

Abstract
Group A Streptococccus (GAS) is a major human pathogen that causes significant morbidity and mortality. Zinc is an essential trace element required for GAS growth, however, zinc can be toxic at excess concentration. The bacterial strategies to maintain zinc sufficiency without incurring zinc toxicity plays a crucial role in host-GAS interactions and has significant impact on GAS pathogenesis. Host deploys nutritional immune mechanisms to retard GAS growth by causing either zinc deprivation or zinc poisoning. However, GAS overcomes the zinc-dependent host defenses and survives in the hostile environment by employing complex adaptive strategies. In this review, we describe the different host immune strategies that employ either zinc limitation or zinc toxicity in different host environments to control GAS infection. We also discuss the molecular mechanisms and machineries used by GAS to evade host nutritional defenses and establish successful infection. Emerging evidence suggest that the metal transporters are major GAS virulence factors as they compete against host nutritional immune mechanisms to acquire or expel metals and promote bacterial survival in the host. Thus, identification of GAS molecules and elucidation of the mechanisms by which GAS combats host-mediated alterations in zinc availability may lead to novel interference strategies targeting GAS metal acquisition systems.
Graphical abstract
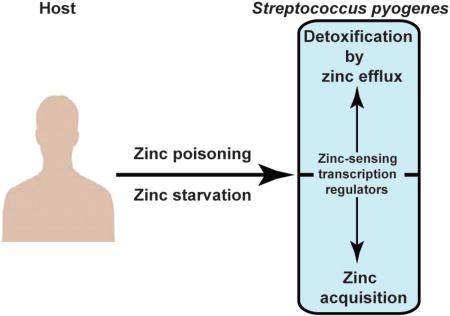
The ability of Streptococcus pyogenes to resist host-mediated zinc starvation or poisoning is critical for bacterial pathogenesis.
Introduction
Streptococcus pyogenes, also known as group A streptococcus (GAS), is a human-only pathogen that infects diverse anatomical sites and causes a variety of diseases. Globally, GAS causes ~700 million cases of pharyngitis, ~111 million cases of impetigo, and 18 million cases of invasive diseases annually.1,2 As a result, GAS infections are one of the top ten infectious causes of mortality worldwide. Furthermore, untreated, recurring mild infections can lead to development of rheumatic heart disease (RHD), the leading cause of preventable pediatric heart disease.3,4 Although antibiotics are effective in treating milder infections, management of invasive infections and RHD requires surgical intervention, prolonged treatment with antibiotics, and systemic immunoglobulin therapy.5,6 The situation is further exacerbated by the continuing rise in antibiotic resistance, reemergence of GAS epidemics, and lack of a human vaccine.7–16 Despite extensive efforts to develop a GAS vaccine for nearly a century, major roadblocks, such as the risk of triggering RHD by GAS antigens, antigenic variation among the 200 different emm serotypes, and lack of cross-serotypic coverage have prevented the realization of this goal.5,11,12,17–20 Thus, the critical need for an effective GAS vaccine remains unfulfilled and rigorous investigations into bacterial signaling pathways involved in host-GAS interactions during infection are imperative to identify novel GAS vaccine candidates.
GAS is a versatile pathogen, colonizing different host environments that vary in nutrient availability, host defense mechanisms, and tissue anatomical barriers. Thus, the ability to sense fluctuating host environments and acquire nutrients is crucial for GAS survival in vivo. As with other organisms, zinc is an essential micronutrient for GAS survival.21 Due to its unique chemical properties, zinc serves as an efficient structural and catalytic cofactor for several bacterial proteins. Zinc is required for proper folding and/or function of metalloproteins involved in fundamental cellular processes, such as transcription, translation, DNA replication, DNA repair, metabolic enzymes, and virulence. In addition, GAS also encodes several zinc-containing virulence factors that are critical for bacterial virulence.22,23 However, excess zinc can be deleterious to bacterial growth. Zinc is at the top of the Irving-Williams series of the stability of metal complexes, indicating that it can form tighter and more stable interactions with metal ligands compared to other transition metals. Thus, excess zinc can bind to non-cognate metalloproteins and render them dysfunctional. In streptococcus pneumoniae, at toxic levels, zinc binds to manganese transporter PsaA, and impairs bacterial oxidative stress defense by preventing manganese acquisition.24,25 Similarly, under surplus growth conditions, zinc binds adventitiously to non-cognate sites in several important bacterial enzymes and inhibits the catalytic activity of metalloenzymes.26–28
The bacterial need to maintain the delicate balance between zinc deprivation and zinc intoxication is targeted by host immune defenses to control microbial growth.29 To inhibit bacterial growth, the host employs various innate immune effector molecules to impose either zinc deprivation or zinc intoxication on the invading pathogen by a process referred to as nutritional immunity.29,30 Bacterial pathogens counter the host-induced zinc immune strategies by modulating the expression of specific sets of genes that promote their survival under conditions of zinc limitation or excess.31 The primary response to zinc limitation or zinc toxicity involves upregulation of zinc uptake or efflux system, respectively. In addition, pathogens also evolved additional molecular strategies to reprogram their metabolic needs, which facilitate their survival under zinc stress growth conditions.31 Typically, the expression of bacterial adaptive responses to opposing zinc stress conditions is coupled with two different zinc-sensing transcription regulators. The highly nuanced bacterial metalloregulators sense the alterations in zinc levels, and orchestrate the expression of genes involved in bacterial responses accordingly. In this review, we discuss the host nutritional immune mechanisms that impose zinc limitation or toxicity on GAS during infection. We also describe the bacterial counterstrategies that aid GAS to overcome host nutritional defenses and contribute to bacterial pathogenesis. The identification of GAS molecular arsenals against host defenses and the characterization of their roles are expected to reveal new prophylactic or therapeutic opportunities to combat GAS infections.
