium) as we previously described via nanoprecipitation17. 100 mg PLGA-PEG was dissolved with 10 mg DEDC in 2 mL acetonitrile and added dropwise to 4 mL water with 100 μL 10% Pluronic F-68 (Gibco; Grand Island, NY). Alternatively, we formulated MNPs encapsulating a random double-stranded DNA duplex 5′-AGTCGTCAGTACGATGCAGAC/3Cy5/3′ with a molecular weight of 13,523.1 g/mol. After stirring for 2 hours, particles were centrifuged at 7356 RCF for 15 minutes and washed before lyophilization in a 2% sucrose solution.
Freeze-dried particles were analyzed for size by dynamic light scattering (DLS) in phosphate-buffered saline and ζ-potential in water by electrophoretic light scattering (ELS) (Malvern; Worcestershire, United Kingdom). Total DEDC encapsulation was measured by UV-Vis absorbance (Jasco; Easton, MD).
Serum Stability Assay
Nanoparticle stability was measured in complete mouse serum obtained from healthy C57BL/6 mice (Charles River; Troy, NY). 1 mg/mL MNPs were suspended in 500 μL serum and incubated at room temperature. At 2, 4, 6, 24, 48, and 72 hours, 100 μL of the sample was removed for DLS measurement and subsequently replaced into the sample. 10 mg/mL MNPs were suspended in 100 μL serum and incubated at room temperature. At 2, 4, 6, 24, 48, and 72 hours, the sample was centrifuged at 4286 RCF for 15 minutes, the supernatant was removed, and the pellet was resuspended in serum and incubated at room temperature. Absorbance measurements of the dye in the supernatant and pellet were obtained using a Tecan Infinite M1000Pro (Mannedorf, Switzerland) in a 96-well plate at 650 nm. Dye release (%) was calculated as the amount of dye in each sample divided by total dye combined from supernatants at each time point and 72 hour pellet measurements.
Administration Route Investigation
All animal experiments were approved by and carried out in accordance with IACUC guidelines at Memorial Sloan Kettering Cancer Center and in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Healthy female 4–8 week hairless mice with intact immune systems (Crl:SKH1-HrHr) were used (Charles River. Mice were fed irradiated 5V75 alfalfa-free diet (LabDiet; St. Louis, MO) to reduce fluorescent imaging background. Groups of 3 mice each were dosed with 25 mg/kg MNPs via the following administration routes: oral gavage (per os, PO), intravenous via the retroorbital vein (RO), intravenous via the tail vein (IV), subcutaneous flank (SQ), and intraperitoneal (IP). Live mice were imaged five at a time (one per group) to determine whole-animal biodistribution at the following post-injection time-points: 30 minutes, 4 hours, 24 hours, 48 hours, and 72 hours. Imaging was performed using an IVIS Spectrum Preclinical In Vivo Imaging System (Perkin Elmer; Waltham, MA) using 640/680 nm excitation/emission filters. Following the 72 hour imaging time-point, mice were euthanized and the following organs were harvested and fluorescently imaged: heart, lungs, liver, spleen, and kidneys. Organs and regions of interest (ROI) in live mice were selected using Living Image Software v4.3 (Perkin Elmer) to quantify average fluorescence efficiency per square centimeter in each ROI. Organ fluorescence measurements were calculated as the fluorescence efficiency normalized by the fluorescence efficiency of PBS-injected control organs. Mean and standard deviation of organ fluorescence were calculated for each group of 3 mice.
Dose Investigation
Healthy female 4–8 hairless mice with intact immune systems were used (Crl:SKH1-HrHr). Three mice were injected IV with 25 mg/kg DEDC MNPs and one mouse was injected with phosphate buffered saline (PBS) as a vehicle control. Separately, three mice were injected IV with 5 mg/kg DEDC MNPs and one mouse was injected with PBS as a control. Imaging was performed as described above at 30 minutes, 24 hours, 48 hours, and 72 hours post injection. Mice were euthanized after 72 hours with organs extracted, imaged, and analyzed as above. Background-subtracted average fluorescence efficiency was reported. Where denoted, normalized total fluorescence efficiency was reported to account for the full volume of each organ. Normalized total fluorescence efficiency was obtained by dividing each organ’s average fluorescence by the total weight of that organ and subtracting that of a control animal. Results were compared with the results from a 50 mg/kg IV administration of anionic MNPs as previously described17. For investigations with Cy5-dsDNA MNPs, three mice were injected with nanoparticles at 50 mg/kg IV with a single PBS-injected control mouse. The mice were euthanized 24 hours after injection; organs were extracted, imaged, and analyzed as above.
In Vivo Imaging
Healthy Cx3crgfp/+ C57BL/6 mice, with green fluorescent protein (GFP)-expressing renal macrophages were used20, 21. An intravital confocal microscopy imaging setup was used to perform superficial renal cortex imaging21. Mice were IV injected via the tail vein with either 25 mg/kg DEDC MNPs or a matched dose of free DEDC. Mice were imaged at 72 hours post-injection.
Perfused Animal Ex Vivo imaging
Healthy 4–8 week hairless mice with intact immune systems were used. One mouse was injected with 25 mg/kg MNPs and one with a matched dose of free DEDC. At 72 hours post-injection, cardiac perfusions were performed with 4% paraformaldehyde (PFA). Organs were removed and fixed in 4% PFA, dehydrated, and embedded in paraffin. 5 μm sections were placed onto glass slides, de-paraffinized and prepared for immunofluorescence imaging. Slides were stained with 4′,6-diamidino-2-phylindole (DAPI) to stain nuclei and then stained with either an anti-CD31 antibody for endothelial cells (Dianova; Hamburg, Germany) or anti-E-cadherin to stain epithelial cells (BD Bioscience; San Jose, CA). Slides were imaged with an Olympus IX51 inverted light microscope (Olympus; Center Valley, PA) and an Olympus XM10 monochrome camera after excitation with a X-Cite 120Q lamp (Lumen Dynamics; Ontario, Canada). Appropriate filter cubes for DAPI, AlexaFluor488, and Cy5 were used with consistent exposure times for each channel and analyzed in ImageJ (NIH; Bethesda, MD) with consistent brightness values.
