

Abstract
Postaxial limb hypoplasia (PALH) is a group of nonhereditary diseases with congenital lower limb deficiency affecting the fibular ray, including fibular hemimelia (FH), proximal femoral focal deficiency (PFFD), and tarsal coalition (TC). The etiology and the developmental biology of the anomaly are still not fully understood. Here, we review the previous classification systems, present the clinical features, and discuss the developmental biology of PALH.
Keywords: postaxial hypoplasia, fibular hemimelia, tarsal coalition, proximal femoral focal deficiency, skeletal development, Wnt signaling
Introduction
Postaxial limb hypoplasia (PALH) is a group of nonhereditary diseases with congenital lower limb deficiency. It affects the ipsilateral hip, femur, knee, tibia/fibula, ankle, and foot. PALH includes fibular hemimelia (FH), proximal femoral focal deficiency (PFFD), and tarsal coalition (TC).1 FH has been known for over 300 years and was first described by Gollier in 1698.2 FH is characterized by the partial or total absence of the fibula. Patients with FH often require surgical reconstruction.3 However, if the ankle joint cannot be formed in FH patients, the only treatment option is amputation.4 PFFD affects the pelvis, particularly the hip bone, and the proximal femur.5 The disorder may affect one side or both, with the hip becoming deformed and the leg shortened. TC is an abnormal bridge of tissue that connects two separate tarsal bones.6 The onset of symptoms related to TC usually occurs at about 9–17 years of age, with a peak incidence occurring at 10–14 years of age. When the bridging link becomes bone, a limitation of motion results, and the onset of pain is initiated. The bones of the tarsus are the most posterior bones of the foot. These bones form the two major foot joints: the subtalar and midtarsal joints. These joints allow complex motions to occur in the feet, which are necessary for activities such as traveling over uneven terrain and creating a gait for the function of the knees, hips, and back. The basic information concerning the three types of PALH is summarized in Table 1. The etiology of PALH appears to be related to developmental defects that occur during the embryonic period, but no genetic factor has been identified.
Table 1.
The classification, etiology, and clinical features of the three types of PALH disorders.
| Commonly used classification system | Etiology | Clinical features | |
|---|---|---|---|
| FH | The Achterman and Kalamchi classification | Unknown | Mild hypoplasia to complete absence of the fibula, with variable shortening of the tibia |
| PFFD | The Aiken classification | Unknown | Variable degrees of shortening or absence of the femoral head, with associated dysplasia of the acetabulum and femoral shaft |
| TC | The Downey classification | Unknown | The fusion of two or more bones of the hindfoot |
FH, fibular hemimelia; PFFD, proximal femoral focal deficiency; TC, tarsal coalition.
Fibular hemimelia
FH is a congenital deficiency or absence of the fibula. It may be associated with anatomical abnormalities in the lower limbs.7 It is rare but still one of the most common congenital deficiencies of the long bones, with an estimated incidence of 7.4–49 per 1 million live births.2,8–10
More than 40 years ago, Thompson et al.11 used the term “congenital absence of the fibula” to describe the anomaly that bears its name, which emphasizes only one aspect of a complex of congenital malformations. However, FH often involves femoral and tibial hypoplasia, hypoplasia of the lateral femoral condyle and anteromedial bowing of the tibia, valgus or varus deformity of the ankle and deficiency of the lateral ray of the foot, ball-and-socket ankle joint deformity, and tarsal coalition of the foot.4,12–14 The hypoplasia of the lateral femoral condyle and anteromedial bowing of the tibia could lead to genu valgum or knock-knee, and valgus or varus deformity of the ankle and deficiency of the lateral ray of the foot may preclude plantigrade walking. The term “postaxial hypoplasia” may be more appropriate in describing this disease as compared with traditional terms FH or “fibular hypoplasia,” since it mainly affects lower extremities.1
Most reported cases of FH are sporadic. The virtual lack of inheritable recurrence suggests that FH may be caused by mosaic somatic mutations. The presence of fibular hypoplasia is sufficient to define FH as a developmental defect. However, the etiology and the developmental biology of the anomaly are still not fully understood.
Classification of FH
There are currently many different classification systems for FH. In 1952, Coventry and Johnson2 gave the earliest classification of FH, describing the severity of the deficiency and the associated findings. The commonly used classification system to describe the limb hypoplasia was introduced by Achterman and Kalamchi in 1979.15 This classification divided FH into two types: type I FH described limbs in which a portion of the fibula was present, and type II FH described limbs where there was a complete absence of the fibula. The type I group was further subdivided according to the extent of the deficiency.15 In type IA, the proximal fibular epiphysis is distal to the level of the tibial growth plate and is often smaller than the normal side, while the distal fibular growth plate is proximal to the dome of the talus. In type IB, there is a partial absence of the fibula: proximally the fibula is absent for 30–50% of its length, while distally it is present but does not support the ankle.15
A new classification system was recently suggested by Stanitski et al.16 on the basis of the morphology of the fibula and ankle, hindfoot coalition, and foot ray deficits. In this classification system, three types of fibula have been suggested: (1) nearly normal fibula; (2) small or miniature fibula, regardless of the position of the fibula in the limb; and (3) complete absence of the fibula. The three types of tibiotalar joint and distal tibial epiphyseal morphology are categorized: H = horizontal; V = valgus (triangular distal tibial epiphysis); and S = spherical (ball-and-socket). The lowercase “c” is used to describe the tarsal coalition, and the number of foot rays (medial to lateral) is numerically quantified (1–5).16 This classification system reflects the broad expression of this disorder and the unpredictable interrelationships between the foot and ankle.16 This classification includes additional data on the status of the foot and ankle and can help in clinical decisions about limb salvage or ablation.
Most recently, the Paley classification17 became the first classification of FH to be designed with reconstructive surgery options in mind. The Paley classification divided FH into three types: type 1––stable ankle; type 2––dynamic valgus; and type 3––fixed equino-valgus. Type 3 can be subdivided into three groups: type 3A––the fixed equino-valgus deformity is due to a malorientation of the ankle joint; type 3B––there is a malunited subtalar coalition; and type 3C––co-existence of both distal tibial malorientation and malunited subtalar coalition. Each type has a different surgical treatment, so it is a reconstructive surgery classification.
Clinical features of FH
We retrospectively reviewed the records and radiographs of patients with FH treated in Shanghai Sixth People’s Hospital, China between 2014 and 2016. A total of 15 patients (seven females and eight males) with 15 affected limbs (six left and nine right legs affected) were identified. The clinical features of each patient are described in Table 2. There were no major malformations in organ systems other than limbs in these patients. Ages ranged from 3 to 47 years, with an average age of 13.9 years at their first visit. The radiographs were evaluated, along with the clinical data, to determine the presence of the following: lateral femoral condylar hypoplasia, ball-and-socket ankle joint malformation, tarsal coalition, and forefoot ray deletion. Using the Achterman and Kalamchi classification system, six patients (Patient No. 1–6) had type IA (Fig. 1A), one (No. 7) type IB (Fig. 1B), and eight (No. 8–15) type II FH (Fig. 1C). Almost all patients had hypoplasia of the lateral femoral condyle (Fig. 1, white arrows) and tarsal coalition (Fig. 2A) with different severity. Four patients (No. 8–10 and 13) with type II FH had anteromedial bowing of the tibia (Fig. 2B). The ball-and-socket ankle joint (Fig. 2C) was observed in eight patients older than 11 years of age, while, in seven patients below 6 years of age, the ankle was poorly developed (Fig. 2B). There were four patients (No. 8–10 and 12) with anteromedial bowing of the tibia. Four limbs (No. 1–3 and 5) had five rays, but patient No. 5 had hypoplasia of the fifth ray. One limb (No. 10) had four rays, eight (No. 4, 6–9, 12–14) had three rays, and two (No. 11 and 15) had two rays. Most limbs with type IA FH had five-ray feet, which were only detected in type IA FH patients. Stanitski et al.13 reported that three-ray feet occurred in none of the type IA or type IB fibulae but 100% of type II fibulae. However, one patient (No. 4) with type IA FH had a three-ray foot in our clinics.