Host-imposed zinc limitation during GAS infection
The host employs several nutritional immune mechanisms to control bacterial growth by limiting metal availability. Phagosomal iron or manganese depletion by the divalent metal-proton exporter, NRAMP1, is a well-documented host antimicrobial strategy.32–34 Similarly, other host immune effectors such as lactoferrin, transferrin, hepcidin, and lipocalin also scavenge iron from the bacterial colonization surfaces and limit bacterial proliferation.35–38 Eukaryotic hosts also possess mechanisms to impose zinc limitation on bacterial pathogens. The members of the S100 family are major players in imposing host-mediated zinc limitation at bacterial infection sites.39–42 Among the various S100 proteins, calprotectin (CP) is the best-characterized antimicrobial zinc chelator.43–47 CP is a heterodimer of S100A8 and S100A9 proteins, which is present abundantly at various bacterial colonization surfaces in the host.45,47–49 CP is produced primarily by myeloid cells, which include neutrophils, monocytes, and macrophages.50 CP constitutes approximately 50% of the cytosolic protein content of neutrophils and it is recruited to the infection sites as part of the first line of host innate defenses.43,44,47 CP is released from the neutrophils and the extracellular CP exerts its antimicrobial activity against several human pathogens by calcium-dependent chelation of zinc and manganese.30,42,46–48,51–56 Structurally, each subunit of CP has two calcium-binding EF-hand motifs and occupancy of calcium at each EF-hand motif is necessary for transition metal binding by CP.45,46 In addition, CP heterodimer has two metal binding sites located at the intersubunit dimerization interface.45,46 Although both sites can bind zinc with affinities in the picomolar to nanomolar range, only site 2 along with additional metal ligands from the C-terminal tail of S100A9 participate in manganese sequestration.46,57
GAS encounters CP during infection and needs to overcome CP-mediated metal sequestration to establish successful colonization.49 CP was present in sub milligram quantities at GAS colonization surfaces in two different mouse models of invasive infection, indicating that CP participates in host defense against GAS infection (Fig. 1).49 During in vitro growth, CP chelates transition metals including zinc from the growth medium (Fig. 1). When grown in the presence of CP, GAS senses CP-mediated zinc sequestration and responds by upregulating expression of the genes involved in GAS adaptive responses to zinc limitation (discussed below).49 Although CP inhibits GAS growth at higher concentrations, GAS outcompetes CP and sustains growth at concentrations comparable to CP levels observed at infection sites. These observations indicate that GAS possesses molecular arsenals to overcome CP-mediated zinc sequestration and survives in the host. Interestingly, growth of several streptococcal pathogens was similarly inhibited by CP during growth in vitro, suggesting that other streptococcal pathogens may also engage in battle against CP during infection.49 However, S. pneumoniae was a notable exception as pneumococci displayed higher levels of resistance against CP compared to other streptococcal pathogens.49 Intriguingly, two different studies investigating the interactions between pneumococci and CP proposed conflicting roles for CP.58, 59 In one study, increased pneumococcal survival and virulence was observed in CP-deficient mice compared to WT mice, suggesting a protective role for CP against pneumococci in mouse model of intranasal infection.59 In another study using an identical mouse model of pneumococcal infection, the absence of CP in the knockout mouse caused defects in bacterial survival, disease dissemination, and virulence, prompting to speculate that CP facilitates pneumococcal infection.58 Additional studies are required to clarify the role of CP in host defense against pneumococcal invasion. Furthermore, investigations into the roles of pneumococcal zinc transporters in bacterial resistance against CP and in bacterial survival in wild type or CP-deficient mice will likely reveal the contribution of zinc chelation properties of CP to host defense against pneumococcal invasion.
Figure 1.

Zinc at host-GAS interface. During invasive infection, neutrophils release calprotectin (CP) at GAS colonization surfaces. CP scavenges zinc from the invading pathogen and retards GAS growth. The CP-imposed zinc scarcity (left panel) is sensed by zinc-sensing metalloregulator, AdcR, and the zinc-free AdcR relieves the repression of adc regulon. The adc regulon is comprised of zinc uptake system (AdcABC), zinc mobilization system (AdcAII, PhtD, and PhtY), and zinc recycling mechanisms (rpsN.2, Adh1, and Adh3). Gene expression of adc regulon facilitates zinc acquisition and aids GAS survival under zinc limiting conditions. When phagocytosed, GAS encounters neutrophil-induced phagosomal zinc intoxication (right panel). During zinc toxicity, the zinc-metallated AdcR dimer (AdcR2:Zn4) binds to target promoter sequences and causes the repression of adc regulon. Conversely, the zinc-bound TetR family regulator, GczA (GczA2:Zn4), interacts with target promoter and activates the transcription of zinc efflux pump czcD. The zinc efflux by CzcD detoxifies GAS cytosol of excess zinc and promotes GAS survival during zinc intoxication.
GAS adaptive responses to zinc limitation
To overcome zinc limitation, GAS employs a three-pronged countermeasure comprising increased zinc uptake, increased mobilization of bacterial zinc storage, and reduced zinc utilization (Fig. 1).60 The increased zinc acquisition is primarily achieved by upregulating the expression of a tripartite ABC-type zinc importer, adcABC. The AdcABC importer is comprised of an extracellular zinc-binding subunit, AdcA, an inner membrane permease that fuses AdcA and AdcC, AdcB, and cytoplasmic ATPase, AdcC (Fig. 1). The AdcABC-like zinc uptake machineries are widely distributed and highly conserved among gram-positive bacteria.
In addition to AdcA, GAS has a second extracellular zinc-binding protein, AdcAII (also known as Lmb or Lsp), that shares significant amino acid sequence similarity with AdcA (Fig. 1). AdcA has two domains, namely, an N-terminal ZnuA-like domain that is involved in extracellular zinc acquisition, and a C-terminal ZinT-like domain that is predicted to facilitate periplasmic zinc acquisition.61,62 AdcAII has the ZnuA-like domain but lacks the ZinT-like domain. Although AdcAII has been implicated in zinc acquisition during zinc limiting conditions, the exact mechanism by which AdcAII contributes to GAS zinc homeostasis remains unknown.63 Several possible roles for AdcAII have been proposed including zinc import and zinc mobilization. To function as a zinc importer, AdcAII must have the structural elements to associate with AdcBC and form the active tripartite transporter. Since AdcAII lacks the ZinT-like domain and the role of ZinT-like domain of AdcA in the assembly of AdcABC is yet to be elucidated, the likely role of AdcAII as a zinc importer requires further investigation. Alternatively, AdcAII has been shown to interact with a family of surface-exposed proteins that are unique to streptococci, the polyhistidine triad proteins (Pht).64,65 The Pht proteins, located at the cell wall, are characterized by the presence of His-XX-His-X-His (His-triad) motifs and each Pht protein contains 4–5 copies of His-triad motifs.66 Streptococcal genomes encode varying numbers of pht genes; pneumococci encode 4 pht genes and GAS encodes 2 zinc-dependent pht paralogs (phtD and phtY) (Fig. 1). Each His-triad motif binds a zinc atom and a single Pht molecule can bind 4–5 zinc atoms.67,68 Given that the pht genes are highly upregulated during zinc limitation and Pht proteins possess the ability to bind several zinc atoms, the Pht proteins are considered cell wall-anchored zinc scavengers.49,60 Interestingly, in pneumococci, AdcAII is co-localized with Pht proteins on the bacterial cell surface. Thus, the direct interactions between AdcAII and Pht as well as the directional transfer of zinc from Pht proteins to AdcAII suggest a plausible role of AdcAII in zinc mobilization from zinc storage.64–66 However, the details of molecular events downstream of zinc transfer from Pht to AdcAII and the significance of the Pht-AdcAII intermolecular zinc transfer to GAS zinc homeostasis remain largely unknown. Further investigations are required to elucidate the functional roles of AdcAII and Pht proteins in GAS adaptive responses to zinc limitation.