Long-term Biodistribution and Safety
Healthy 4–8 week female BALB/c mice (BALB/cAnNCrl) were used (Charles River). Twelve mice were IV injected with 25 mg/kg DEDC MNPs and separated into groups of three. One group was euthanized at each of the following time-points: 1 day, 3 days, 7 days, and 28 days post-injection. Separately, three mice were injected with a PBS vehicle control and sacrificed on day 1. Prior to sacrifice, blood was collected retroorbitally and urine was collected following excretion (urine collection failed at day 28). Organs were collected, imaged, and analyzed as described above. A complete renal panel was performed on serum samples, including: blood urea nitrogen (BUN), creatinine, total protein, albumin, globulin, phosphorous, calcium, total CO2, sodium, potassium, chloride, and anion gap. Urine chemistry was performed to obtain BUN, creatinine, and micro total protein. Whole blood was used to perform a complete blood count for each mouse, including: white blood cells, neutrophils, lymphocytes, monocytes, eosinophils, red blood cells (RBCs), nucleated RBCs, hemoglobin, hematocrit, mean corpuscular volume, mean corpuscular hemoglobin, mean corpuscular hemoglobin concentration, red blood cell distribution width, reticulocytes, and platelets.
Histological analyses were performed on tissues of 4–8 week SKH1 mice injected with 50 mg/kg DEDC MNPs and sacrificed at three and seven days post-injection. As a control, mice were injected with an equal amount of PBS and sacrificed at seven days post-injection. Kidneys, lungs, spleen, heart, and liver were obtained and fixed in 4% paraformaldehyde overnight. Fixed organs were dehydrated and paraffin-embedded before 5 μm sections were placed on glass slides. Paraffin was removed and slides were stained with haematoxylin and eosin (H&E) for basic histology. Immunohistochemistry was performed using a Discovery XT processor (Ventana Medical Systems; Oro Valley, AZ) by first blocking for 30 minutes with 10% normal rabbit serum in PBS + 2% BSA. A 2 μg/mL aliquot of an anti-F4/80 antibody (Abcam; Cambridge, UK; Cat # ab6640) was applied to sections and incubated for 3 hours, followed by a 60 minute incubation with biotinylated rabbit anti-rat IgG (Vector Labs; Burlingame, CA; Cat BA-400) at 1:200 dilution. The assay was performed with a DAB detection kit (Ventana Medical Systems) according to manufacturer instructions. Slides were counterstained with hematoxylin (Ventana Medical Systems). Slides were imaged with an Olympus IX51 inverted light microscope (Olympus; Center Valley, PA) outfitted with an Olympus DP73 digital color camera.
RESULTS
Mesoscale Nanoparticle Formulation
Mesoscale nanoparticles were constructed from a diblock copolymer consisting of poly(lactic-co-glycolic acid) and polyethylene glycol (PLGA-PEG) as previously described17, 19. The nanoprecipitation method was used to formulate MNPs encapsulating the fluorescent dye 3,3′-diethylthiadicarbocyanine iodide (DEDC)22. As prior work found that anionic, cationic, and neutral MNPs exhibited approximately the same renal selectivity, we focused on the investigation of anionic MNPs17. Particle size was confirmed to be an average of 347.6 ± 21.0 nm in phosphate-buffered saline (PBS) by dynamic light scattering. Particle size was stable for up to 48 hours in complete mouse serum (SI Figure S1A). Particle surface charge as determined electrophoretic light scattering was −19.0 ± 0.3 mV in deionized water. The MNPs contained 0.27 mg DEDC/g of total particle mass. Under 20% of the dye was released in complete mouse serum in six hours, and under 50% was released in 3 days (SI Figure S1B).
Renal Selectivity by Administration Route and Dose
We first investigated the optimal route of nanoparticle administration to maximize relative renal localization. The choice of administration route for selective particle localization is a particularly important issue in the study of nanomaterials and drug delivery23–25. For these experiments, 4–8 week healthy female hairless (Crl:SKH1-HrHr) mice with intact immune systems were used. Three mice per group were dosed with 25 mg/kg nanoparticles containing the DEDC dye via the following common administration routes: oral gavage (per os, PO), retroorbital intravenous (RO), intravenous via the tail vein (IV), subcutaneous flank (SQ), or intraperitoneal (IP). Fluorescence of the nanoparticles was measured in vivo via an IVIS Spectrum In Vivo Imaging System using 640 nm excitation and 680 nm emission filters at the following time points post-injection: 30 minutes, 4 hours, 24 hours, 48 hours, and 72 hours (SI Figures S2–3). Following 72 hours, mice were euthanized. Organs were harvested and imaged using the dye fluorescence to quantify particle biodistribution (SI Figure S4).
These experiments revealed that the most efficient renal localization occurred using intravenous administration methods. The data indicate that MNPs injected IV via the tail vein accumulated in the kidneys 30 minutes following injection, and they persisted over the course of the 72 hour experiment (Figure 1A–B; SI Figure S2–3). Given the relative stability of the particles and slow rate of dye release in mouse serum (SI Figure S1), we conclude that the overwhelming majority of the renal fluorescence signal was due to MNP-encapsulated dye. Intravenous injection via the retroorbital vein (RO) also predominantly resulted in renal accumulation of the particles, but there was significant variability as compared to the tail vein route (Figure 1C). This variability may be explained by leakage or related phenomena at the injection site, as evinced by a strong fluorescence signal at the ocular site of administration throughout the duration of the experiment (SI Figure S2). As expected, PO administration resulted in rapid localization to the stomach which dissipated by 24 hours, likely via fecal excretion24 (SI Figure S3). Interestingly, we did find some fluorescence in the kidneys in mice injected SQ and IP, although the levels were lower than by intravenous routes, as measured by ex vivo fluorescence (Figure 1C, SI Figures S2–3).
Figure 1.
