

Summary
Fibroblast Growth Factors (FGFs) participate in organ development and tissue maintenance as well as the control of vascular function. The paracrine-acting FGFs are stored in the extracellular matrix and their release is controlled by a secreted FGF-binding protein (FGF-BP, FGFBP1, BP1) that modulates FGF receptor (FGFR) signaling. A genetic polymorphism in the human FGFBP1 gene was associated with higher gene expression and an increased risk of familial hypertension. Here we report on the effects of inducible BP1 expression in a transgenic mouse model. Induction of BP1 expression in adult animals leads to a sustained rise in mean arterial pressure by >30 mm Hg. The hypertensive effect of BP1 expression is prevented by candesartan, an angiotensin II (AngII) receptor antagonist, or by tempol, an inhibitor of reactive oxygen species. In vivo, BP1 expression sensitizes peripheral resistance vessels to AngII constriction by 20-fold but does not alter adrenergic vasoconstriction. FGFR kinase inhibition reverses the sensitization to AngII. Also, constriction of isolated renal afferent arterioles by AngII is enhanced after BP1 expression and blocked by FGFR kinase inhibition. Furthermore, AngII-mediated constriction of renal afferent arterioles is abolished in FGF2−/− mice but can be restored by add-back of FGF2 plus BP1 proteins. In contrast to AngII, adrenergic constriction is not affected in the FGF2−/− model. Proteomics and gene expression analysis of kidney tissues after BP1 induction show that MAP kinase signaling via MKK4, p38 and JNK integrates the crosstalk of the FGFR and AngII pathways and thus impact vascular tone and blood pressure.
Keywords: blood pressure, fibroblast growth factor, angiotensin II, reactive oxygen species, vascular resistance
Introduction
The family of fibroblast growth factors (FGFs) encompasses eighteen FGF receptor ligands and seven distinct receptor proteins with a wide expression range. They have distinct roles in embryonic development, in adult organ homeostasis and vascular adaptation as well as in a wide range of diseases1. Genome-wide association studies in hypertensive populations have shown a potential role of molecules in the FGF pathway (reviewed in2). Polymorphisms in the FGF5 locus were associated with blood pressure regulation and hypertension in large populations of European3, 4 and of Japanese ancestry5. Variations in the FGF1 locus correlated with familial hypertension and with the upregulation of FGF1 expression in kidneys6, 7. Also, genomic analysis revealed that a polymorphism in the FGFBP1 locus (= FGF Binding Protein 1; BP1) was associated with familial hypertension and hypertensive subjects showed increased expression of BP1 mRNA and protein in renal tissues8. Studies in hypertensive rats corroborated a contribution of the FGFBP1 genomic locus to glomerular damage and to hypertension9. Still, we lack information about mechanisms by which BP1 may control blood pressure and how it fits into the known paradigms of blood pressure control10.
Secreted FGF binding proteins (BPs) shuttle paracrine-acting FGFs from their extracellular matrix storage sites to their receptors and thus enhance their signaling11, 12. BP1 (originally named HBp1713 and FGF-BP) is the best characterized of the three known members of the family14, and interacts via its C-terminus15 with FGF1, 2, 7, 10 and 22 in a reversible, noncovalent manner13, 16–18. Depletion of endogenous BP1 reduced FGF2 release and blunted tumor growth and angiogenesis of human cancer cells19 and resulted in distinct developmental defects during chick embryogenesis20. On the other hand BP1 is upregulated in angioproliferative Kaposi Sarcoma21, contributes to an angiogenic phenotype of cultured endothelial cells22 and controls angiogenesis and wound healing in adult mice23, 24.
Here we evaluate how the upregulation of BP1 may cause hypertension. For this we used a transgenic mouse model with conditional BP1 gene expression under tetracycline control23. We found that induction of BP1 expression results in sustained hypertension and FGFR signaling-dependent increased vascular sensitivity to the vasoconstrictor angiotensin II (AngII) via reactive oxygen species and MAP kinase pathway signaling.
Methods
Conditional transgenic animal model
Animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee. Transgenic animals carrying a human BP1 cDNA under the control of a tetracycline response element (TRE-BP1) were described23.
Statistical methods and data analyses
Data analyses are described in the online-only supplement. P-values <0.05 were considered as significant.
Further details and Methods see http://hyper.ahajournals.org, the online-only supplement.