Another key strategy that GAS employs to overcome zinc limitation involves reduced cellular zinc utilization and zinc recycling (Fig. 1). This is achieved by replacing the relatively abundant zinc-containing proteins with their zinc-free functional paralogs. GAS encodes two paralogs for 30S ribosomal protein S14, a zinc-containing S14 and its zinc-free paralog, rpsN.2. During zinc limitation, expression of rpsN.2 is induced, and the zinc-free rpsN.2 replaces its zinc-containing counterpart from the ribosome.60 Similarly, GAS also has three genes encoding alcohol dehydrogenase (adh) enzymes and only one adh is zinc-dependent.60 During zinc limitation, the expression of genes encoding iron-dependent adh, and adh with reduced zinc requirements for catalytic activity are upregulated.60 The cellular consequences of this strategy include: i) ensuring that basic cellular processes such as translation remain functional under zinc-limiting conditions, ii) reducing the bacterial zinc requirement by utilization of zinc-free paralogs, and iii) recycling the zinc released from zinc-containing proteins for essential zinc-dependent cellular processes during zinc limitation.
The genes encoding adcA, adcC, adcAII and phtD are significantly upregulated in GAS grown either in the presence of CP or under zinc limiting growth conditions, suggesting that these genes contribute to GAS resistance against CP-mediated zinc limitation.49,69 A similar induction of adc regulon also occurs during murine subcutaneous infection,69 indicating that GAS encounters host-mediated zinc limitation during invasive infection. In accordance with this, inactivation of adcCB caused defective survival during subcutaneous infection and significantly attenuated GAS virulence in two different mouse models of invasive infection.49 Furthermore, the ΔadcCB mutant exhibited defective growth in the presence of CP, suggesting that zinc acquisition by adcABC contributes to bacterial resistance against CP-mediated zinc limitation.49 These observations underscore the critical contribution of AdcABC to bacterial survival in the host and GAS pathogenesis. This is not surprising as zinc transporters are implicated in CP resistance and bacterial virulence in several human pathogens including Staphylococcus aureus, Acinetobacter baumannii, Salmonella typhimurium, and Helicobacter pylori.46–48,53,54 Although genes encoding adcA, adcAII, phtD, phtY and rpsN.2 have been implicated in GAS zinc homeostasis, their individual contributions to zinc acquisition, CP resistance, survival in vivo, and GAS pathogenesis are largely unknown. Further investigations are required to elucidate the functions of these molecules in zinc acquisition in vivo and their roles in GAS pathogenesis at various host anatomic sites.
Metalloregulation during zinc limitation
GAS adaptive responses to zinc limitation are controlled by a zinc-sensing MarR family transcription regulator, Adhesin competence repressor (AdcR) (Fig. 1). Under zinc sufficient growth conditions, AdcR controls the expression of 70 genes with 31 genes repressed and 39 genes upregulated.60 AdcR directly represses transcription of 15 genes by binding to the operator sequences (adc motifs) in the target promoters, and these genes are primarily involved in zinc acquisition and zinc mobilization.60 Under zinc replete growth conditions, the zinc-bound AdcR binds to adc motifs and represses transcription (Fig. 1). Conversely, when GAS encounters zinc limitation, the apo-AdcR dissociates from the target promoters and causes derepression of gene expression (Fig. 1).60 Genetic inactivation of adcR resulted in constitutive expression of the adc regulon and significantly attenuated GAS virulence in mouse model of systemic infection.60 These data suggest that dysregulation of adaptive responses to zinc limitation is disadvantageous to bacterial pathogenesis, and zinc sensing and gene regulation by AdcR during infection is critical for GAS virulence.
Structural and biochemical characterization of AdcR from GAS and pneumococci identified two zinc-sensing sites in AdcR.49,60,70,71 The metal-binding sites are ideally located in the interdomain region between the DNA-binding and dimerization domains of AdcR to induce zinc-dependent allosteric changes that promote AdcR-DNA interactions. The side chains of E24 and H42 from the DNA-binding domain, and H108 and H112 from the dimerization domain coordinate zinc at site 1, whereas the side chains of C30 and E41 from the DNA-binding domain and E107 from the dimerization domain form the second zinc binding site.49 The metal-binding ligands from both sites of AdcR are highly conserved, indicating the likelihood of similar metal sensing events in AdcR paralogs from other streptococci. Mutations of the metal ligands at either site 1 or 2 impaired metal binding and attenuated GAS virulence in mouse model of systemic infection, suggesting that both sites participate in zinc sensing and gene regulation by AdcR during infection.49, 60 Metal binding studies indicated that site 1 is the primary zinc-sensing site with picomolar affinity and site 2 is the secondary zinc site with nanomolar affinity. Consistent with this, the loss of zinc at site 1 resulted in complete loss of repression, whereas defective metal binding at site 2 caused only partial loss of repression.49 These observations support a model in which AdcR senses fluctuations in zinc levels and mediates gradual derepression of the target genes. Under zinc-replete conditions, zinc occupancy at both metal-binding sites leads to full repression of the adc regulon. When GAS encounters mild zinc deficiency, zinc dissociates from the lower affinity site, site 2, and leads to partial derepression of the target genes. When the zinc deficiency becomes severe, loss of zinc from both sites results in full derepression of adc regulon. This mechanism allows the pathogen to monitor fluctuating zinc concentrations, and mount a measured response in accordance with the severity of zinc deficiency.
Host imposed zinc toxicity during GAS infection
Emerging evidence indicates that the host also exploits the chemical toxicity of transition metals to poison the pathogens and mediate bacterial killing. Host-mediated copper toxicity against bacterial pathogens is a well-documented antimicrobial mechanism72–75. The first evidence that host immune effectors employ zinc intoxication to inhibit bacterial growth emerged from investigations of the interactions between macrophages and the intracellular pathogen, Mycobacterium tuberculosis.76, 77 Upon mycobacterial invasion, macrophages mobilize zinc around the internalized bacteria and use phagosomal zinc intoxication to impair intracellular bacterial survival.76 Similar studies on the interactions between neutrophils and GAS suggest that neutrophils may employ macrophage-like zinc poisoning as an antimicrobial strategy against GAS (Fig. 1).78 Compared to resting neutrophils, GAS internalization by neutrophils evokes a surge in neutrophil intracellular zinc concentration, and a GAS-encoded zinc efflux pump confers resistance against neutrophil-induced phagosomal zinc toxicity (Fig. 1).78
Although these data suggest that neutrophils employ zinc intoxication as an antimicrobial strategy against GAS during phagocytosis, several questions in regard to the host-induced zinc intoxication mechanism require further investigation. Specifically, the neutrophil signaling pathways that induce zinc influx into the phagosome, the neutrophil cellular source of zinc that is shuttled into the phagosome, and the neutrophil transporters involved in zinc import remain unknown. In mycobacterium-containing macrophages, reactive oxygen species generated by oxidative burst in macrophages caused the release of zinc from intracellular sources.76 The free zinc was further transported into the phagosomes by unknown importers. However, it remains to be seen whether neutrophils utilize similar intracellular sources to mobilize zinc and impose zinc intoxication on GAS.