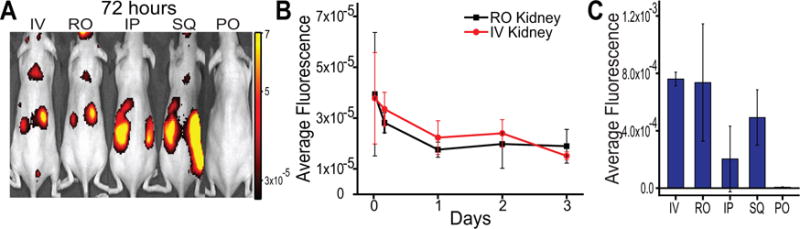
Investigation of route of nanoparticle administration. A) In vivo near-infrared fluorescence images of nanoparticle-encapsulated dye in mice 72 hours following administration via different administration routes: IV=intravenous tail vein; RO=intravenous retroorbital; IP=intraperitoneal; SQ=subcutaneous flank; PO=per os, oral gavage. B) Fluorescence efficiency of renal nanoparticle localization at indicated timepoints following injection, for intravenous administration routes. Data represent mean ± standard deviation, N = 3. C) Average fluorescence efficiency of mouse kidneys, measured after sacrifice 72 hours after animals were administered with nanoparticles via different routes. Data are background subtracted and represent mean ± standard deviation, N = 3.
We next sought to assess the relative selectivity of IV-administered MNPs to the kidneys. We hypothesized that non-specific accumulation in other organs may be reduced by decreasing the total dose of particles. To investigate this, we injected three 4–8 week healthy female hairless mice bearing intact immune systems with nanoparticles containing fluorescent dye via tail vein intravenous administration. Mice were injected with 25 mg/kg nanoparticles (MNPs) or PBS and imaged 72 hours post-injection via fluorescence (IVIS) in vivo (Figure 2A). At 72 hours post-injection, mice were euthanized, and organs were harvested and imaged via IVIS (SI Figure S5). The intensity in each organ was compared to that from a 50 mg/kg IV administration of MNPs (Figure 2B)17. The results revealed that the decreased dose resulted in a 26-fold greater accumulation in the kidneys than in any other organ (Figure 2C). Whether we measured the per-area fluorescence of each organ (Figure 2B) or the total fluorescence normalized by organ weight (SI Figure S6A), to account for the 3-dimensionality of the organs, we found similar selectivity (SI Figure S6B). This accumulation is significantly greater than the 5-fold greater accumulation we found with this anionic MNP particle formulation at 50 mg/kg. As expected, the total renal fluorescence was half of the 50 mg/kg administration; however the accumulation in other organs was proportionately much less (Figure 2B). To further explore this phenomenon, we injected three mice each with 5 mg/kg MNPs. We found that, at this dose, we were still able to observe significant renal localization (SI Figure S7). However, the renal targeting efficiency was not as great as at the dose of 25 mg/kg (Figure 2C). Lower concentrations approached the limit of fluorescence imaging above background autofluorescence. For a dose of 25 mg/kg, we found 26 to 94-fold greater accumulation of nanoparticles in the kidneys than any other measured organ, as compared to 5 to 27-fold greater kidney localization with the 50 mg/kg dose, and 4 to 17-fold greater localization with the 5 mg/kg dose (Figure 2C).
Figure 2.
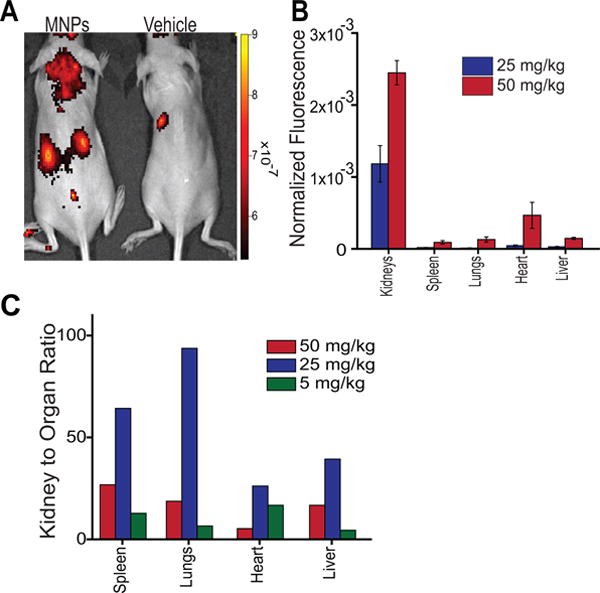
Dependence of nanoparticle dose on renal targeting efficiency. A) Dorsal in vivo fluorescence images of mice injected IV with 25 mg/kg nanoparticles or PBS vehicle control. B) Average fluorescence efficiency per organ of mice injected with 25 mg/kg or 50 mg/kg MNPs measured 72 hours after nanoparticle administration. Data are background subtracted and represent mean ± standard deviation. N = 3 for each MNP dose and N = 1 for control. C) Comparison of fluorescence intensity in the kidneys to other organs upon IV injection of 50, 25, or 5 mg/kg MNPs. Data represent the quotient of normalized average whole-kidney fluorescence and that of the specified organ.
To further assess the renal targeting of the MNP system, we formulated a 385.7 ± 2.7 nm nanoparticle encapsulating a Cy5-labeled double-stranded DNA. We administered a dose of 50 mg/kg, to ensure sufficient signal due to reduced total fluorescence encapsulation, intravenously to three mice. We found significant renal localization via fluorescence organ imaging ex vivo (SI Figure S8). These experiments suggest that the renal targeting capability of MNPs is not dependent upon the encapsulated cargo, and that this system has the capability to deliver hydrophobic or hydrophilic cargo, whether small molecules (DEDC dye) or larger biomolecules (Cy5-labeled dsDNA) to the kidneys.