Results
Conditional expression of BP1 in vivo
We had found that BP1 transgene expression results in embryonic lethality due to vascular leakage25. Thus, we established a conditional transgenic mouse model where BP1 transgene expression is repressed by tetracycline (“OFF”) and induced by switching animals to a regular diet (“ON”)23 thus avoiding the negative impact of embryonic gene expression. In vivo regulation of conditional BP1 mRNA and protein expression in kidneys of transgenic animals was confirmed by quantitative RT-PCR (qRT-PCR) and staining of formalin-fixed, paraffin-embedded kidneys from BP1 OFF and ON transgenic animals (Figure 1A,B). Inducible BP1 mRNA and protein expression was also confirmed in heart and lung tissues by qRT-PCR and by Western blot analysis that showed the BP1 protein migrating at the predicted mass of 34 kDa after induction of expression (Figure S1). Overall, we found that BP1 mRNA was inducible by 3- to 5-fold.
Figure 1.
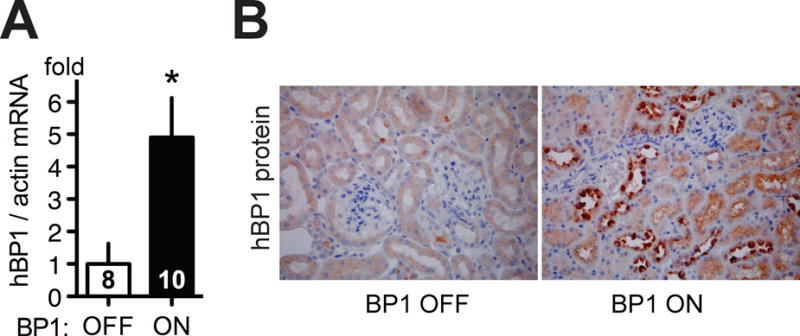
Conditional BP1 transgene expression in kidneys in vivo. A, BP1 mRNA expression in kidneys of BP1 OFF and ON animals by quantitative RT-PCR. Expression was normalized to endogenous beta-actin mRNA (means ± SEM; n = 8 and 10 animals per group). *, P< 0.05, BP1 ON versus OFF. B, Detection of BP1 protein by immunohistochemistry in kidneys from BP1 OFF and ON mice (total n=3). Size bar: 50 μm. Note: Positive immunoreactivity results in dark brown staining. (Note: Figure S1 shows BP1 protein and mRNA expression in heart and lung tissues)
BP1 expression and blood pressure control
To test the effect of conditional expression of BP1 mean arterial pressure (MAP) was monitored by telemetry in conscious transgenic mice. Under control conditions (=BP1 OFF) there is a circadian rhythm of MAP that varied by 20 mm Hg between periods of activity (night time) and rest (day time). A sinus wave function described the day/night variation of the data (Figure 2A). Analysis of a 10 day period before and after induction of the BP1 transgene in the same animals showed a rise of MAP during the activity phase by >30 mm Hg (Figure 2C) and an almost doubling of the MAP changes between activity and rest periods (Figure 2A). This overall increase in MAP coincided with a significant decrease in the heart rate and a dampened amplitude of circadian regulation (Figure 2B). The increase of blood pressure occurred within two days of a switch to the tetracycline-free, regular diet in parallel with the induction of the BP1 transgene (Figure 2C). Switching non-transgenic littermates from the tetracycline-containing to the regular diet did not change MAP (Figure 2C, circles versus triangles).
Figure 2.