Table 2.
The clinical features of Chinese patients with fibular hemimelia.
| Patient No. | Sex | Age | Side | Achterman and Kalamchi classification system | Hypoplasia of the lateral femoral condyle | Anteromedial bowing of the tibia | Ankle morphology | Tarsal coalition | Number of foot rays |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Male | 4 | Left | Type IA | Yes | No | Dysplasia of the ankle | Yes | Five |
| 2 | Male | 11 | Left | Type IA | Yes | No | Balland-socket type | Yes | Five |
| 3 | Female | 21 | Right | Type IA | Yes | No | Balland-socket type | Yes | Five |
| 4 | Male | 23 | Right | Type IA | Yes | No | Balland-socket type | Yes | Three |
| 5 | Female | 23 | Left | Type IA | Yes | No | Balland-socket type | Yes | Five but hypoplasia of the fifth ray |
| 6 | Male | 19 | Right | Type IA | Yes | No | Balland-socket type | Yes | Three |
| 7 | Female | 23 | Right | Type IB | Yes | No | Balland-socket type | Yes | Three |
| 8 | Female | 3 | Right | Type II | Yes | Yes | Dysplasia of the ankle | Not fully developed | Three |
| 9 | Male | 3 | Right | Type II | Yes | Yes | Dysplasia of the ankle | Not fully developed | Three |
| 10 | Female | 4 | Left | Type II | Yes | Yes | Dysplasia of the ankle | Yes | Four |
| 11 | Male | 6 | Left | Type II | Yes | No | Dysplasia of the ankle | Yes | Two |
| 12 | Male | 6 | Right | Type II | Yes | Yes | Dysplasia of the ankle | Yes | Three |
| 13 | Male | 12 | Left | Type II | Yes | No | Balland-socket type | Yes | Three |
| 14 | Female | 47 | Right | Type II | Yes | No | Balland-socket type | Yes | Three |
| 15 | Female | 3 | Right | Type II | Yes | No | Dysplasia of the ankle | Not fully developed | Two |
Figure 1.
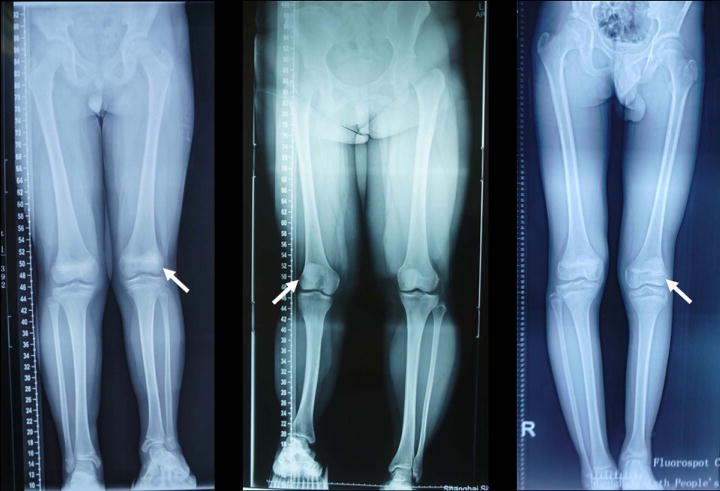
According to the Achterman and Kalamchi classification, (A) represents type IA FH, (B) represents type IB FH, and (C) represents type II FH. Note that all three patients had hypoplasia of the lateral femoral condyle (white arrows) with different degrees of severity.
Figure 2.
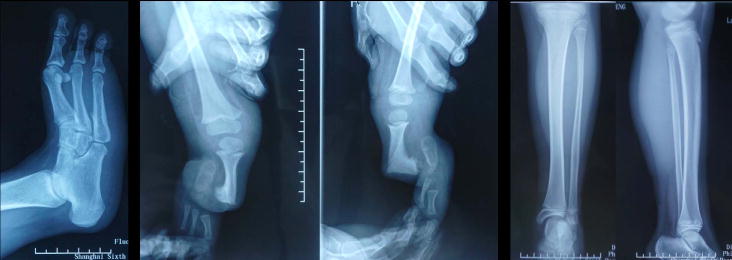
(A) Tarsal coalition. (B) Anteromedial bowing of the tibia. Also note that the ankle was poorly developed. (C) Ball-and-socket ankle joint.
Proximal femoral focal deficiency
Proximal femoral focal deficiency (PFFD) is a musculoskeletal malformation in the proximal portion of the femur that is a result of early disturbance of growing mesenchymal homeostasis.17–19 The first PFFD case, reported in 1974 by Kelly, delineated a deformity of the right leg of an infant.20 The classification of PFFD was described by Torode and Gillespie in 1991.21 In the following decades, continuous case reports were published illustrating that the epidemiology of PFFD ranges from 1 in 50,000 live births (1995) to 0.11–0.2/10,000 live births (2015).17,18 Specifically, the ratio of the incidence of PFFD is greater in males than females (1.5:1); furthermore, PFFD more often presents unilaterally than bilaterally.17,22 The clinical diagnosis of PFFD has depended on imaging techniques available at the time; these techniques have varied to a great extent, from radiography in the 19th century to emerging ultrasonography in the 20th century. By this progress, the categorizations of PFFD are divided subjectively.19,23 The Aiken classification system of PFFD has been well accepted. This classification system separates PFFD on the basis of the presence of anatomic features of the femoral head, acetabulum, and femoral segment.17,18,24 According to the radiographic appearance on plain X-ray, the Aiken classification delineated PFFD into four types (A–D). Type A presents as a short femur with a femoral head and acetabulum; there is a possibility of subtrochanteric and pseudoarthrosis. Type B refers to a short femur with bony tuft; the femoral head is present, and there is a moderately dysplastic acetabulum. Type C is described as a short and tapered femur with an absent/very small femoral head, and the acetabulum is severely dysplastic. The most severe situation is type D, where the femoral head and acetabulum is missing, and the femur is short and deformed.17,18,24 Other classifications were devised with more groupings or with the aid of advanced diagnostic methods. For example, Amstutz categorized PFFD into five types by dividing type A of the Aiken classification into two subgroups on the basis of the presence of subtrochanteric pseudoarthrosis and coxa vara.18 Advanced diagnostic methods, such as sonographic technique, improve the detection of these features of PFFD during the prenatal examination.18,22,25,26
To date, treatments for PFFD have included two methods: surgical and nonsurgical. Nonsurgical treatments include prosthesis fitting around the plantar-flexed foot as assistance.17 Surgical approaches vary depending on the individual case, including Van Ness rotationplasty, Torode-Gillespie rotationplasty, the knee fusion technique of King, and Syme’s amputation technique.19 The etiology of PFFD remains unclear. However, some hypotheses have been proposed, such as abnormal neural crest development, defects in chondrocyte maturation, teratogen exposure, such as thalidomide and misoprostol, poorly controlled diabetes, viral infection, radiation exposure, and focal ischemia.26–29 Epps26 suggested that sclerotome subtraction as a result of injury to the neural crest could be one theory related to limb deformities. The neural crest is responsible for the formation of the precursor of the peripheral sensory nerves for the fourth and fifth lumbar vertebrae. By analyzing a fetus at 21 weeks with unilateral PFFD, Boden et al.27 found that PFFD may be related to unsuccessful proximal growth plate migration, the immature arrangement of proliferative and hypertrophic chondrocytes, abnormal septal architecture truncating the immature hypertrophic zone, and irregular vascular invasion of primary trabecular. Hamanishi30 studied 70 patients with 91 congenital short femora and illustrated that either different etiological factors or different timing of drug insult to the fetus in the thalidomide group resulted in the defects. The thalidomide group had femur–tibia–radius anomalies, while the non-thalidomide group had femur–fibula–ulna anomalies. It was suggested that, in the non-thalidomide group, a multifactorial genetic background is related to hypoplasia of the femur. Marchese et al.29 reported a 5-month-old female orphan with an absence of the proximal femur, evident fibular hypoplasia, and abnormal metatarsals and phalanges and suggested that misoprostol elicited these defects through vascular disruption by uterine contraction during the first trimester of gestation. Lin22 and Mailath-Pokorny25 reported a case in which a 30-year-old woman with over 19 weeks’ gestation had a fetus with an isolated unilateral short femur on ultrasound examination. The fetus presented with a normal karyotype via amniocentesis, suggesting the possibility that epigenetic modification may be involved in the development of PFFD.