GAS adaptive responses to zinc toxicity
To mitigate the host-mediated metal toxicity, bacteria typically recruit efflux systems to expel the excess metal from the cytosol. Most bacterial pathogens encode zinc efflux systems, and inactivation of zinc exporters attenuates bacterial virulence, suggesting that pathogens encounter host-induced zinc poisoning during infection.76, 78 Two GAS-encoded transporters were implicated in bacterial resistance to zinc toxicity: a P1B4-type ATPase, PerR-controlled metal transporter A (PmtA), and a cation diffusion facilitator family zinc exporter, CzcD. Although PmtA was originally thought to be a zinc efflux pump,69 recent studies showed that PmtA aids GAS antioxidant defense by exporting excess iron out of the bacterial cytosol.79 Given that pmtA expression was specifically induced by iron excess, and inactivation of pmtA resulted in increased bacterial sensitivity to iron stress and elevated levels of intracellular iron, it was further confirmed that iron, not zinc, is the physiological ligand for PmtA.79 However, similar studies with CzcD indicated that zinc is its cognate ligand (Fig. 1).78 Consistent with its role as a zinc efflux pump, expression of czcD was induced during zinc intoxication, and inactivation of czcD resulted in increased intracellular accumulation of zinc.78 Interestingly, the expression of czcD was also upregulated during growth in the presence of neutrophils, suggesting that GAS encounters neutrophil-induced zinc poisoning.78 Consistent with this, the ΔczcD mutant displayed reduced ability to survive in the presence of neutrophils, indicating that czcD contributes to GAS resistance against neutrophil-mediated zinc cytotoxicity. A similar protective role for the zinc efflux pump, CtpC, in mycobacterial resistance against macrophage-induced zinc poisoning was observed.76 Inactivation of ctpC led to elevated intracellular zinc levels, loss of cell integrity, and bacterial cell death.76 These observations suggest that zinc poisoning by host immune effectors may be a broad-spectrum strategy against internalized intracellular and extracellular pathogens. These studies also emphasize the critical contribution of zinc efflux pumps to bacterial virulence as they promote bacterial survival by conferring resistance against host-mediated zinc poisoning during infection.
Metalloregulation during zinc toxicity
Gene expression of czcD is under the control of the divergently transcribed metalloregulator, group A streptococcal czcD activator (gczA). GczA belongs to the TetR family of transcription regulators that activates the transcription of czcD in a zinc-dependent fashion (Fig. 1).78,80,81 Deletion of gczA abolished czcD expression and the ΔgczA mutant was defective in zinc efflux, hypersensitive to zinc stress, more susceptible to neutrophil-mediated cytotoxicity, and significantly attenuated for GAS virulence.78 These findings suggest that zinc stress sensing by GczA is critical to bacterial survival in the host and GAS pathogenesis.
Although the biochemical basis for metal binding and gene regulation by GczA has not been elucidated, investigations of GczA paralogs from pneumococci and S. agalactiae provided insights into the likely mechanism of zinc sensing and gene regulation by GczA.80,81 The promoter sequences of pneumococcal czcD have two binding sites for the pneumococcal GczA paralog, SczA.80 The interaction between SczA and the operator sequences proximal to czcD start codon is zinc-independent.80 As a result, during zinc limitation, apo-SczA binds to the proximal site and negatively influences czcD expression. Conversely, during zinc excess, the zinc-metallated SczA occupies the distal site and mediates transcription activation of czcD.80 Although the czcD promoter sequence in GAS has the distal GczA binding site, nucleotide sequences similar to proximal site does not exist. Thus, such a dual regulatory mechanism for GczA is unlikely. However, as observed in pneumococci, it is likely that the GczA activates czcD expression from the distal site in a zinc-dependent manner.
The crystal structure of SczA from S. agalactiae has the typical TetR fold with an N-terminal DNA-binding domain containing the helix-turn-helix motif, and a C-terminal dimerization/metal-binding domain. Although the structure has a non-cognate nickel atom per subunit, spectroscopic studies indicated that SczA binds two zinc atoms per subunit.81 Based on the amino acid sequence conservation and mutational analysis of conserved residues, the metal ligands coordinating zinc at both sites were identified. Zinc at the high affinity site is coordinated by metal ligands H67, E114, E116, H117, and H118, whereas the second zinc site is composed of the side chains of H68, H84, and H88.81 Importantly, the metal ligands at both zinc-sensing sites are critical for zinc-dependent transcription activation of czcD, suggesting that both sites are functional in vivo. Thus, it is likely that zinc occupancy at both sites of SczA is required to induce the allosteric changes needed to transition from the apo-SczA conformation to the transcriptionally competent conformational state. Given that all the identified metal ligands are absolutely conserved between SczA and GczA, it is likely that GczA has similar zinc sensing mechanisms to induce czcD expression in response to zinc toxicity.
Concluding remarks
Bacterial infection is a multistage process, which includes the initial entry into the host, colonization at the primary site of infection, localized disease manifestation, systemic dissemination, and spread of infection to distal secondary sites. During the course of infection, zinc levels in the host vary and pathogens may encounter a gamut of zinc levels ranging from zinc scarcity to toxicity. Thus, to establish successful infection, the invading pathogens must be proficient in sensing the ever-changing alterations in zinc levels in the host and recruiting zinc stress-specific molecular arsenals to combat nutritional defense mechanisms. Recent literature indicates that host employs opposing zinc immune strategies to control GAS growth at two distinct stages of invasive infection: i) zinc limitation during extracellular growth and ii) zinc poisoning upon internalization by neutrophils. Although these results provide snapshots of host-GAS interactions, key questions about the spatial and temporal distribution of host zinc immune mechanisms during GAS infection remain to be addressed. Investigations into niche- and infection stage-specific host zinc immune mechanisms are required to understand the full extent of host-GAS exchange, and to develop host-based antimicrobial strategies.