Evaluation of Renal Tissue Accumulation
We conducted intravital and ex vivo microscopy to assess renal distribution of the nanoparticles. Transgenic Cx3crgfp/+ C57BL/6 mice, with green fluorescent protein (GFP)-expressing renal macrophages20, 21, were imaged intravitally via confocal microscopy. The mice were injected with fluorescent MNPs or an equal amount of unencapsualted dye. After 72 hours, a kidney was exposed in an anesthetized mouse and the superficial renal cortex was imaged under parameters developed previously21. The results showed bright near-infrared fluorescence in the tubular epithelial cells in mice receiving MNPs, but little-to-no near-infared emission in the mice administered unencapsulated dye (Figure 3A–B). The fluorescence from nanoparticles appeared to be localized to the tubules with almost no emission detected in the regions of the GFP-expressing macrophages. Due to the lack of fluorescent signal in the blood vessels or lumen of the tubules, it also appeared that there was no accumulation at these sites. We confirmed this localization via fluorescence microscopy on fluorescently-stained renal tissue sections ex vivo. Perfused SKH1 mice were used to reduce autofluorescence from red blood cells26 (Figure 3C–D). These results confirmed primarily tubular localization of the mesoscale nanoparticles.
Figure 3.
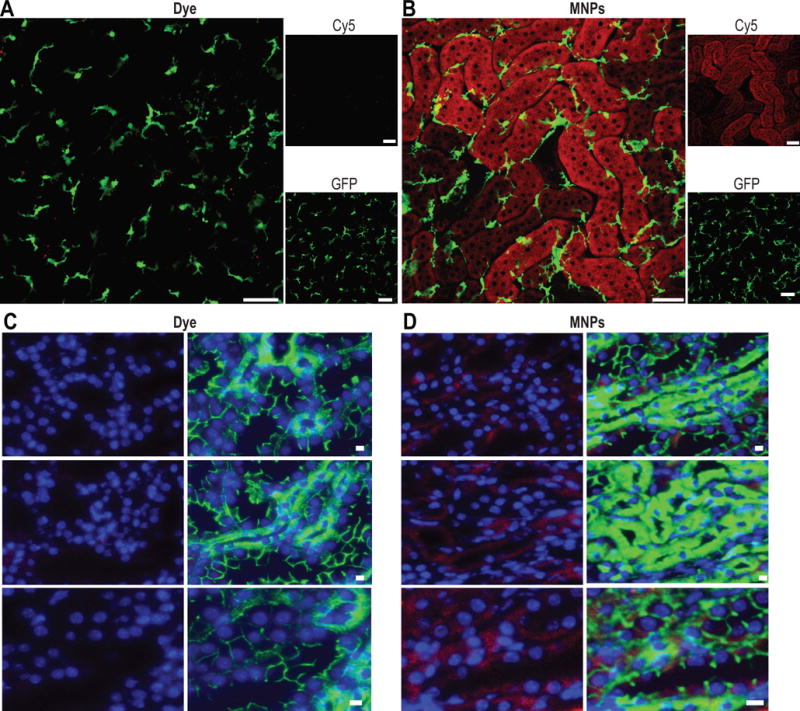
Renal tissue imaging. Intravital microscopy of mice injected IV with A) fluorescent dye (equal to amount found in particles) or B) 25 mg/kg MNPs. Animals were imaged at 72 hours following injection. Red denotes dye fluorescence (free or in nanoparticles; imaged using Cy5 filter); green denotes GFP-expressing renal macrophages. Scale bars are 50 μm for panels A and B. C) Immunofluorescence imaging of fixed tissues from perfused mice injected with C) dye alone (equal to amount found in particles) or D) 25 mg/kg MNPs. Red denotes dye fluorescence (free or in nanoparticles); green denotes E-cadherin staining (strong staining for proximal tubular epithelial cells, weak staining for distal tubular epithelial cells); blue denotes DAPI nuclear stain. Scale bars are 10 μm for panels C and D.
Pharmacological Dose Safety Evaluation
Finally, we sought to investigate the safety profile of the nanoparticles. Using 4–8 week old female BALB/c mice (BALB/cAnNCrl), we injected nanoparticles and sacrificed at day 1, 3, 7, or 28 post-injection. The vehicle control was sacrificed at 7 days (N = 3). We found that the nanoparticles persisted in the kidneys for up to 28 days (Figure 4A). Average fluorescence decayed over this time span, however prior work found that nanoparticles are no longer apparent in the kidney after two months17. Renal function at the proposed pharmacological dose was measured via blood urea nitrogen and creatinine, as well as total protein, globulin, albumin, and ion levels in the serum and urine protein levels, which were measured from biofluids collected at the time of sacrifice. These experiments found no significant changes in these markers and therefore no adverse effects on renal function over the one-month period measured following injection (Figure 4B–C, SI Figure S9 A–B). We also investigated liver function in mice 28 days after injection with MNPs and found no changes in major serum markers, including alkaline phosphatase (ALP), alanine aminotransferase (ALT), and aspartate aminotransferase (AST), among others (SI Figure S10). We further investigated the systemic safety profile via a complete blood count. Platelets, white blood cells, red blood cells, hematocrit, and hemoglobin were assessed, among other metrics. These experiments showed no biologically-significant results and few statistically-significant differences in the assessments between any of the nanoparticle-administered groups and the control group (Figure 4D, SI Figure 9C). These biochemical measurements were corroborated by histological analysis of renal tissue, which showed no pathological damage or local inflammation due to particle administration (Figure 4E, SI Figure S11). Additionally, anti-F4/80 staining revealed no increased presence of macrophages in the kidneys of mice injected with MNPs 28 days following injection (SI Figure S12). We also assessed the histology of the lungs, heart, liver, and spleen, finding no inflammation or other signs of damage (SI Figure S11).
Figure 4.