Tarsal coalition
TC refers to a pedal anomaly in which fusion of two or more bones of the hindfoot occurs, resulting in absent or restricted movement between bones.6,31–36 Buffon is considered to be the first to describe TC, in 1769.31,32,34 Later, Coventry37 published an initial study in 1950 that illustrated the idea of hindfoot abnormality with TC. The reported epidemiology of TC varied from the 19th century to the 20th century. Before the millennium, the incidence of TC is suggested to have been 1–2%,31,32 while, in 2003, Solomon et al.38 concluded that the incidence of TC could be as high as 12.7% after studying 100 cadaveric feet. The most common coalitions are the talocalcaneal coalition and calcaneal navicular fusion; the coalitions are 50% bilateral and have a slight male preponderance.6,34 Unlike PFFD patients who come to the clinic for limb correction, TC patients usually present with symptoms, such as vague aching hindfoot pain, subtalar stiffness, peroneal spasm, and sinus tarsi syndrome.6,32,34 These symptoms are elicited by interruption and restriction of rotation and gliding of subtalar joints, thus leading to a compensation of involved hindfoot and ligaments, which can ultimately result in a peroneal spastic flatfoot or traction spur.31,34 Initial clinical diagnosis of TC primarily depends on radiographic analysis and nuclear scintigraphy (bone scintigraphy).33,34 With the advent of three-dimensional imaging techniques (CT and MRI), clear stereoscopic images can be taken to provide better characterization and facilitate diagnosis. Radiographic features, such as talar beaking, C sign, and drunken waiter sign, support the clinical diagnosis of TC.6,31,32,34
The classification of TC is mainly based on the inheritance of the disease and anatomic defects. Specifically, Tachdjian and Buckholtz divided TC by anatomic occurrence of the coalitions and concluded that medial talocalcaneal coalitions are the most common form of TC, while the second most common are calcaneal navicular coalitions.6,31 Jacobs, Schlefman, and Sakellariou classified TC on the basis of the type of connective tissue involved with uniting the involved osseous segments.6,31,32 For example, when the union of the coalition between two or more tarsal bones is attributed to cartilaginous tissue, it is termed cartilaginous/synchondrosis. Perlman and Wertheimer classified TC based on etiology as congenital or acquired disease.31 Cowell and Harris classified TC as complete, incomplete, or rudimentary coalition on the basis of the motion at the joint.6 Downey and Lawrence categorized TC by taking the status of osseous morphological changes related to the middle talocalcaneal joint as extra-articular– or intra-articular–type coalitions.31,35
The treatment of TC is more focused on symptomatic relief and is dependent on the degenerative extent.6,31,32,34 Patients often complain of pain, talar joint sprain, or severe peroneal spasm. The standard nonsurgical treatments are assistant therapies (orthosis, non-steroidal anti-inflammatory medication, peroneal nerve block, cast/immobilization, physical therapy, etc.). Surgical treatments, including resection and arthrodesis, are performed when non-operative treatments are not sufficient.6,31,34 Zaw et al.34 pointed out a salvage surgery (triple arthrodesis) for the treatment of patients with TC and persistent pain. Regarding the etiology of TC, acquired TC in the adult population may be ascribed to trauma and posttraumatic complications (infection, neoplasia).31,32,34 The possible etiologies of congenital TC have also been proposed, such as unsuccessful mesenchyme differentiation, delayed ossified accessory ossicles, and autosomal dominant inheritance.6,31,32,34 Jack and Leboucq both illustrated the idea that TC may be related to the failure of segmentation and differentiation of primitive/embryonic mesenchyme.6,31,32,34 The delayed ossified accessory ossicles were proposed by Pfitzner in 1896 with the observation that accessory ossicles presented at the location of tarsal bone development.32,34 Interestingly, Harris found a talocalcaneal coalition in the fetus and confirmed Jack and Leboucq’s hypothesis to some extent; while denying Pfitzner’s hypothesis.32,34 By analyzing three generations of the calcaneal navicular and talocalcaneal coalitions in the same family, Wray and Herndon suggested an autosomal dominant trait with reduced penetrance.6,34 Leonard39 concluded that tarsal coalition is a unifactorial disorder of autosomal dominant inheritance with full penetrance, after reviewing 98 first-degree relatives of 31 index patients with confirmed tarsal coalitions. Plotkin40 considered the inheritance of calcaneal navicular coalition as part of a complex polygenic system responsible for overall limb development by studying monozygotic twins.
Development of lower limb
The etiology of PALH remains largely unknown. To determine the mechanisms of PALH defects, it is necessary to understand the developmental biology of the limb, in particular, the limb patterning and the genetic network during embryonic skeletal development.
The vertebrate limb develops from a small bud of undifferentiated mesoderm cells encased in ectoderm.41 The appearance of limb buds, which are small projections of fetal mesenchymal condensation on the ventrolateral aspect of the trunk, occurs during the 4th to 5th weeks of gestation.13 The development of the bud has intrinsic polarity and occurs along three different axes: proximal–distal (PD), anterior–posterior (AP), and dorsoventral (DV). The complex patterning process is regulated by three main signaling centers––the apical ectodermal ridge (AER), the zone of polarizing activity (ZPA), and the progress zone (PZ).42,43 The AER is a structure formed from ectodermal cells at the distal end of the bud; the ZPA is located in the mesenchyme on the posterior rim of the bud; and the PZ is located adjacent to the AER.42,43 Each signaling center controls patterning in one of the axes of the limb, and they are all essential for development since they provide the growth factors necessary for patterning the limb. The functions of these signaling centers are interdependent, and adequate interactions between them are essential for proper morphological limb development.