GAS deploys an array of molecular ammunitions to combat and overcome the host zinc immune mechanisms. However, current understanding of the individual and collective contribution of GAS molecules in bacterial response to host zinc defenses is incomplete. Future research into bacterial adaptive strategies to host immune mechanisms, and the contributions of microbial coping mechanisms to GAS virulence will further the understanding of underlying bacterial resistance mechanisms to host nutritional immune mechanisms. Another promising area of investigation includes exploring new vaccine strategies targeting zinc transporters. Several traits of zinc transporters and mobilization systems such as their contribution to GAS virulence, relative abundance during infection, localization at bacterial surface, and accessibility to host immune cells make them attractive candidates for protein-based GAS vaccines. Consistent with this, immunization with the extracellular subunit of AdcA elicits the production of protective antibodies and confers protection against infections caused by diverse GAS M protein serotypes.49 Similar characterization of PhtY validated the utility of PhtY as an effective immunogen to prevent GAS infections.82 Since dysregulation of GAS adaptive responses to zinc limitation or toxicity is detrimental to bacterial virulence,49, 60, 78 chemical biology approaches to impair metalloregulatory systems may be another fertile area of research to develop new antimicrobials to treat GAS infections.
Significance to Metallomics.
Streptococcus pyogenes, also known as Group A Streptococcus (GAS), is a strict human pathogen that causes significant disease burden worldwide. Zinc is at the forefront of the battle between the host and GAS during infection and key in determining the final disease outcome. Impaired zinc acquisition or detoxification caused decreased GAS survival and attenuated bacterial virulence. Analyses of the host nutritional defenses showed that the eukaryotic host employs zinc starvation or intoxication to control GAS proliferation during infection. This review describes the role of zinc at host-GAS interface, the molecular details of host-GAS interactions and their contributions to GAS pathogenesis.
Acknowledgments
This work was supported in part by the National Institute of Health grant (1R01AI109096-01A1) to M.K.
Footnotes
Conflicts of interest
There are no conflicts of interest to declare.
References
- 1.Ralph AP, Carapetis JR. In: Host-Pathogen Interactions in Streptococcal Diseases. Chhatwal SG, editor. Vol. 280. Springer Berlin Heidelberg; Berlin, Heidelberg: 2013. p. 1. [DOI] [Google Scholar]
- 2.Sanyahumbi AS, Colquhoun S, Wyber R, Carapetis JR. In: Streptococcus pyogenes: Basic Biology to Clinical Manifestations. Ferretti J, Stevens D, Fishcetti V, editors. University of Oklahoma Health Sciences Center; 2016. [PubMed] [Google Scholar]
- 3.Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. The Lancet Infectious Diseases. 2005;5:685. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 4.Ralph AP, Carapetis JR. Group A Streptococcal Diseases and Their Global Burden. Curr. Top. Microbiol. Immunol. 2013;368:1. doi: 10.1007/82_2012_280. [DOI] [PubMed] [Google Scholar]
- 5.Steer AC, Carapetis JR. Prevention and treatment of rheumatic heart disease in the developing world. Nat. Rev. Cardiol. 2009;6:689. doi: 10.1038/nrcardio.2009.162. [DOI] [PubMed] [Google Scholar]
- 6.Waddington CS, Snelling TL, Carapetis JR. Management of invasive group A streptococcal infections. J. Infect. 2014;69(Supplement 1):S63. doi: 10.1016/j.jinf.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 7.Rathore MH, Jenkins SG. Group A beta-hemolytic Streptococcus: issue of resistance. Ped. Infec. Dis. J. 1993;12:354. doi: 10.1097/00006454-199304000-00023. [DOI] [PubMed] [Google Scholar]
- 8.Gerber MA. Antibiotic resistance in group A streptococci. Ped. Clin. North Amer. 1995;42:539. doi: 10.1016/s0031-3955(16)38978-7. [DOI] [PubMed] [Google Scholar]
- 9.Perez-Trallero E, Urbieta M, Montes M, Ayestaran I, Marimon JM. Emergence of Streptococcus pyogenes strains resistant to erythromycin in Gipuzkoa, Spain. Europ. J. Clin. Microbiol & Infect. Dis. 1998;17:25. doi: 10.1007/BF01584359. [DOI] [PubMed] [Google Scholar]
- 10.de Dassel JL, Ralph AP, Carapetis JR. Controlling acute rheumatic fever and rheumatic heart disease in developing countries: are we getting closer? Curr. Opin. Pediatr. 2015;27:116. doi: 10.1097/MOP.0000000000000164. [DOI] [PubMed] [Google Scholar]
- 11.Sheel M, Moreland NJ, Fraser JD, Carapetis J. Development of Group A streptococcal vaccines: an unmet global health need. Expert Rev. Vaccines. 2016;15:227. doi: 10.1586/14760584.2016.1116946. [DOI] [PubMed] [Google Scholar]
- 12.Steer AC, Carapetis JR, Dale JB, Fraser JD, Good MF, Guilherme L, Moreland NJ, Mulholland EK, Schodel F, Smeesters PR. Status of research and development of vaccines for Streptococcus pyogenes. Vaccine. 2016;34:2953. doi: 10.1016/j.vaccine.2016.03.073. [DOI] [PubMed] [Google Scholar]
- 13.Beres SB, Carroll RK, Shea PR, Sitkiewicz I, Martinez-Gutierrez JC, Low DE, McGeer A, Willey BM, Green K, Tyrrell GJ, Goldman TD, Feldgarden M, Birren BW, Fofanov Y, Boos J, Wheaton WD, Honisch C, Musser JM. Molecular complexity of successive bacterial epidemics deconvoluted by comparative pathogenomics. Proc. Natl Acad. Sci. U.S.A. 2010;107:4371. doi: 10.1073/pnas.0911295107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fittipaldi N, Beres SB, Olsen RJ, Kapur V, Shea PR, Watkins ME, Cantu CC, Laucirica DR, Jenkins L, Flores AR, Lovgren M, Ardanuy C, Liñares J, Low DE, Tyrrell GJ, Musser JM. Full-Genome Dissection of an Epidemic of Severe Invasive Disease Caused by a Hypervirulent, Recently Emerged Clone of Group A Streptococcus. Am. J. Pathol. 2012;180:1522. doi: 10.1016/j.ajpath.2011.12.037. [DOI] [PubMed] [Google Scholar]