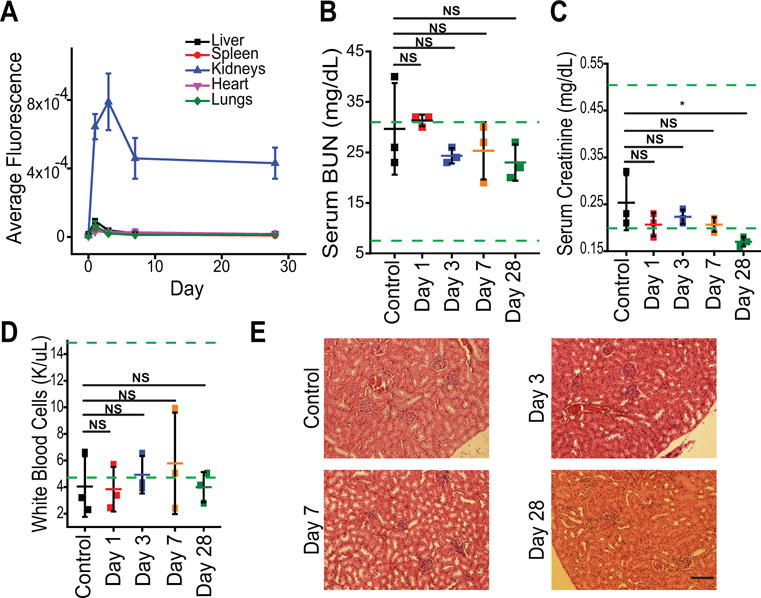
Nanoparticle safety studies. A) Average organ fluorescence measured following sacrifice of mice at each time point after IV tail vein injection of 25 mg/kg MNPs. Data represent mean ± standard deviation,. N = 3. B) Serum blood urea nitrogen of mice sacrificed at Days 1, 3, 7, and 28 post-injection. C) Serum creatinine of mice injected with 25 mg/kg MNPs. * = p > 0.05. D) White blood cell count of mice injected with 25 mg/kg MNPs. For B–D, control represents mice injected with PBS vehicle and sacrificed at Day 1 post-injection. Each point represents data from an individual mouse; center bar represents mean ± standard deviation, N = 3. Green lines denote guideline normal values published by Charles River for male BALB/C mice 8–10 weeks in age. Statistical significance was calculated using a one-way ANOVA with Tukey post-test; NS = p > 0.05. E) Representative renal H&E stained images of mice injected with MNPs and sacrificed at the denoted time-points. Scale bar = 100 μm.
DISCUSSION
In this work, we investigated a nanoparticle technology which targets the kidneys with high selectivity to the tubules. The need for kidney-targeted medicines is based on the relatively poor pharmacokinetic profiles of investigative therapeutics for renal disease, which include a short renal retention time, low accumulation at the site of disease, and systemic off-target effects27. Drug delivery technologies may be used to control the release of therapies in renal tissues, potentially allowing for less-frequent administration and improved compliance28, 29. We investigated the localization and safety of mesoscale nanoparticle technology as it relates to renal, systemic, and hematological parameters.
We found that localization of mesoscale nanoparticles to the kidneys was not contingent upon IV administration, but that this was the most optimal route. In prior work we found that nanoparticle localization to the kidneys is dependent on the relatively large size and hydrophilic PEG surface chemistry of the particle, but not the surface charge17. Thus, for proper localization to the kidney, vascular access is necessary and is most efficient via direct vascular injection, as other routes of administration may hinder access to the vasculature due to the relatively large particle size30. Although other works show that oral administration of certain nanoparticle formulations increased drug absorbance31, we did not see evidence of renal localization of the nanoparticles via this route. This work suggests that the mesoscale nanoparticles must be administered via intravenous administration to result in bioavailability in the kidneys.
The highly selective accumulation reported here is unique to this nanoparticle system, as renal selectivity of nanomaterials is uncommon17. Reports of other nanoparticle systems that predominantly localize in the kidneys found no more than two-fold selectivity of localization in the kidneys32, 33. While studies found some nanoparticle localization in specific renal structures, they largely achieved minimal-to-moderate selectivity to the kidneys34, 35. The current work found renal uptake of mesoscale nanoparticles with 26–94-fold higher efficiency than other organs, which is an improvement from a previous finding of 5–7 fold selectivity17. We additionally found that the renal localization was not dependent on the cargo encapsulated within the particle, portending the use of this particle system for delivery of large or small hydrophobic or hydrophilic molecules.
We found that when an equal amount of free dye (not encapsulated in MNPs) was administered, little fluorescence signal was found in the kidneys. This suggests that signal found in the kidneys in each experiment presented here was due to renal accumulation of the nanoparticles and not accumulation of the dye itself.
We found that the mesoscale nanoparticles localized specifically within tubular epithelial cells. Although this is likely due to a physiologic phenomenon, as there were no targeting moieties (such as peptides, antibodies, etc.) on the nanoparticles, analogous findings are rare in the literature. Nanoparticle targeting of renal proximal tubules has been shown via glomerular filtration of carbon nanotubes that translocate to the nucleus of these cells36 and deliver oligonucleotide therapeutics for AKI treatment37. Another study found a similar localization for chitosan/siRNA nanoparticles, although this material was hypothesized to disintegrate at the glomerulus and be separately reabsorbed by tubular epithelial cells38. We believe, however, that the large size (~350–400 nm) of MNPs precludes glomerular filtration (10 nm cutoff)18, 39 and suggests transcytosis across the peritubular capillary endothelium17, 40, which is supported by the data presented here. Further investigation is warranted to investigate the details of this mechanism.
We investigated the safety of these materials at the renal, hematological, and systemic levels with nanoparticle residence in the kidneys up to one month. The material system is chemically similar to several other nanoparticle classes that do not target the kidneys41, 42. The core material, PLGA, is FDA-approved and has been used as a clinical biomaterial for decades in sutures43, 44, drug depots45, and in nanomaterials46 due to its controlled hydrolytic degradation into lactic and glycolic acids which are harmlessly metabolized by the body44, 47. While the widely-used and FDA-approved PEG, which coats the surface of the MNPs, may have minor immunogenicity48, 49, this is most often seen with longer PEG chains than those used here (5 kDa) and has caused no ill-effects clinically due to its relative low abundance in administered formulations50. In prior work, we found that PEG is necessary to target the kidneys and avoid complement protein binding and particle opsonization, which results in localization to the liver due to uptake by resident macrophages and the mononuclear phagocyte system (MPS)17, 51. The effects of these materials in the kidneys, however, were heretofore unknown. Using complete renal serum and urine panels, and histology, we found no negative consequences of selective targeting to the kidneys, long-term renal localization, or cargo release on renal function. Further, we could find no evidence of inhibition of liver function, inflammation, or hematological problems.