The PD axis
The AER is the specialized and thickened ectoderm rimming the distal edge of the limb bud and is required for PD elongation.44 The AER is formed through a complex and not completely understood process that starts with the induction of the AER precursor cells, which are marked by their expression of fibroblast growth factor 8 (FGF8).45 Limb bud outgrowth and the patterning of mesenchymal progenitors are coordinately regulated by epithelial–mesenchymal interactions between the AER and mesenchyme. Early findings showed that removal of the AER resulted in limb truncation.46 Recently, the “two-signal model” was formulated, which proposes that two opposing signals, such as retinoic acid (RA) secreted from the flank mesenchyme and FGF secreted by the AER, pattern the skeletal elements along the PD axis.47 This model indicates that AER FGFs play an instructive role in regulating the expression of specific genes and thereby identities along the PD axis.47
The AP axis
Patterning along the AP axis is controlled by sonic hedgehog (Shh) in the ZPA.48,49 Grafting a ZPA to the anterior distal margin of a chicken limb bud causes mirror-image duplications of skeletal elements, and restriction of Shh expression in the ZPA can mimic the effects of ZPA grafts. These findings suggest that Shh mimics the function of ZPA and plays a key role in the AP patterning of the limb bud.50,51 Shh acts as a morphogen diffusing from the ZPA, establishing a concentration gradient across the digit-forming field.52,53 Experimental manipulation in chicks and genetic analysis in mice suggested that Shh participates in the early establishment of AP-axis polarity and controls the proliferative expansion of specified mesenchymal progenitors.54,55 Deficiency of Shh results in ulnar ray deficiency in the upper limbs and fibular ray deficiency in the lower limbs.56,57 Misexpression of Shh could lead to its ectopic activation at the anterior border of the bud, and this is considered to be at the root of many pre-axial polydactylies.
The expression of Shh needs to be tightly regulated at different levels, since alterations in the strength, duration, or location of Shh expression could have profound phenotypic consequences.58 First, normal Shh expression in the ZPA is regulated by an enhancer designated zone of polarizing activity regulatory sequence (ZRS).59,60 Recently, Williamson et al.61 showed that Shh and ZRS co-localize to the ZPA, and constrained chromatin configuration optimizes the opportunity for the active enhancer to locate and facilitate the expression of Shh. Point mutations in ZRS can induce anterior, ectopic Shh expression and can cause preaxial polydactyly, triphalangeal thumb, Werner mesomelic syndrome, or tibial hypoplasia.60,62–67 Duplications and even triplications of the ZRS have been implicated in polysyndactyly disease, including triphalangeal thumb-polysyndactyly syndrome and Haas-type polysyndactyly syndrome.66,68,69 Second, the activation and posterior restriction of Shh expression in the ZPA are maintained by mutual antagonism between the transcription factors HAND2 and GLI3.41 Initially, HAND2 and GLI3 are expressed throughout the presumptive limb field, and, during the onset of limb bud outgrowth, direct cross-regulation results in posterior restriction of HAND2 and anterior restriction of GLI3.70 This mutually antagonistic interaction results in the activation and posterior restriction of Shh expression in the limb bud mesenchyme.70 Recently, Deimling et al.71 identified a ~ 5-kb deletion within the Shh repressor GLI3 in two patients with bilateral tibial hemimelia. This deletion results in a truncated GLI3 protein that failed to repress anterior ectopic Shh expression.71 Third, in the established limb bud, the Shh/Gremlin1 (GREM1)/FGF feedback loop coordinates Shh signaling by the mesenchymal ZPA with FGF signaling by AER.72 Shh maintains FGF expression in the AER through the upregulation of bone morphogenetic protein (BMP)73 and GREM1,74,75 and FGFs produced by the AER maintain Shh expression in the ZPA.76,77 Finally, Wnt7a secreted by the dorsal ectoderm is required for Shh expression,78,79 which is discussed below.
The DV axis
The DV axis determines the development of dorsal (e.g., the nails and the thin hairy skin) and ventral (e.g., the thick, hairless skin of the palm and sole) structures in the hands and feet.56 The development of ventral structures is regulated by engrailed-1 (En1) (present in the ventral ectoderm), and the development of dorsal structures is controlled by WNT7a and LMX1b (present in the dorsal ectoderm and mesoderm, respectively) (80, 81). The expression of WNT7a is limited to the dorsal ectoderm, and it is repressed by En1 in the ventral ectoderm. Inactivation of En1 in mice results in double-dorsal limbs, probably because of the successive misexpression of WNT7a in the ventral ectoderm.82 WNT7a has two functions: it defines the DV axis by inducing LMX1b via the noncanonical Ca2+-mediated pathway,83,84 which is responsible for the development of dorsal structures. WNT7a also maintains Shh activity in the ZPA79 via the canonical β-catenin pathway,85 which is responsible for the development of the ulnar/fibular ray. Mutations in LMX1B result in nail–patella syndrome, a human condition characterized by loss of dorsal features,86 indicating that LMX1B participates in DV patterning. Mice lacking WNT7a activity exhibit loss of dorsal features and lack posterior digits, demonstrating that WNT7a is required for both DV and AP patterning.78
Potential signaling involvement in PALH
PALH is likely caused by a disturbance of the AP axis that determines the development of ulnoradial structures in the upper limbs and fibula tibial structures in the lower limbs. Shh is considered the essential controller of this axis. Indeed, as discussed previously, deficiency of Shh results in ulnar ray deficiency in the upper limbs and fibular ray deficiency in the lower limbs.56,57 However, it is not known how Shh and the regulatory factors in the AP axis act in the pathogenesis of PALH. Moreover, it is important to understand that the three axes are not independent and that they interact with each other. Therefore, a review of syndromes with fibular aplasia/hypoplasia can provide additional clues to the pathogenesis of PALH.
Al-Awadi, Raas-Rothschild, Schinzel phocomelia (AARRS) and Fuhrmann syndromes were considered to be distinct limb-malformation disorders. Fuhrmann syndrome is characterized by bowing of the femurs, aplasia or hypoplasia of the fibula, and poly-, syn-, and oligodactyly.87 In contrast, AARRS is much more severe and characterized by severe malformations of upper and lower limbs (including hypoplastic femora and absent ulnae and fibulae) with severely hypoplastic pelvis and abnormal genitalia.88 Human genetic studies have suggested that the both diseases are caused by mutations in the same gene––WNT7A; a partial loss of WNT7a function caused Fuhrmann syndrome, whereas the more severe limb truncation phenotypes were observed in AARRS, which resulted from functionally null mutations of WNT7A.89 All patients with WNT7A mutations have evidence of posterior hypoplasia in addition to the loss of dorsal features in the limbs, and this is explained by WNT7a effects on Shh via β-catenin signaling. The findings also implicate the importance of β-catenin signaling pathway in the pathogenesis of PALH.
Du Pan syndrome, characterized by a combination of brachydactyly and bilateral fibular hypo/aplasia, is the disease with growth differentiation factor 5 gene (GDF5) mutations.90 GDF5 belongs to the superfamily of BMPs binding with high affinity to the BMP receptor 1B (BMPR-1B).91,92 Affected individuals are homozygotes or compound heterozygotes for loss-of-function mutations in the highly conserved mature domain of GDF5, which results in an impaired BMP signaling through the BMPR-1B.90 Szczaluba et al.93 identified heterozygous mutations in GDF5 in a family with dominant Du Pan syndrome. A possible synergistic effect of the cis-acting mutations located in the active domain of the mature GDF5 protein is likely to be responsible for the clinical expression of the disorder. Recently, Stange et al.94 identified and characterized a hypomorphic BMPR-1B missense mutation that causes Du Pan syndrome. Several BMPs could interact with Shh signaling and have a weak polarizing activity to enhance the polarizing effect of a brief Shh application.52,95 These findings indicate that BMP signaling also plays a role in the pathogenesis of PALH.