- 15.Christopher CB, Randall JO, Nahuel F, Monica LM, Peter LF, Robert N, Mohammed M, William BW, James MM. Spread of Virulent Group A Streptococcus type emm59 from Montana to Wyoming, USA. Emerg. Infect. Dis. 2014;20:658. doi: 10.3201/eid2004.130564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nasser W, Beres SB, Olsen RJ, Dean MA, Rice KA, Long SW, Kristinsson KG, Gottfredsson M, Vuopio J, Raisanen K, Caugant DA, Steinbakk M, Low DE, McGeer A, Darenberg J, Henriques-Normark B, Van Beneden CA, Hoffmann S, Musser JM. Evolutionary pathway to increased virulence and epidemic group A Streptococcus disease derived from 3,615 genome sequences. Proc. Natl. Acad. Sci. U.S.A. 2014;111:E1768. doi: 10.1073/pnas.1403138111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McNeil SA, Halperin SA, Langley JM, Smith B, Warren A, Sharratt GP, Baxendale DM, Reddish MA, Hu MC, Stroop SD, Linden J, Fries LF, Vink PE, Dale JB. Safety and Immunogenicity of 26-Valent Group A Streptococcus Vaccine in Healthy Adult Volunteers. Clin. Infect. Dis. 2005;41:1114. doi: 10.1086/444458. [DOI] [PubMed] [Google Scholar]
- 18.Guilherme L, Faé K, Oshiro S, Kalil J. Crossreactivity of M-protein-reactive antibodies with human proteins. Exp. Rev. Mol. Med. 2005;7 [Google Scholar]
- 19.Dale JB, Beachey EH. Multiple, heart-cross-reactive epitopes of streptococcal M proteins. J. Exp. Med. 1985;161:113. doi: 10.1084/jem.161.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dale JB, Penfound TA, Chiang EY, Walton WJ. New 30-valent M protein-based vaccine evokes cross-opsonic antibodies against non-vaccine serotypes of group A streptococci. Vaccine. 2011;29:8175. doi: 10.1016/j.vaccine.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andreini C, Banci L, Bertini I, Rosato A. Zinc through the three domains of life. J. Proteome Res. 2006;5:3173. doi: 10.1021/pr0603699. [DOI] [PubMed] [Google Scholar]
- 22.Baker MD, Gendlina I, Collins CM, Acharya KR. Crystal structure of a dimeric form of streptococcal pyrogenic exotoxin A (SpeA1) Protein Sci. 2004;13:2285. doi: 10.1110/ps.04826804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krishnan KC, Mukundan S, Figueroa JAL, Caruso JA, Kotb M. Metal-mediated modulation of streptococcal cysteine protease activity and its biological implications. Infect. Immun. 2014;82:2992. doi: 10.1128/IAI.01770-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McDevitt CA, Ogunniyi AD, Valkov E, Lawrence MC, Kobe B, McEwan AG, Paton JC. A molecular mechanism for bacterial susceptibility to zinc. PLoS Pathog. 2011;7:e1002357. doi: 10.1371/journal.ppat.1002357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eijkelkamp BA, Morey JR, Ween MP, Cheryl-lynn YO, McEwan AG, Paton JC, McDevitt CA. Extracellular zinc competitively inhibits manganese uptake and compromises oxidative stress management in Streptococcus pneumoniae. PLoS one. 2014;9:e89427. doi: 10.1371/journal.pone.0089427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ong C-lY, Walker MJ, McEwan AG. Zinc disrupts central carbon metabolism and capsule biosynthesis in Streptococcus pyogenes. Sci. Rep. 2015;5:10799. doi: 10.1038/srep10799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maret W. Inhibitory zinc sites in enzymes. Biometals. 2013;26:197. doi: 10.1007/s10534-013-9613-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valko M, Morris H, Cronin M. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005;12:1161. doi: 10.2174/0929867053764635. [DOI] [PubMed] [Google Scholar]
- 29.Becker KW, Skaar EP. Metal limitation and toxicity at the interface between host and pathogen. FEMS Microbiol. Rev. 2014;38:1235. doi: 10.1111/1574-6976.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kehl-Fie TE, Skaar EP. Nutritional immunity beyond iron: a role for manganese and zinc. Curr. Opin. Chem. Biol. 2010;14:218. doi: 10.1016/j.cbpa.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chandrangsu P, Rensing C, Helmann JD. Metal homeostasis and resistance in bacteria. Nat. Rev. Microbiol. 2017;15:338–350. doi: 10.1038/nrmicro.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cellier MF, Courville P, Campion C. Nramp1 phagocyte intracellular metal withdrawal defense. Microbes Infect. 2007;9:1662. doi: 10.1016/j.micinf.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 33.Jabado N, Jankowski A, Dougaparsad S, Picard V, Grinstein S, Gros P. Natural resistance to intracellular infections: Nramp1 functions as a pH-dependent manganese transporter at the phagosomal membrane. J. Exp. Med. 2000;192:1237. doi: 10.1084/jem.192.9.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peracino B, Wagner C, Balest A, Balbo A, Pergolizzi B, Noegel AA, Steinert M, Bozzaro S. Function and Mechanism of Action of Dictyostelium Nramp1 (Slc11a1) in Bacterial Infection. Traffic. 2006;7:22. doi: 10.1111/j.1600-0854.2005.00356.x. [DOI] [PubMed] [Google Scholar]
- 35.Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 36.Raffatellu M, George MD, Akiyama Y, Hornsby MJ, Nuccio S-P, Paixao TA, Butler BP, Chu H, Santos RL, Berger T. Lipocalin-2 resistance confers an advantage to Salmonella enterica serotype Typhimurium for growth and survival in the inflamed intestine. Cell Host Microbe. 2009;5:476. doi: 10.1016/j.chom.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Drakesmith H, Prentice AM. Hepcidin and the iron-infection axis. Science. 2012;338:768. doi: 10.1126/science.1224577. [DOI] [PubMed] [Google Scholar]
- 38.Skaar EP. The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog. 2010;6:e1000949. doi: 10.1371/journal.ppat.1000949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gilston BA, Skaar EP, Chazin WJ. Binding of transition metals to S100 proteins. Sci. China Life Sci. 2016;59:792. doi: 10.1007/s11427-016-5088-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zackular JP, Chazin WJ, Skaar EP. Nutritional Immunity: S100 Proteins at the Host-Pathogen Interface. J. Biol. Chem. 2015;290:18991. doi: 10.1074/jbc.R115.645085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cunden LS, Gaillard A, Nolan EM. Calcium ions tune the zinc-sequestering properties and antimicrobial activity of human S100A12. Chem. Sci. 2016;7:1338. doi: 10.1039/c5sc03655k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haley KP, Delgado AG, Piazuelo MB, Mortensen BL, Correa P, Damo SM, Chazin WJ, Skaar EP, Gaddy JA. The Human Antimicrobial Protein Calgranulin C Participates in Control of Helicobacter pylori Growth and Regulation of Virulence. Infect. Immun. 2015;83:2944. doi: 10.1128/IAI.00544-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sohnle PG, Collins-Lech C, Wiessner JH. Antimicrobial activity of an abundant calcium-binding protein in the cytoplasm of human neutrophils. J. Infect. Dis. 1991;163:187. doi: 10.1093/infdis/163.1.187. [DOI] [PubMed] [Google Scholar]