While this work found selective renal targeting and renal tubular localization of mesoscale nanoparticles, further work is necessary to realize its clinical potential. One potential limitation of this system is its specific site of localization within the kidney, which may render it less capable of targeting components of the glomerulus or other parts of the kidney52, 53. Furthermore, although the basic materials used in this nanoparticle system are used in various clinical applications, a full toxicological evaluation will be necessary in other animals and humans using MNPs loaded with a therapeutic cargo. Finally, a full evaluation of the mechanism of tubular localization is warranted to further the value of this system. However, a clear path exists to realize the clinical significance of this nanotechnology.
PERSPECTIVES
Here, we present an investigation of a technology that may have implications for the treatment of renal disease. We found that mesoscale nanoparticles localize in the kidneys with 26–94-fold efficiency over other organs, and that they specifically target the tubules. We also found that the material exhibits no inhibition of renal function or systemic toxicity. Therapeutic delivery by renal-selective nanoparticles portends advances in drug development for kidney diseases, including the use of new therapeutic agents, improved therapeutic efficacy, reduced systemic toxicities, and the control of drug release.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What is new?
Mesoscale nanoparticles selectively target the kidneys (26-fold greater than other organs)
Intravital imaging of renal tubule nanoparticle localization
Safety of renal-specific nanoparticle accumulation
What is relevant?
The renal localization and safety of these particles portends treatment of kidney-related diseases
Proximal tubular localization may facilitate treatment of AKI and CKD, both causes of and caused by hypertension
Long-term cargo release may allow for reduced therapeutic dosing intervals, increasing patient adherence
Summary
We found that renal-selective (26-fold more than other organs) mesoscale nanoparticles target the renal tubules, degrade over the course of a month, and are safe both renally and systemically.
Acknowledgments
The authors would like to acknowledge the Antitumor Assessment Core Facility, the Molecular Cytology Core Facility, the Center of Comparative Medicine and Pathology, and the Small Animal Imaging Core Facility at Memorial Sloan Kettering Cancer Center. The authors would like to thank Amanda Kulick for assistance with administration route experiments and Efstathios Stamatiades for assistance with preliminary intravital imaging experiments.
Sources of Funding
This work was supported by the NIH New Innovator Award (DP2-HD075698), the NIH Cancer Center Support Grant (P30 CA008748), the Pershing Square Sohn Cancer Research Alliance, the Honorable Tina Brozman Foundation for Ovarian Cancer Research, the Louis V. Gerstner Jr. Young Investigator’s Fund, the Frank A. Howard Scholars Program, the Alan and Sandra Gerry Metastasis Research Initiative, the Center for Molecular Imaging and Nanotechnology at Memorial Sloan Kettering Cancer Center, Cycle for Survival, the Anna Fuller Fund, the Expect Miracles Foundation - Financial Services Against Cancer, Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, the Imaging and Radiation Sciences Program, and the Experimental Therapeutics Center at Memorial Sloan Kettering Cancer Center. RMW was supported by the Ovarian Cancer Research Fund Mentored Investigator Award [#370463] and the American Heart Association Postdoctoral Fellowship [17POST33650043]. HT was supported by the National Cancer Institute of the NIH under Award Number R25CA020449.
Footnotes
MS ID: HYPE201709843R1
Disclosure: All authors declare no financial conflicts of interest.
References
- 1.Collins AJ, Foley RN, Chavers B, et al. ‘United states renal data system 2011 annual data report: Atlas of chronic kidney disease & end-stage renal disease in the united states. American journal of kidney diseases. 2012;59(A7):e1–420. doi: 10.1053/j.ajkd.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 2.United States Rrenal Data System Annual data report: Atlas of chronic kidney disease and end stage renal disease in the united states. Bethesda, MD: National institutes of health, national institute of diabetes and digestive and kidney diseases; 2013. [Google Scholar]
- 3.Kaufman J, Dhakal M, Patel B, Hamburger R. Community-acquired acute renal failure. American journal of kidney diseases. 1991;17:191–198. doi: 10.1016/s0272-6386(12)81128-0. [DOI] [PubMed] [Google Scholar]
- 4.Schiffl H. Renal recovery from acute tubular necrosis requiring renal replacement therapy: A prospective study in critically ill patients. Nephrology, dialysis, transplantation. 2006;21:1248–1252. doi: 10.1093/ndt/gfk069. [DOI] [PubMed] [Google Scholar]
- 5.Uchino S, Kellum JA, Bellomo R, et al. Acute renal failure in critically ill patients: A multinational, multicenter study. JAMA. 2005;294:813–818. doi: 10.1001/jama.294.7.813. [DOI] [PubMed] [Google Scholar]
- 6.McCarthy JT. Prognosis of patients with acute renal failure in the intensive-care unit: A tale of two eras. Mayo Clinic proceedings. 1996;71:117–126. doi: 10.4065/71.2.117. [DOI] [PubMed] [Google Scholar]
- 7.de Mendonca A, Vincent JL, Suter PM, Moreno R, Dearden NM, Antonelli M, Takala J, Sprung C, Cantraine F. Acute renal failure in the icu: Risk factors and outcome evaluated by the sofa score. Intensive Care Med. 2000;26:915–921. doi: 10.1007/s001340051281. [DOI] [PubMed] [Google Scholar]
- 8.Metnitz PG, Krenn CG, Steltzer H, Lang T, Ploder J, Lenz K, Le Gall JR, Druml W. Effect of acute renal failure requiring renal replacement therapy on outcome in critically ill patients. Critical care medicine. 2002;30:2051–2058. doi: 10.1097/00003246-200209000-00016. [DOI] [PubMed] [Google Scholar]