Waardenburg anophthalmia syndrome is characterized by ocular anomalies ranging from mild microphthalmia to anophthalmia and limb defects.96 The distal abnormalities of the limbs include the absence of the fifth metatarsals and toes, oligodactyly, syndactyly, synostosis of the fourth and fifth metacarpals, and hypoplasia of the fibula with short femur or tibia.96 These defects could be caused by homozygous mutation in the SMOC1 gene.97–99 Although the molecular mechanism for this is unknown, SMOC1 has been shown to function as a BMP antagonist.100 These findings suggest that proper BMP antagonism is essential for postaxial development at the embryonic stage.
Summary
In summary, PALH, which includes FH, PFFD, and TC, is a group of nonhereditary diseases with congenital lower limb deficiency. The etiology of PALH remains unknown, although it appears to be related to developmental defects that occur during the embryonic period. We reviewed the previous classification systems, presented the clinical features, and discussed the potential signaling involvement in PALH.
Acknowledgments
This work was supported by National Institutes of Health Grants R01 AR054465 and R01 AR070222 to D.C. and F31 AR070002 to J.L.H. This work was also partially supported by the National Natural Science Foundation of China (Grant # 81371999) to D.C. This work was also partially supported by a grant from the Shenzhen Science and Technology Innovation Committee (Grant # JCYJ20160331114205502) to D.C. and a grant from Shenzhen Development and Reform Committee for Shenzhen Engineering Laboratory of Orthopaedic Regenerative Technologies to D.C.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Stevens PM, Arms D. Postaxial hypoplasia of the lower extremity. J Pediatr Orthop. 2000;20(2):166–72. [PubMed] [Google Scholar]
- 2.Coventry MB, Johnson EW., Jr Congenital absence of the fibula. J Bone Joint Surg Am. 1952;34 A(4):941–55. [PubMed] [Google Scholar]
- 3.Paley D. Surgical reconstruction for fibular hemimelia. J Child Orthop. 2016;10(6):557–83. doi: 10.1007/s11832-016-0790-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oberc A, Sulko J. Fibular hemimelia - diagnostic management, principles, and results of treatment. J Pediatr Orthop B. 2013;22(5):450–6. doi: 10.1097/BPB.0b013e32836330dd. [DOI] [PubMed] [Google Scholar]
- 5.Bryant DD, 3rd, Epps CH., Jr Proximal femoral focal deficiency: evaluation and management. Orthopedics. 1991;14(7):775–84. doi: 10.3928/0147-7447-19910701-08. [DOI] [PubMed] [Google Scholar]
- 6.Jacobs AM, Sollecito V, Oloff L, Klein N. Tarsal coalitions: an instructional review. J Foot Surg. 1981;20(4):214–21. [PubMed] [Google Scholar]
- 7.Fordham LA, Applegate KE, Wilkes DC, Chung CJ. Fibular Hemimelia: More Than Just An Absent Bone. Semin Musculoskelet Radiol. 1999;3(3):227–38. doi: 10.1055/s-2008-1080068. [DOI] [PubMed] [Google Scholar]
- 8.Froster UG, Baird PA. Congenital defects of lower limbs and associated malformations: a population based study. Am J Med Genet. 1993;45(1):60–4. doi: 10.1002/ajmg.1320450116. [DOI] [PubMed] [Google Scholar]
- 9.Syvanen J, Nietosvaara Y, Ritvanen A, Koskimies E, Kauko T, Helenius I. High risk for major nonlimb anomalies associated with lower-limb deficiency: a population-based study. J Bone Joint Surg Am. 2014;96(22):1898–904. doi: 10.2106/JBJS.N.00155. [DOI] [PubMed] [Google Scholar]
- 10.Rogala EJ, Wynne-Davies R, Littlejohn A, Gormley J. Congenital limb anomalies:frequency and aetiological factors. Data from the Edinburgh Register of the Newborn (1964–68) J Med Genet. 1974;11(3):221–33. doi: 10.1136/jmg.11.3.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thompson TC, Straub LR, Arnold WD. Congenital absence of the fibula. J Bone Joint Surg Am. 1957;39-A(6):1229–37. [PubMed] [Google Scholar]
- 12.Mishima K, Kitoh H, Iwata K, Matsushita M, Nishida Y, Hattori T, et al. Clinical Results and Complications of Lower Limb Lengthening for Fibular Hemimelia: A Report of Eight Cases. Medicine (Baltimore) 2016;95(21):e3787. doi: 10.1097/MD.0000000000003787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bedoya MA, Chauvin NA, Jaramillo D, Davidson R, Horn BD, Ho-Fung V. Common Patterns of Congenital Lower Extremity Shortening: Diagnosis, Classification, and Follow-up. Radiographics. 2015;35(4):1191–207. doi: 10.1148/rg.2015140196. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez-Ramirez A, Thacker MM, Becerra LC, Riddle EC, Mackenzie WG. Limb length discrepancy and congenital limb anomalies in fibular hemimelia. J Pediatr Orthop B. 2010;19(5):436–40. doi: 10.1097/BPB.0b013e32832d5d7d. [DOI] [PubMed] [Google Scholar]
- 15.Achterman C, Kalamchi A. Congenital deficiency of the fibula. J Bone Joint Surg Br. 1979;61-B(2):133–7. doi: 10.1302/0301-620X.61B2.438260. [DOI] [PubMed] [Google Scholar]
- 16.Stanitski DF, Stanitski CL. Fibular hemimelia: a new classification system. J Pediatr Orthop. 2003;23(1):30–4. [PubMed] [Google Scholar]
- 17.Paley D. Surgical reconstruction for fibular hemimelia. J Child Orthop. 2016;10(6):557–83. doi: 10.1007/s11832-016-0790-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stormer SV. Proximal femoral focal deficiency. Orthop Nurs. 1997;16(5):25–31. [PubMed] [Google Scholar]
- 19.D’Ambrosio V, Pasquali G, Squarcella A, Marcoccia E, De Filippis A, Gatto S, et al. Prenatal diagnosis of proximal focal femoral deficiency: Literature review of prenatal sonographic findings. J Clin Ultrasound. 2016;44(4):252–9. doi: 10.1002/jcu.22306. [DOI] [PubMed] [Google Scholar]
- 20.Kalaycioglu A, Aynaci O. Proximal focal femoral deficiency, contralateral hip dysplasia in association with contralateral ulnar hypoplasia and clefthand: a case report and review of literatures of PFFD and/or FFU. Okajimas Folia Anat Jpn. 2001;78(2–3):83–9. doi: 10.2535/ofaj1936.78.2-3_83. [DOI] [PubMed] [Google Scholar]
- 21.Kelly TE. Proximal focal femoral deficiency (familial) Birth Defects Orig Artic Ser. 1974;10(12):508–9. [PubMed] [Google Scholar]
- 22.Torode IP, Gillespie R. The classification and treatment of proximal femoral deficiencies. Prosthet Orthot Int. 1991;15(2):117–26. doi: 10.3109/03093649109164646. [DOI] [PubMed] [Google Scholar]