- 44.Sohnle PG, Collins-Lech C, Wiessner JH. The zinc-reversible antimicrobial activity of neutrophil lysates and abscess fluid supernatants. J. Infect. Dis. 1991;164:137. doi: 10.1093/infdis/164.1.137. [DOI] [PubMed] [Google Scholar]
- 45.Korndörfer IP, Brueckner F, Skerra A. The crystal structure of the human (S100A8/S100A9) 2 heterotetramer, calprotectin, illustrates how conformational changes of interacting α-helices can determine specific association of two EF-hand proteins. J. Mol. Biol. 2007;370:887. doi: 10.1016/j.jmb.2007.04.065. [DOI] [PubMed] [Google Scholar]
- 46.Damo SM, Kehl-Fie TE, Sugitani N, Holt ME, Rathi S, Murphy WJ, Zhang Y, Betz C, Hench L, Fritz G, Skaar EP, Chazin WJ. Molecular basis for manganese sequestration by calprotectin and roles in the innate immune response to invading bacterial pathogens. Proc Natl Acad Sci U.S.A. 2013;110:3841. doi: 10.1073/pnas.1220341110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, Torres VJ, Anderson KL, Dattilo BM, Dunman PM, Gerads R, Caprioli RM, Nacken W, Chazin WJ, Skaar EP. Metal chelation and inhibition of bacterial growth in tissue abscesses. Science. 2008;319:962. doi: 10.1126/science.1152449. [DOI] [PubMed] [Google Scholar]
- 48.Liu JZ, Jellbauer S, Poe AJ, Ton V, Pesciaroli M, Kehl-Fie TE, Restrepo NA, Hosking MP, Edwards RA, Battistoni A, Pasquali P, Lane TE, Chazin WJ, Vogl T, Roth J, Skaar EP, Raffatellu M. Zinc sequestration by the neutrophil protein calprotectin enhances Salmonella growth in the inflamed gut. Cell Host Microbe. 2012;11:227. doi: 10.1016/j.chom.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Makthal N, Nguyen K, Do H, Gavagan M, Chandrangsu P, Helmann JD, Olsen RJ, Kumaraswami M. A Critical Role of Zinc Importer AdcABC in Group A Streptococcus-Host Interactions During Infection and Its Implications for Vaccine Development. EBioMedicine. 2017;21:131. doi: 10.1016/j.ebiom.2017.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hessian PA, Edgeworth J, Hogg N. MRP-8 and MRP-14, two abundant Ca (2+)-binding proteins of neutrophils and monocytes. J. Leukoc. Biol. 1993;53:197. [PubMed] [Google Scholar]
- 51.Amich J, Vicentefranqueira R, Mellado E, Ruiz-Carmuega A, Leal F, Calera JA. The ZrfC alkaline zinc transporter is required for Aspergillus fumigatus virulence and its growth in the presence of the Zn/Mn-chelating protein calprotectin. Cell. Microbiol. 2014;16:548. doi: 10.1111/cmi.12238. [DOI] [PubMed] [Google Scholar]
- 52.Diaz-Ochoa VE, Lam D, Lee CS, Klaus S, Behnsen J, Liu JZ, Chim N, Nuccio SP, Rathi SG, Mastroianni JR, Edwards RA, Jacobo CM, Cerasi M, Battistoni A, Ouellette AJ, Goulding CW, Chazin WJ, Skaar EP, Raffatellu M. Salmonella Mitigates Oxidative Stress and Thrives in the Inflamed Gut by Evading Calprotectin-Mediated Manganese Sequestration. Cell Host Microbe. 2016;19:814. doi: 10.1016/j.chom.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gaddy JA, Radin JN, Loh JT, Piazuelo MB, Kehl-Fie TE, Delgado AG, Ilca FT, Peek RM, Cover TL, Chazin WJ, Skaar EP, Scott Algood HM. The host protein calprotectin modulates the Helicobacter pylori cag type IV secretion system via zinc sequestration. PLoS Pathog. 2014;10:e1004450. doi: 10.1371/journal.ppat.1004450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hood MI, Mortensen BL, Moore JL, Zhang Y, Kehl-Fie TE, Sugitani N, Chazin WJ, Caprioli RM, Skaar EP. Identification of an Acinetobacter baumannii Zinc Acquisition System that Facilitates Resistance to Calprotectin-mediated Zinc Sequestration. PLoS. Pathog. 2012;8:e1003068. doi: 10.1371/journal.ppat.1003068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lusitani D, Malawista SE, Montgomery RR. Calprotectin, an abundant cytosolic protein from human polymorphonuclear leukocytes, inhibits the growth of Borrelia burgdorferi. Infect Immun. 2003;71:4711. doi: 10.1128/IAI.71.8.4711-4716.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hayden JA, Brophy MB, Cunden LS, Nolan EM. High-Affinity Manganese Coordination by Human Calprotectin Is Calcium-Dependent and Requires the Histidine-Rich Site Formed at the Dimer Interface. J. Am. Chem. Soc. 2013;135:775. doi: 10.1021/ja3096416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brunjes Brophy M, Nakashige TG, Gaillard A, Nolan EM. Contributions of the S100A9 C-Terminal Tail to High-Affinity Mn(II) Chelation by the Host-Defense Protein Human Calprotectin. J. Am. Chem. Soc. 2013;135:17804. doi: 10.1021/ja407147d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Achouiti A, Vogl T, Endeman H, Mortensen BL, Laterre PF, Wittebole X, van Zoelen MA, Zhang Y, Hoogerwerf JJ, Florquin S, Schultz MJ, Grutters JC, Biesma DH, Roth J, Skaar EP, van 't Veer C, de Vos AF, van der Poll T. Myeloid-related protein-8/14 facilitates bacterial growth during pneumococcal pneumonia. Thorax. 2014;69:1034. doi: 10.1136/thoraxjnl-2014-205668. [DOI] [PubMed] [Google Scholar]
- 59.De Filippo K, Neill DR, Mathies M, Bangert M, McNeill E, Kadioglu A, Hogg N. A new protective role for S100A9 in regulation of neutrophil recruitment during invasive pneumococcal pneumonia. FASEB J. 2014;28:3600. doi: 10.1096/fj.13-247460. [DOI] [PubMed] [Google Scholar]