- 9.Coresh J, Astor BC, Greene T, Eknoyan G, Levey AS. Prevalence of chronic kidney disease and decreased kidney function in the adult us population: Third national health and nutrition examination survey. American journal of Kidney diseases. 2003;41:1–12. doi: 10.1053/ajkd.2003.50007. [DOI] [PubMed] [Google Scholar]
- 10.Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. The New England journal of medicine. 2004;351:1296–1305. doi: 10.1056/NEJMoa041031. [DOI] [PubMed] [Google Scholar]
- 11.De Caestecker M, Humphreys BD, Liu KD, Fissell WH, Cerda J, Nolin TD, Askenazi D, Mour G, Harrell FE, Pullen N. Bridging translation by improving preclinical study design in aki. Journal of the American Society of Nephrology. 2015 doi: 10.1681/ASN.2015070832. ASN. 2015070832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Molitoris BA. Therapeutic translation in acute kidney injury: The epithelial/endothelial axis. The Journal of clinical investigation. 2014;124:2355–2363. doi: 10.1172/JCI72269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zarjou A, Sanders PW, Mehta RL, Agarwal A. Enabling innovative translational research in acute kidney injury. Clinical and translational science. 2012;5:93–101. doi: 10.1111/j.1752-8062.2011.00302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levey AS, Coresh J. Chronic kidney disease. The Lancet. 2012;379:165–180. doi: 10.1016/S0140-6736(11)60178-5. [DOI] [PubMed] [Google Scholar]
- 15.Hostetter TH. Prevention of the development and progression of renal disease. Journal of the American Society of Nephrology. 2003;14:S144–S147. doi: 10.1097/01.asn.0000070150.60928.06. [DOI] [PubMed] [Google Scholar]
- 16.Riegersperger M, Sunder‐Plassmann G. How to prevent progression to end stage renal disease. Journal of renal care. 2007;33:105–107. doi: 10.1111/j.1755-6686.2007.tb00053.x. [DOI] [PubMed] [Google Scholar]
- 17.Williams RM, Shah J, Ng BD, Minton DR, Gudas LJ, Park CY, Heller DA. Mesoscale nanoparticles selectively target the renal proximal tubule epithelium. Nano Letters. 2015;15:2358–2364. doi: 10.1021/nl504610d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams RM, Jaimes EA, Heller DA. Nanomedicines for kidney diseases. Kidney International. 2016;90:740–745. doi: 10.1016/j.kint.2016.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng J, Teply BA, Sherifi I, Sung J, Luther G, Gu FX, Levy-Nissenbaum E, Radovic-Moreno AF, Langer R, Farokhzad OC. Formulation of functionalized plga–peg nanoparticles for in vivo targeted drug delivery. Biomaterials. 2007;28:869–876. doi: 10.1016/j.biomaterials.2006.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor cx3cr1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stamatiades EG, Tremblay M-E, Bohm M, Crozet L, Bisht K, Kao D, Coelho C, Fan X, Yewdell WT, Davidson A, Heeger PS, Diebold S, Nimmerjahn F, Geissmann F. Immune monitoring of trans-endothelial transport by kidney-resident macrophages. Cell. 2016;166:991–1003. doi: 10.1016/j.cell.2016.06.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Astete CE, Sabliov CM. Synthesis and characterization of plga nanoparticles. Journal of Biomaterials Science, Polymer Edition. 2006;17:247–289. doi: 10.1163/156856206775997322. [DOI] [PubMed] [Google Scholar]
- 23.Cheng CJ, Tietjen GT, Saucier-Sawyer JK, Saltzman WM. A holistic approach to targeting disease with polymeric nanoparticles. Nature Reviews Drug Discovery. 2015;14:239–247. doi: 10.1038/nrd4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bednarski M, Dudek M, Knutelska J, Nowiński L, Sapa J, Zygmunt M, Nowak G, Luty-Błocho M, Wojnicki M, Fitzner K. The influence of the route of administration of gold nanoparticles on their tissue distribution and basic biochemical parameters: In vivo studies. Pharmacological Reports. 2015;67:405–409. doi: 10.1016/j.pharep.2014.10.019. [DOI] [PubMed] [Google Scholar]
- 25.Zhang X-D, Wu H-Y, Wu D, Wang Y-Y, Chang J-H, Zhai Z-B, Meng A-M, Liu P-X, Zhang L-A, Fan F-Y. Toxicologic effects of gold nanoparticles in vivo by different administration routes. International journal of nanomedicine. 2010;5:771–781. doi: 10.2147/IJN.S8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barclay GR, Tura O, Samuel K, Hadoke PW, Mills NL, Newby DE, Turner ML. Systematic assessment in an animal model of the angiogenic potential of different human cell sources for therapeutic revascularization. Stem cell research & therapy. 2012;3:1. doi: 10.1186/scrt114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jo SK, Rosner MH, Okusa MD. Pharmacologic treatment of acute kidney injury: Why drugs haven’t worked and what is on the horizon. Clinical journal of the American Society of Nephrology. 2007;2:356–365. doi: 10.2215/CJN.03280906. [DOI] [PubMed] [Google Scholar]
- 28.Burnier M, Pruijm M, Wuerzner G, Santschi V. Drug adherence in chronic kidney diseases and dialysis. Nephrology Dialysis Transplantation. 2015;30:39–44. doi: 10.1093/ndt/gfu015. [DOI] [PubMed] [Google Scholar]