- 23.Lin TH, Chung CH, Shih JC, Lin CH, Lee CN, Su YN. Prenatal diagnosis of proximal femoral focal deficiency: a case report and literature review. Taiwan J Obstet Gynecol. 2013;52(2):267–9. doi: 10.1016/j.tjog.2013.04.020. [DOI] [PubMed] [Google Scholar]
- 24.Chomiak J, Horak M, Masek M, Frydrychova M, Dungl P. Computed tomographic angiography in proximal femoral focal deficiency. J Bone Joint Surg Am. 2009;91(8):1954–64. doi: 10.2106/JBJS.H.00902. [DOI] [PubMed] [Google Scholar]
- 25.Kayser R, Mahlfeld K, Grasshoff H, Merk HR. Proximal focal femoral deficiency – a rare entity in the sonographic differential diagnosis of developmental dysplasia of the hip. Ultraschall Med. 2005;26(5):379–84. doi: 10.1055/s-2005-858064. [DOI] [PubMed] [Google Scholar]
- 26.Mailath-Pokorny M, Timor-Tritsch IE, Monteagudo A, Mittal K, Konno F, Santos R. Prenatal diagnosis of unilateral proximal femoral focal deficiency at 19 weeks’ gestation: case report and review of the literature. Ultrasound Obstet Gynecol. 2011;38(5):594–7. doi: 10.1002/uog.8995. [DOI] [PubMed] [Google Scholar]
- 27.Epps CH., Jr Proximal femoral focal deficiency. J Bone Joint Surg Am. 1983;65(6):867–70. [PubMed] [Google Scholar]
- 28.Boden SD, Fallon MD, Davidson R, Mennuti MT, Kaplan FS. Proximal femoral focal deficiency. Evidence for a defect in proliferation and maturation of chondrocytes. J Bone Joint Surg Am. 1989;71(8):1119–29. [PubMed] [Google Scholar]
- 29.Panting AL, Williams PF. Proximal femoral focal deficiency. J Bone Joint Surg Br. 1978;60(1):46–52. doi: 10.1302/0301-620X.60B1.627578. [DOI] [PubMed] [Google Scholar]
- 30.Marchese JW, Mullen MG, Doherty JH., Jr Proximal femoral focal deficiency and fibular hemimelia associated with misoprostol use: a case report. Clin Dysmorphol. 2012;21(4):229–30. doi: 10.1097/MCD.0b013e3283590a95. [DOI] [PubMed] [Google Scholar]
- 31.Hamanishi C. Congenital short femur. Clinical, genetic and epidemiological comparison of the naturally occurring condition with that caused by thalidomide. J Bone Joint Surg Br. 1980;62(3):307–20. doi: 10.1302/0301-620X.62B3.7410462. [DOI] [PubMed] [Google Scholar]
- 32.Pachuda NM, Lasday SD, Jay RM. Tarsal coalition: etiology, diagnosis, and treatment. J Foot Surg. 1990;29(5):474–88. [PubMed] [Google Scholar]
- 33.Sakellariou A, Claridge RJ. Tarsal coalition. Orthopedics. 1999;22(11):1066–73. doi: 10.3928/0147-7447-19991101-16. discussion 73-4; quiz 10. [DOI] [PubMed] [Google Scholar]
- 34.Thometz J. Tarsal coalition. Foot Ankle Clin. 2000;5(1):103–18. vi. [PubMed] [Google Scholar]
- 35.Zaw H, Calder JD. Tarsal coalitions. Foot Ankle Clin. 2010;15(2):349–64. doi: 10.1016/j.fcl.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 36.Lawrence DA, Rolen MF, Haims AH, Zayour Z, Moukaddam HA. Tarsal Coalitions: Radiographic, CT, and MR Imaging Findings. HSS J. 2014;10(2):153–66. doi: 10.1007/s11420-013-9379-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Denning JR. Tarsal Coalition in Children. Pediatr Ann. 2016;45(4):e139–43. doi: 10.3928/00904481-20160309-01. [DOI] [PubMed] [Google Scholar]
- 38.Coventry MB. Flatfoot, with special consideration of tarsal coalition. Minn Med. 1950;33(11):1091–7. passim. [PubMed] [Google Scholar]
- 39.Solomon LB, Ruhli FJ, Taylor J, Ferris L, Pope R, Henneberg M. A dissection and computer tomograph study of tarsal coalitions in 100 cadaver feet. J Orthop Res. 2003;21(2):352–8. doi: 10.1016/S0736-0266(02)00131-6. [DOI] [PubMed] [Google Scholar]
- 40.Leonard MA. The inheritance of tarsal coalition and its relationship to spastic flat foot. J Bone Joint Surg Br. 1974;56B(3):520–6. [PubMed] [Google Scholar]
- 41.Plotkin S. Case presentation of calcaneonavicular coalition in monozygotic twins. J Am Podiatr Med Assoc. 1996;86(9):433–8. doi: 10.7547/87507315-86-9-433. [DOI] [PubMed] [Google Scholar]
- 42.Zuniga A. Next generation limb development and evolution: old questions, new perspectives. Development. 2015;142(22):3810–20. doi: 10.1242/dev.125757. [DOI] [PubMed] [Google Scholar]
- 43.Mariani FV, Martin GR. Deciphering skeletal patterning: clues from the limb. Nature. 2003;423(6937):319–25. doi: 10.1038/nature01655. [DOI] [PubMed] [Google Scholar]
- 44.Tickle C. How the embryo makes a limb: determination, polarity and identity. J Anat. 2015;227(4):418–30. doi: 10.1111/joa.12361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fernandez-Teran M, Ros MA. The Apical Ectodermal Ridge: morphological aspects and signaling pathways. Int J Dev Biol. 2008;52(7):857–71. doi: 10.1387/ijdb.072416mf. [DOI] [PubMed] [Google Scholar]
- 46.Haro E, Delgado I, Junco M, Yamada Y, Mansouri A, Oberg KC, et al. Sp6 and Sp8 transcription factors control AER formation and dorsal-ventral patterning in limb development. PLoS Genet. 2014;10(8):e1004468. doi: 10.1371/journal.pgen.1004468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saunders JW., Jr Developmental control of three-dimensional polarity in the avian limb. Ann N Y Acad Sci. 1972;193:29–42. doi: 10.1111/j.1749-6632.1972.tb27821.x. [DOI] [PubMed] [Google Scholar]
- 48.Mercader N, Leonardo E, Piedra ME, Martinez AC, Ros MA, Torres M. Opposing RA and FGF signals control proximodistal vertebrate limb development through regulation of Meis genes. Development. 2000;127(18):3961–70. doi: 10.1242/dev.127.18.3961. [DOI] [PubMed] [Google Scholar]
- 49.Pearse RV, 2nd, Tabin CJ. The molecular ZPA. J Exp Zool. 1998;282(6):677–90. [PubMed] [Google Scholar]
- 50.Harfe BD, Scherz PJ, Nissim S, Tian H, McMahon AP, Tabin CJ. Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell. 2004;118(4):517–28. doi: 10.1016/j.cell.2004.07.024. [DOI] [PubMed] [Google Scholar]
- 51.Capdevila J, Izpisua Belmonte JC. Patterning mechanisms controlling vertebrate limb development. Annu Rev Cell Dev Biol. 2001;17:87–132. doi: 10.1146/annurev.cellbio.17.1.87. [DOI] [PubMed] [Google Scholar]
- 52.Johnson RL, Riddle RD, Laufer E, Tabin C. Sonic hedgehog: a key mediator of anterior-posterior patterning of the limb and dorso-ventral patterning of axial embryonic structures. Biochem Soc Trans. 1994;22(3):569–74. doi: 10.1042/bst0220569. [DOI] [PubMed] [Google Scholar]
- 53.Drossopoulou G, Lewis KE, Sanz-Ezquerro JJ, Nikbakht N, McMahon AP, Hofmann C, et al. A model for anteroposterior patterning of the vertebrate limb based on sequential long- and short-range Shh signalling and Bmp signalling. Development. 2000;127(7):1337–48. doi: 10.1242/dev.127.7.1337. [DOI] [PubMed] [Google Scholar]