- 60.Sanson M, Makthal N, Flores AR, Olsen RJ, Musser JM, Kumaraswami M. Adhesin competence repressor (AdcR) from Streptococcus pyogenes controls adaptive responses to zinc limitation and contributes to virulence. Nucl. Acids Res. 2015;43:418. doi: 10.1093/nar/gku1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gabbianelli R, Scotti R, Ammendola S, Petrarca P, Nicolini L, Battistoni A. Role of ZnuABC and ZinT in Escherichia coliO157:H7 zinc acquisition and interaction with epithelial cells. BMC Microbiol. 2011;11:36. doi: 10.1186/1471-2180-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Petrarca P, Ammendola S, Pasquali P, Battistoni A. The Zur-Regulated ZinT Protein Is an Auxiliary Component of the High-Affinity ZnuABC Zinc Transporter That Facilitates Metal Recruitment during Severe Zinc Shortage. J. Bacteriol. 2010;192:1553. doi: 10.1128/JB.01310-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tedde V, Rosini R, Galeotti CL. Zn2+ Uptake in Streptococcus pyogenes: Characterization of adcA and lmb Null Mutants. PLos one. 2016;11:e0152835. doi: 10.1371/journal.pone.0152835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Galeotti CL, Bove E, Pezzicoli A, Nogarotto R, Norais N, Pileri S, Lelli B, Falugi F, Balloni S, Tedde V, Chiarot E, Bombaci M, Soriani M, Bracci L, Grandi G, Grifantini R. Surface Interactome in Streptococcus pyogenes. Mol. Cell. Proteomics. 2012;11:M111.015206. doi: 10.1074/mcp.M111.015206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Plumptre CD, Eijkelkamp BA, Morey JR, Behr F, Couñago RM, Ogunniyi AD, Kobe B, O'Mara ML, Paton JC, McDevitt CA. AdcA and AdcAII employ distinct zinc acquisition mechanisms and contribute additively to zinc homeostasis in Streptococcus pneumoniae. Mol. Microbiol. 2014;91:834. doi: 10.1111/mmi.12504. [DOI] [PubMed] [Google Scholar]
- 66.Bersch B, Bougault C, Roux L, Favier A, Vernet T, Durmort C. New insights into histidine triad proteins: solution structure of a Streptococcus pneumoniae PhtD domain and zinc transfer to AdcAII. PLoS one. 2013;8:e81168. doi: 10.1371/journal.pone.0081168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Loisel E, Chimalapati S, Bougault C, Imberty A, Gallet B, Di Guilmi AM, Brown J, Vernet T, Durmort C. Biochemical characterization of the histidine triad protein PhtD as a cell surface zinc-binding protein of pneumococcus. Biochemistry. 2011;50:3551. doi: 10.1021/bi200012f. [DOI] [PubMed] [Google Scholar]
- 68.Plumptre CD, Ogunniyi AD, Paton JC. Polyhistidine triad proteins of pathogenic streptococci. Trends Microbiol. 2012;20:485. doi: 10.1016/j.tim.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 69.Brenot A, Weston BF, Caparon MG. A PerR-regulated metal transporter (PmtA) is an interface between oxidative stress and metal homeostasis in Streptococcus pyogenes. Mol. Microbiol. 2007;63:1185. doi: 10.1111/j.1365-2958.2006.05577.x. [DOI] [PubMed] [Google Scholar]
- 70.Guerra AJ, Dann CE, 3rd, Giedroc DP. Crystal structure of the zinc-dependent MarR family transcriptional regulator AdcR in the Zn(II)-bound state. J. Am. Chem. Soc. 2011;133:19614. doi: 10.1021/ja2080532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reyes-Caballero H, Guerra AJ, Jacobsen FE, Kazmierczak KM, Cowart D, Koppolu UMK, Scott RA, Winkler ME, Giedroc DP. The Metalloregulatory Zinc Site in Streptococcus pneumoniae AdcR, a Zinc-activated MarR Family Repressor. J. Mol. Biol. 2010;403:197. doi: 10.1016/j.jmb.2010.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shafeeq S, Yesilkaya H, Kloosterman TG, Narayanan G, Wandel M, Andrew PW, Kuipers OP, Morrissey JA. The cop operon is required for copper homeostasis and contributes to virulence in Streptococcus pneumoniae. Mol. Microbiol. 2011;81:1255. doi: 10.1111/j.1365-2958.2011.07758.x. [DOI] [PubMed] [Google Scholar]
- 73.Ward SK, Abomoelak B, Hoye EA, Steinberg H, Talaat AM. CtpV: a putative copper exporter required for full virulence of Mycobacterium tuberculosis. Mol. Microbiol. 2010;77:1096. doi: 10.1111/j.1365-2958.2010.07273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.White C, Lee J, Kambe T, Fritsche K, Petris MJ. A Role for the ATP7A Copper-transporting ATPase in Macrophage Bactericidal Activity. J. Biol. Chem. 2009;284:33949. doi: 10.1074/jbc.M109.070201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wolschendorf F, Ackart D, Shrestha TB, Hascall-Dove L, Nolan S, Lamichhane G, Wang Y, Bossmann SH, Basaraba RJ, Niederweis M. Copper resistance is essential for virulence of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 2011;108:1621. doi: 10.1073/pnas.1009261108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Botella H, Peyron P, Levillain F, Poincloux R, Poquet Y, Brandli I, Wang C, Tailleux L, Tilleul S, Charrière GM, Waddell Simon J, Foti M, Lugo-Villarino G, Gao Q, Maridonneau-Parini I, Butcher Philip D, Castagnoli Paola R, Gicquel B, de Chastellier C, Neyrolles O. Mycobacterial P(1)-Type ATPases Mediate Resistance to Zinc Poisoning in Human Macrophages. Cell Host Microbe. 2011;10:248. doi: 10.1016/j.chom.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Botella H, Stadthagen G, Lugo-Villarino G, de Chastellier C, Neyrolles O. Metallobiology of host–pathogen interactions: an intoxicating new insight. Trends Microbiol. 2012;20:106. doi: 10.1016/j.tim.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 78.Ong C-lY, Gillen CM, Barnett TC, Walker MJ, McEwan AG. An Antimicrobial Role for Zinc in Innate Immune Defense Against Group A Streptococcus. J. Infect. Dis. 2014;209:1500. doi: 10.1093/infdis/jiu053. [DOI] [PubMed] [Google Scholar]
- 79.VanderWal AR, Makthal N, Pinochet-Barros A, Helmann JD, Olsen RJ, Kumaraswami M. Iron efflux by PmtA is critical for oxidative stress resistance and contributes significantly to group A streptococcus virulence. Infect. Immun. 2017;85:e00091. doi: 10.1128/IAI.00091-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kloosterman TG, Van Der Kooi-Pol MM, Bijlsma JJE, Kuipers OP. The novel transcriptional regulator SczA mediates protection against Zn2+ stress by activation of the Zn2+-resistance gene czcD in Streptococcus pneumoniae. Mol. Microbiol. 2007;65:1049. doi: 10.1111/j.1365-2958.2007.05849.x. [DOI] [PubMed] [Google Scholar]
- 81.Martin JE, Edmonds KA, Bruce KE, Campanello GC, Eijkelkamp BA, Brazel EB, McDevitt CA, Winkler ME, Giedroc DP. The zinc efflux activator SczA protects Streptococcus pneumoniae serotype 2 D39 from intracellular zinc toxicity. Mol. Microbiol. 2017;104:636. doi: 10.1111/mmi.13654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kunitomo E, Terao Y, Okamoto S, Rikimaru T, Hamada S, Kawabata S. Molecular and biological characterization of histidine triad protein in group A streptococci. Microbes Infect. 2008;10:414. doi: 10.1016/j.micinf.2008.01.003. [DOI] [PubMed] [Google Scholar]