- 29.Ghimire S, Castelino RL, Lioufas NM, Peterson GM, Zaidi STR. Nonadherence to medication therapy in haemodialysis patients: A systematic review. PloS one. 2015;10:e0144119. doi: 10.1371/journal.pone.0144119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barua S, Mitragotri S. Challenges associated with penetration of nanoparticles across cell and tissue barriers: A review of current status and future prospects. Nano today. 2014;9:223–243. doi: 10.1016/j.nantod.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Fan J, Liu Y, Zhao B, Jia Z-R, Zhang Q. Bioavailability and pharmacokinetics of sorafenib suspension, nanoparticles and nanomatrix for oral administration to rat. Int J Pharm. 2011;419:339–346. doi: 10.1016/j.ijpharm.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 32.Shenoy D, Little S, Langer R, Amiji M. Poly (ethylene oxide)-modified poly (β-amino ester) nanoparticles as a ph-sensitive system for tumor-targeted delivery of hydrophobic drugs: Part 2. In vivo distribution and tumor localization studies. Pharmaceutical research. 2005;22:2107–2114. doi: 10.1007/s11095-005-8343-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fischer NO, Weilhammer DR, Dunkle A, Thomas C, Hwang M, Corzett M, Lychak C, Mayer W, Urbin S, Collette N. Evaluation of nanolipoprotein particles (nlps) as an in vivo delivery platform. PloS one. 2014;9:e93342. doi: 10.1371/journal.pone.0093342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nair AV, Keliher EJ, Core AB, Brown D, Weissleder R. Characterizing the interactions of organic nanoparticles with renal epithelial cells in vivo. ACS nano. 2015;9:3641–3653. doi: 10.1021/acsnano.5b00428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi CHJ, Zuckerman JE, Webster P, Davis ME. Targeting kidney mesangium by nanoparticles of defined size. Proc Natl Acad Sci. 2011;108:6656–6661. doi: 10.1073/pnas.1103573108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruggiero A, Villa CH, Bander E, Rey DA, Bergkvist M, Batt CA, Manova-Todorova K, Deen WM, Scheinberg DA, McDevitt MR. Paradoxical glomerular filtration of carbon nanotubes. Proc Natl Acad Sci. 2010;107:12369–12374. doi: 10.1073/pnas.0913667107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alidori S, Akhavein N, Thorek DL, Behling K, Romin Y, Queen D, Beattie BJ, Manova-Todorova K, Bergkvist M, Scheinberg DA. Targeted fibrillar nanocarbon rnai treatment of acute kidney injury. Science translational medicine. 2016;8:331ra339–331ra339. doi: 10.1126/scitranslmed.aac9647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao S, Hein S, Dagnæs-Hansen F, Weyer K, Yang C, Nielsen R, Christensen EI, Fenton RA, Kjems J. Megalin-mediated specific uptake of chitosan/sirna nanoparticles in mouse kidney proximal tubule epithelial cells enables aqp1 gene silencing. Theranostics. 2014;4:1039. doi: 10.7150/thno.7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zuckerman JE, Davis ME. Targeting therapeutics to the glomerulus with nanoparticles. Advances in chronic kidney disease. 2013;20:500–507. doi: 10.1053/j.ackd.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 40.Williams SK, Greener DA, Solenski NJ. Endocytosis and exocytosis of protein in capillary endothelium. Journal of cellular physiology. 1984;120:157–162. doi: 10.1002/jcp.1041200208. [DOI] [PubMed] [Google Scholar]
- 41.Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. Plga-based nanoparticles: An overview of biomedical applications. J Controlled Release. 2012;161:505–522. doi: 10.1016/j.jconrel.2012.01.043. [DOI] [PubMed] [Google Scholar]
- 42.Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces. 2010;75:1–18. doi: 10.1016/j.colsurfb.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 43.Cutright DE, Beasley JD, Perez B. Histologic comparison of polylactic and polyglycolic acid sutures. Oral Surgery, Oral Medicine, Oral Pathology. 1971;32:165–173. doi: 10.1016/0030-4220(71)90265-9. [DOI] [PubMed] [Google Scholar]
- 44.Hines DJ, Kaplan DL. Poly (lactic-co-glycolic) acid– controlled-release systems: Experimental and modeling insights. Critical Reviews™ in Therapeutic Drug Carrier Systems. 2013;30 doi: 10.1615/critrevtherdrugcarriersyst.2013006475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jeong B, Bae YH, Kim SW. Drug release from biodegradable injectable thermosensitive hydrogel of peg–plga–peg triblock copolymers. J Controlled Release. 2000;63:155–163. doi: 10.1016/s0168-3659(99)00194-7. [DOI] [PubMed] [Google Scholar]
- 46.Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized pt (iv) prodrug-plga–peg nanoparticles. Proc Natl Acad Sci. 2008;105:17356–17361. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Siegel SJ, Kahn JB, Metzger K, Winey KI, Werner K, Dan N. Effect of drug type on the degradation rate of plga matrices. European Journal of Pharmaceutics and Biopharmaceutics. 2006;64:287–293. doi: 10.1016/j.ejpb.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 48.Schlapschy M, Binder U, Börger C, Theobald I, Wachinger K, Kisling S, Haller D, Skerra A. Pasylation: A biological alternative to pegylation for extending the plasma half-life of pharmaceutically active proteins. Protein Engineering, Design & Selection. 2013;26:489–501. doi: 10.1093/protein/gzt023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu Y, Reidler H, Pan J, Milunic D, Qin D, Chen D, Vallejo YR, Yin R. A double antigen bridging immunogenicity elisa for the detection of antibodies to polyethylene glycol polymers. J Pharmacol Toxicol Methods. 2011;64:238–245. doi: 10.1016/j.vascn.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 50.Caliceti P, Veronese FM. Pharmacokinetic and biodistribution properties of poly (ethylene glycol)–protein conjugates. Adv Drug Del Rev. 2003;55:1261–1277. doi: 10.1016/s0169-409x(03)00108-x. [DOI] [PubMed] [Google Scholar]
- 51.Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, Langer R. Biodegradable long-circulating polymeric nanospheres. Science. 1994;263:1600–1603. doi: 10.1126/science.8128245. [DOI] [PubMed] [Google Scholar]
- 52.Liao J, Hayashi K, Horikoshi S, Ushijima H, Kimura J, Tomino Y. Effect of steroid-liposome on immunohistopathology of iga nephropathy in ddy mice. Nephron. 2001;89:194–200. doi: 10.1159/000046067. [DOI] [PubMed] [Google Scholar]
- 53.Suana AJ, Tuffin G, Frey BM, Knudsen L, Muhlfeld C, Rodder S, Marti HP. Single application of low-dose mycophenolate mofetil-ox7-immunoliposomes ameliorates experimental mesangial proliferative glomerulonephritis. J Pharmacol Exp Ther. 2011;337:411–422. doi: 10.1124/jpet.110.176222. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.