- 54.Tickle C. Patterning systems–from one end of the limb to the other. Dev Cell. 2003;4(4):449–58. doi: 10.1016/s1534-5807(03)00095-9. [DOI] [PubMed] [Google Scholar]
- 55.Towers M, Mahood R, Yin Y, Tickle C. Integration of growth and specification in chick wing digit-patterning. Nature. 2008;452(7189):882–6. doi: 10.1038/nature06718. [DOI] [PubMed] [Google Scholar]
- 56.Zhu J, Nakamura E, Nguyen MT, Bao X, Akiyama H, Mackem S. Uncoupling Sonic hedgehog control of pattern and expansion of the developing limb bud. Dev Cell. 2008;14(4):624–32. doi: 10.1016/j.devcel.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Niswander L. Pattern formation: old models out on a limb. Nat Rev Genet. 2003;4(2):133–43. doi: 10.1038/nrg1001. [DOI] [PubMed] [Google Scholar]
- 58.Al-Qattan MM, Al-Sahabi A, Al-Arfaj N. Ulnar ray deficiency: a review of the classification systems, the clinical features in 72 cases, and related developmental biology. J Hand Surg Eur. 2010;35(9):699–707. doi: 10.1177/1753193409358240. [DOI] [PubMed] [Google Scholar]
- 59.Li Y, Zhang H, Litingtung Y, Chiang C. Cholesterol modification restricts the spread of Shh gradient in the limb bud. Proc Natl Acad Sci U S A. 2006;103(17):6548–53. doi: 10.1073/pnas.0600124103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lettice LA, Horikoshi T, Heaney SJ, van Baren MJ, van der Linde HC, Breedveld GJ, et al. Disruption of a long-range cis-acting regulator for Shh causes preaxial polydactyly. Proc Natl Acad Sci U S A. 2002;99(11):7548–53. doi: 10.1073/pnas.112212199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lettice LA, Heaney SJ, Purdie LA, Li L, de Beer P, Oostra BA, et al. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet. 2003;12(14):1725–35. doi: 10.1093/hmg/ddg180. [DOI] [PubMed] [Google Scholar]
- 62.Williamson I, Lettice LA, Hill RE, Bickmore WA. Shh and ZRS enhancer colocalisation is specific to the zone of polarising activity. Development. 2016;143(16):2994–3001. doi: 10.1242/dev.139188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu PF, Guo S, Fan XF, Fan LL, Jin JY, Tang JY, et al. A Novel ZRS Mutation in a Chinese Patient with Preaxial Polydactyly and Triphalangeal Thumb. Cytogenet Genome Res. 2016;149(3):171–5. doi: 10.1159/000448820. [DOI] [PubMed] [Google Scholar]
- 64.Norbnop P, Srichomthong C, Suphapeetiporn K, Shotelersuk V. ZRS 406A>G mutation in patients with tibial hypoplasia, polydactyly and triphalangeal first fingers. J Hum Genet. 2014;59(8):467–70. doi: 10.1038/jhg.2014.50. [DOI] [PubMed] [Google Scholar]
- 65.Girisha KM, Bidchol AM, Kamath PS, Shah KH, Mortier GR, Mundlos S, et al. A novel mutation (g.106737G>T) in zone of polarizing activity regulatory sequence (ZRS) causes variable limb phenotypes in Werner mesomelia. Am J Med Genet A. 2014;164A(4):898–906. doi: 10.1002/ajmg.a.36367. [DOI] [PubMed] [Google Scholar]
- 66.Johnson EJ, Neely DM, Dunn IC, Davey MG. Direct functional consequences of ZRS enhancer mutation combine with secondary long range SHH signalling effects to cause preaxial polydactyly. Dev Biol. 2014;392(2):209–20. doi: 10.1016/j.ydbio.2014.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wieczorek D, Pawlik B, Li Y, Akarsu NA, Caliebe A, May KJ, et al. A specific mutation in the distant sonic hedgehog (SHH) cis-regulator (ZRS) causes Werner mesomelic syndrome (WMS) while complete ZRS duplications underlie Haas type polysyndactyly and preaxial polydactyly (PPD) with or without triphalangeal thumb. Hum Mutat. 2010;31(1):81–9. doi: 10.1002/humu.21142. [DOI] [PubMed] [Google Scholar]
- 68.Furniss D, Lettice LA, Taylor IB, Critchley PS, Giele H, Hill RE, et al. A variant in the sonic hedgehog regulatory sequence (ZRS) is associated with triphalangeal thumb and deregulates expression in the developing limb. Hum Mol Genet. 2008;17(16):2417–23. doi: 10.1093/hmg/ddn141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Klopocki E, Ott CE, Benatar N, Ullmann R, Mundlos S, Lehmann K. A microduplication of the long range SHH limb regulator (ZRS) is associated with triphalangeal thumb-polysyndactyly syndrome. J Med Genet. 2008;45(6):370–5. doi: 10.1136/jmg.2007.055699. [DOI] [PubMed] [Google Scholar]
- 70.Sun M, Ma F, Zeng X, Liu Q, Zhao XL, Wu FX, et al. Triphalangeal thumb-polysyndactyly syndrome and syndactyly type IV are caused by genomic duplications involving the long range, limb-specific SHH enhancer. J Med Genet. 2008;45(9):589–95. doi: 10.1136/jmg.2008.057646. [DOI] [PubMed] [Google Scholar]
- 71.te Welscher P, Fernandez-Teran M, Ros MA, Zeller R. Mutual genetic antagonism involving GLI3 and dHAND prepatterns the vertebrate limb bud mesenchyme prior to SHH signaling. Genes Dev. 2002;16(4):421–6. doi: 10.1101/gad.219202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Deimling S, Sotiropoulos C, Lau K, Chaudhry S, Sturgeon K, Kelley S, et al. Tibial hemimelia associated with GLI3 truncation. J Hum Genet. 2016;61(5):443–6. doi: 10.1038/jhg.2015.161. [DOI] [PubMed] [Google Scholar]
- 73.Benazet JD, Bischofberger M, Tiecke E, Goncalves A, Martin JF, Zuniga A, et al. A self-regulatory system of interlinked signaling feedback loops controls mouse limb patterning. Science. 2009;323(5917):1050–3. doi: 10.1126/science.1168755. [DOI] [PubMed] [Google Scholar]
- 74.Nissim S, Hasso SM, Fallon JF, Tabin CJ. Regulation of Gremlin expression in the posterior limb bud. Dev Biol. 2006;299(1):12–21. doi: 10.1016/j.ydbio.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 75.Khokha MK, Hsu D, Brunet LJ, Dionne MS, Harland RM. Gremlin is the BMP antagonist required for maintenance of Shh and Fgf signals during limb patterning. Nat Genet. 2003;34(3):303–7. doi: 10.1038/ng1178. [DOI] [PubMed] [Google Scholar]
- 76.Michos O, Panman L, Vintersten K, Beier K, Zeller R, Zuniga A. Gremlin-mediated BMP antagonism induces the epithelial-mesenchymal feedback signaling controlling metanephric kidney and limb organogenesis. Development. 2004;131(14):3401–10. doi: 10.1242/dev.01251. [DOI] [PubMed] [Google Scholar]
- 77.Laufer E, Nelson CE, Johnson RL, Morgan BA, Tabin C. Sonic hedgehog and Fgf-4 act through a signaling cascade and feedback loop to integrate growth and patterning of the developing limb bud. Cell. 1994;79(6):993–1003. doi: 10.1016/0092-8674(94)90030-2. [DOI] [PubMed] [Google Scholar]