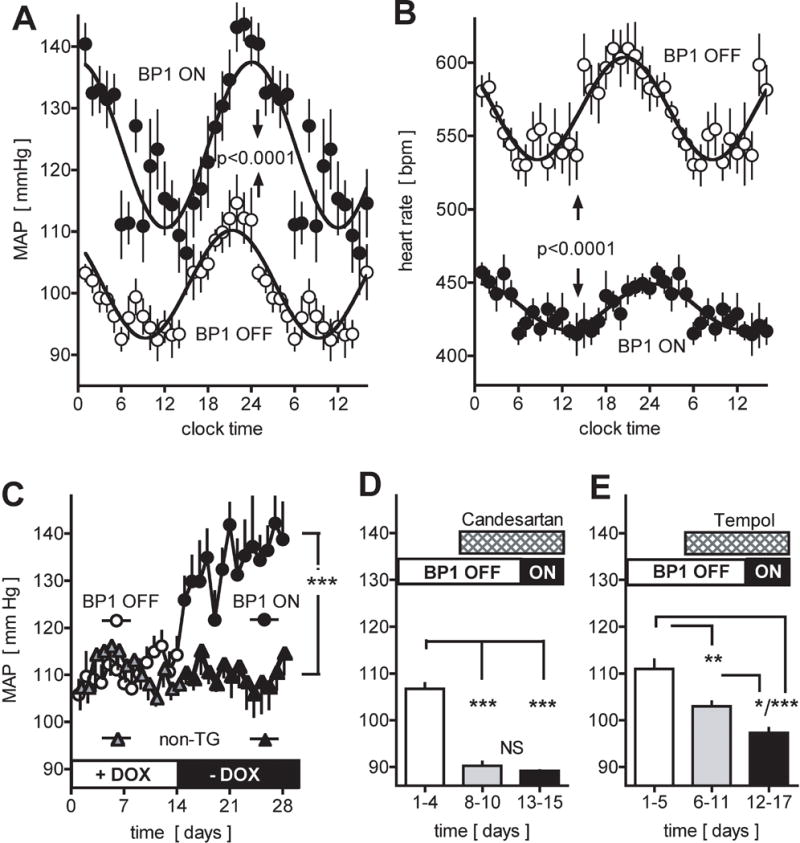
Effect of conditional expression of the BP1 transgene and of inhibitors on mean arterial pressure (MAP) and heart rate (HR). A, B, Circadian changes in MAP (A) and HR (B) by telemetry of conscious mice. Averages for 10 days before (BP1 OFF, open symbols) and 10 days after the induction of the BP1 transgene (BP1 ON, closed symbols) relative to the 24 hour clock time. Dark and light periods are indicated. Mean ± SEM values (for n=10 time points) from one representative animal (total n=6). P<0.0001 by non-linear regression analysis and ANOVA. C, Effect of induction of BP1 transgene expression on night time MAP. Gene expression was induced by switching from a tetracycline containing to a regular chow (+DOX to −DOX; open and filled circles). Non-transgenic littermates (non-TG; triangles) were subjected to the same diet schedule. Mean ± SEM of night time MAP; n = 6 per group. ANOVA analysis of BP1 ON versus OFF or versus control group. ***, P<0.0001. D, E, Effect of treatment with candesartan (D) or tempol (E) on night time MAP of animals with the BP1 transgene OFF or ON. Mean ± SEM; n = 5 per group. *, P<0.05; **, P<0.01; ***, P <0.0001 by ANOVA.
The role of angiotensin II (AngII) and reactive oxygen species (ROS)
The increase in MAP and decrease in heart rate after induction of BP1 expression (Figure 2A,B) is reminiscent of an angiotensin-like vasopressor effect that can depend on ROS signaling26. To assess whether endogenous AngII receptor signaling and ROS contribute to BP1-mediated hypertension, mice were treated with the receptor antagonist Candesartan or the redox-cycling antioxidant nitroxide tempol27. Candesartan reduced basal MAP (Figure 2D, grey bar) and prevented the increase of MAP after BP1 induction (Figure 2D, grey vs black bars). Tempol also reduced basal MAP (Figure 2E grey bars) and prevented the increase in MAP after BP1 induction (Figure 2E grey vs black bars).
Induction of BP1, FGF pathway activation and vessel contractility
To evaluate whether conditional BP1 expression sensitizes resistance vessels in vivo, cremaster arterioles were exposed in anesthetized mice and superfused locally with vasoconstrictor or -dilator ligands. Representative intravital microscopic images of an arteriole at baseline, with AngII (constriction) or acetylcholine (dilation) superfusion is shown in Figure 3A. There was a 20-fold sensitization of the AngII vasoconstrictor response after the induction of BP1 gene expression (EC50 = 230 nM versus 12 nM; Figure 3B; p<0.0001 BP1 OFF versus ON). This sensitization was prevented by pretreatment of animals with the FGFR kinase inhibitor PD17307423 (Figure 3B, filled black versus red circles). Control animals (BP1 OFF) showed only a small and insignificant effect after the FGFR kinase inhibitor (Figure 3B, open black versus red circles).
Figure 3.