- 78.Niswander L, Jeffrey S, Martin GR, Tickle C. A positive feedback loop coordinates growth and patterning in the vertebrate limb. Nature. 1994;371(6498):609–12. doi: 10.1038/371609a0. [DOI] [PubMed] [Google Scholar]
- 79.Parr BA, McMahon AP. Dorsalizing signal Wnt-7a required for normal polarity of D-V and A-P axes of mouse limb. Nature. 1995;374(6520):350–3. doi: 10.1038/374350a0. [DOI] [PubMed] [Google Scholar]
- 80.Yang Y, Niswander L. Interaction between the signaling molecules WNT7a and SHH during vertebrate limb development: dorsal signals regulate anteroposterior patterning. Cell. 1995;80(6):939–47. doi: 10.1016/0092-8674(95)90297-x. [DOI] [PubMed] [Google Scholar]
- 81.Cygan JA, Johnson RL, McMahon AP. Novel regulatory interactions revealed by studies of murine limb pattern in Wnt-7a and En-1 mutants. Development. 1997;124(24):5021–32. doi: 10.1242/dev.124.24.5021. [DOI] [PubMed] [Google Scholar]
- 82.Logan C, Hornbruch A, Campbell I, Lumsden A. The role of Engrailed in establishing the dorsoventral axis of the chick limb. Development. 1997;124(12):2317–24. doi: 10.1242/dev.124.12.2317. [DOI] [PubMed] [Google Scholar]
- 83.Loomis CA, Harris E, Michaud J, Wurst W, Hanks M, Joyner AL. The mouse Engrailed-1 gene and ventral limb patterning. Nature. 1996;382(6589):360–3. doi: 10.1038/382360a0. [DOI] [PubMed] [Google Scholar]
- 84.Riddle RD, Ensini M, Nelson C, Tsuchida T, Jessell TM, Tabin C. Induction of the LIM homeobox gene Lmx1 by WNT7a establishes dorsoventral pattern in the vertebrate limb. Cell. 1995;83(4):631–40. doi: 10.1016/0092-8674(95)90103-5. [DOI] [PubMed] [Google Scholar]
- 85.Kengaku M, Capdevila J, Rodriguez-Esteban C, De La Pena J, Johnson RL, Izpisua Belmonte JC, et al. Distinct WNT pathways regulating AER formation and dorsoventral polarity in the chick limb bud. Science. 1998;280(5367):1274–7. doi: 10.1126/science.280.5367.1274. [DOI] [PubMed] [Google Scholar]
- 86.Adamska M, MacDonald BT, Sarmast ZH, Oliver ER, Meisler MH. En1 and Wnt7a interact with Dkk1 during limb development in the mouse. Dev Biol. 2004;272(1):134–44. doi: 10.1016/j.ydbio.2004.04.026. [DOI] [PubMed] [Google Scholar]
- 87.Dreyer SD, Zhou G, Baldini A, Winterpacht A, Zabel B, Cole W, et al. Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail patella syndrome. Nat Genet. 1998;19(1):47–50. doi: 10.1038/ng0598-47. [DOI] [PubMed] [Google Scholar]
- 88.Fuhrmann W, Fuhrmann-Rieger A, de Sousa F. Poly-, syn- and oligodactylyl, aplasia or hypoplasia of fibula, hypoplasia of pelvis and bowing of femora in three sibs–a new autosomal recessive syndrome. Eur J Pediatr. 1980;133(2):123–9. doi: 10.1007/BF00441580. [DOI] [PubMed] [Google Scholar]
- 89.Al-Awadi SA, Teebi AS, Farag TI, Naguib KM, el-Khalifa MY. Profound limb deficiency, thoracic dystrophy, unusual facies, and normal intelligence: a new syndrome. J Med Genet. 1985;22(1):36–8. doi: 10.1136/jmg.22.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Woods CG, Stricker S, Seemann P, Stern R, Cox J, Sherridan E, et al. Mutations in WNT7A cause a range of limb malformations, including Fuhrmann syndrome and Al-Awadi/Raas-Rothschild/Schinzel phocomelia syndrome. Am J Hum Genet. 2006;79(2):402–8. doi: 10.1086/506332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Faiyaz-Ul-Haque M, Ahmad W, Zaidi SH, Haque S, Teebi AS, Ahmad M, et al. Mutation in the cartilage-derived morphogenetic protein-1 (CDMP1) gene in a kindred affected with fibular hypoplasia and complex brachydactyly (DuPan syndrome) Clin Genet. 2002;61(6):454–8. doi: 10.1034/j.1399-0004.2002.610610.x. [DOI] [PubMed] [Google Scholar]
- 92.Nickel J, Kotzsch A, Sebald W, Mueller TD. A single residue of GDF-5 defines binding specificity to BMP receptor IB. J Mol Biol. 2005;349(5):933–47. doi: 10.1016/j.jmb.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 93.Baur ST, Mai JJ, Dymecki SM. Combinatorial signaling through BMP receptor IB and GDF5: shaping of the distal mouse limb and the genetics of distal limb diversity. Development. 2000;127(3):605–19. doi: 10.1242/dev.127.3.605. [DOI] [PubMed] [Google Scholar]
- 94.Szczaluba K, Hilbert K, Obersztyn E, Zabel B, Mazurczak T, Kozlowski K. Du Pan syndrome phenotype caused by heterozygous pathogenic mutations in CDMP1 gene. Am J Med Genet A. 2005;138(4):379–83. doi: 10.1002/ajmg.a.30969. [DOI] [PubMed] [Google Scholar]
- 95.Stange K, Desir J, Kakar N, Mueller TD, Budde BS, Gordon CT, et al. A hypomorphic BMPR1B mutation causes du Pan acromesomelic dysplasia. Orphanet J Rare Dis. 2015;10:84. doi: 10.1186/s13023-015-0299-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bastida MF, Sheth R, Ros MA. A BMP-Shh negative-feedback loop restricts Shh expression during limb development. Development. 2009;136(22):3779–89. doi: 10.1242/dev.036418. [DOI] [PubMed] [Google Scholar]
- 97.Richieri-Costa A, Gollop TR, Otto PG. Brief clinical report: autosomal recessive anophthalmia with multiple congenital abnormalities–type Waardenburg. Am J Med Genet. 1983;14(4):607–15. doi: 10.1002/ajmg.1320140403. [DOI] [PubMed] [Google Scholar]
- 98.Okada I, Hamanoue H, Terada K, Tohma T, Megarbane A, Chouery E, et al. SMOC1 is essential for ocular and limb development in humans and mice. Am J Hum Genet. 2011;88(1):30–41. doi: 10.1016/j.ajhg.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Abouzeid H, Boisset G, Favez T, Youssef M, Marzouk I, Shakankiry N, et al. Mutations in the SPARC-related modular calcium-binding protein 1 gene, SMOC1, cause waardenburg anophthalmia syndrome. Am J Hum Genet. 2011;88(1):92–8. doi: 10.1016/j.ajhg.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rainger J, van Beusekom E, Ramsay JK, McKie L, Al-Gazali L, Pallotta R, et al. Loss of the BMP antagonist, SMOC-1, causes Ophthalmo-acromelic (Waardenburg Anophthalmia) syndrome in humans and mice. PLoS Genet. 2011;7(7):e1002114. doi: 10.1371/journal.pgen.1002114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Thomas JT, Canelos P, Luyten FP, Moos M., Jr Xenopus SMOC-1 Inhibits bone morphogenetic protein signaling downstream of receptor binding and is essential for postgastrulation development in Xenopus. J Biol Chem. 2009;284(28):18994–9005. doi: 10.1074/jbc.M807759200. [DOI] [PMC free article] [PubMed] [Google Scholar]