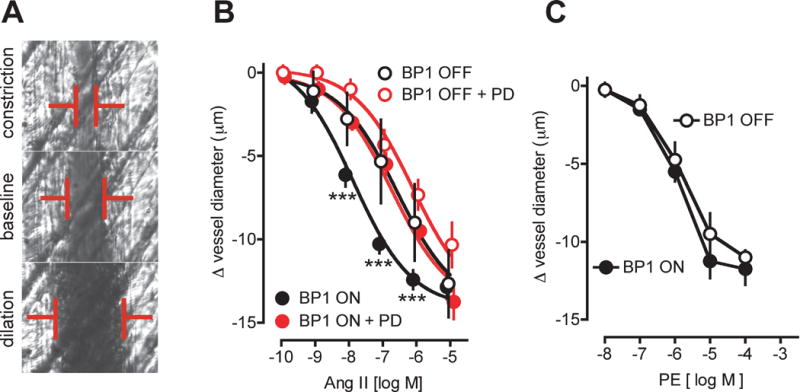
Effect of the induction of BP1 expression on cremaster arteriole contractility in vivo. A, Representative images of a single arteriole in a live animal at baseline, and after constriction or dilation by superfusion with AngII or acetylcholine respectively. The vessel diameter (red bars) is recorded by intravital microphotography. B, Dose-response of AngII on arteriole diameter in mice with BP1 OFF (open symbols) or ON (for 48 hours; closed symbols). The FGFR kinase inhibitor PD173074 (PD) was administered intraperitoneally to a subset of animals (red symbols). Mean ± SEM, n = 4 to 7 animals/group. pEC50 values of 6.64 ± 0.15 (= 230 nM; BP1 OFF) and 7.92 ± 0.13 (= 12 nM; BP1 ON) were calculated from non-linear regression analysis. ***, P<0.0001 BP1 OFF vs. BP1 ON and BP1 ON + PD vs. BP1 ON. C, Dose response of Phenylephrine (PE) on cremaster arteriole diameter in mice with BP1 OFF (open symbols) or ON (for 48 hours; closed symbols). Mean ± SEM, n = 4 animals/group.
We inquired whether BP1 expression also could sensitize the cremaster arterioles to other vasoconstrictive stimuli. We had found previously that adrenergic receptor activation in resistance arterioles, unlike AngII, does not induce ROS generation and induces vasoconstriction that is not enhanced by oxidative stress28. Thus, we selected phenylephrine (PE) as a ligand that activates alpha1-adrenergic receptors. The extent of vasoconstriction by PE was similar to AngII (Figure 3B vs C; BP1 OFF). But, unlike AngII, BP1 expression did not sensitize vessels to PE (Figure 3C). Neither the EC50 (2 μM) nor the maximal effect of PE were different between the BP1 ON and OFF groups (Figure 3C). Thus, conditional expression of BP1 sensitizes arterioles to AngII in vivo. This sensitization is dependent on FGFR signaling.
BP1 impacts contractility of renal afferent arterioles
Isolated vessels provide an approach for the analysis of vascular function that is not affected by systemic cardiovascular regulation in the intact animal. BP1 up-regulation related to human hypertension was found in the kidneys that are key organs in systemic blood pressure regulation. Thus, we evaluated the impact of BP1 expression in isolated renal afferent arterioles that are the major renal resistance vessels29. Figure 4A shows the experimental set-up with a glomerulus and an afferent arteriole mounted onto a perfusion pipette. Addition of AngII to this preparation leads to a concentration-dependent constriction. Afferent arterioles isolated from mice after conditional BP1 transgene expression showed a significantly enhanced effect of AngII (Figure 4B, open vs closed circles). Pretreatment of the vessels with the FGFR kinase inhibitor PD173074 inhibited the effect of AngII (Figure 4B, blue symbol) to a level similar to the response in arterioles from FGF2−/− mice (red, open symbols). Thus, the AngII contractile effect depends on FGFR signaling in isolated vessels as well as in arterioles in vivo (see above, Figure 3B).
Figure 4.
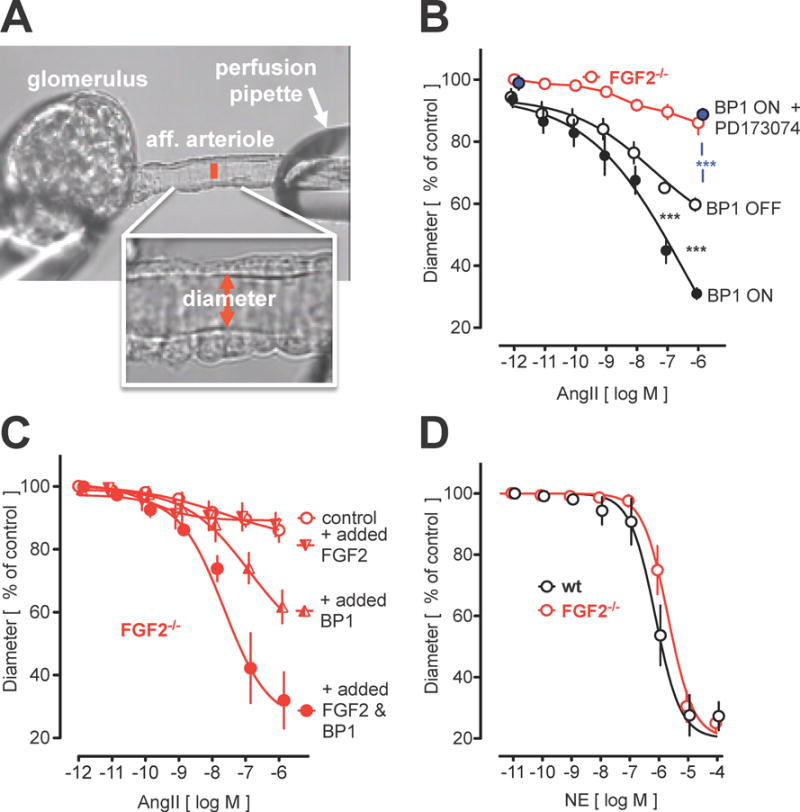
Effect of BP1 on isolated renal afferent arteriole contractility. A, Isolated perfused renal afferent arteriole mounted on a perfusion pipette with diameter recorded. The internal diameter (red bar) is 10 to 11 micrometers under control conditions (=100%). B to D, Impact of BP1 induction and/or FGF2 knockout (FGF2−/−) on angiotensin II (AngII) or norepinephine (NE) contractility. B, Effect of conditional BP1 expression (BP1 ON/OFF). Expression of BP1 increases the AngII effect (open versus filled black circles). Addition of the FGFR kinase inhibitor PD173074 (100 nM) inhibits the effect of AngII (blue). The effect of AngII in FGF2−/− mice is shown for comparison (see panel C). C, Effect of AngII in afferent arterioles from FGF2−/− mice. Add-back of FGF2 plus BP1 proteins (20 ng/ml for 30 min) restores the AngII effect. BP1 alone or FGF2 alone are shown for comparison. D, Effect of NE in afferent arterioles from wt and FGF2−/− mice. Contractility induced by NE is not affected in FGF2−/− mice.
Contribution of FGF2 to the effect of BP1
Kidneys contain amongst the highest concentrations of FGF2 protein that is immobilized in the extracellular matrix30 and can be released by BP1 as outlined in the introduction. To assess a contribution by FGF2 we investigated the efficacy of AngII and the crosstalk with BP1 in renal afferent arterioles from FGF2−/− mice. FGF2−/− mice had shown a reduced vascular tone and lower blood pressure31, 32. We confirmed by telemetric measurements that mean arterial pressure (MAP) in FGF2−/− mice is reduced significantly by 15 mm Hg relative to wt mice (Figure S2a, open vs closed bar). Also, we found that the effect of exogenously administered AngII on MAP was reduced (Figure S2a).
Consistent with the blood pressure effect, AngII failed to induce a contraction in renal afferent arterioles isolated from the FGF2−/− mice (Figure 4B and C, open red symbols). The blockade of AngII effects in vessels from wt mice by an FGFR kinase inhibitor (Figure 4B, red open vs blue symbols) corroborates the crucial role of FGF signaling for AngII efficacy. It is noteworthy that vessels from FGF2−/− mice still contracted in response to other ligands: The effect of norepinephrine (NE) was indistinguishable between FGF2−/− and wt controls (Figure 4D, black vs. red symbols) indicating a selective crosstalk between AngII and FGF signaling. Renal AngII receptor 1 (AT1R) mRNA was not changed upon induction of BP1 expression or in FGF2−/− mice (Figure S3).
Because FGF2 and BP1 are extracellularly acting proteins, we added recombinant proteins to renal afferent arterioles from FGF2−/− mice to assess if this would rescue AngII vasoconstriction. Whilst FGF2 alone did not impact the AngII response most likely due to its capture by the extracellular matrix, the combination of FGF2 & BP1 restored a full contractile response of AngII (Figure 4C).
Gene expression and pathway analysis
An unbiased gene expression analysis by cDNA array was undertaken in kidneys before and two days after induction of BP1, i.e. after the initial rise in blood pressure (see Figure 2C). Upstream regulators were identified using the Ingenuity platform and Gene Set Enrichment Analysis (GSEA) to detect expressions patterns associated with signaling pathways.
The upstream regulator analysis (Figure 5A; complete list in Table S1) showed positive z-scores >+1 and p-values <10−10 for the gene sets controlled by the transcriptional regulators TP53 (Tumor Suppressor P53), NRF2 (=NFE2L2, nuclear factor erythroid-derived 2-like 2) and CREB1 (cAMP responsive element binding protein 1). TP53 modulates cellular stress response genes33, NRF2 regulates anti-oxidant genes and ChIPseq has shown an overlap of the target genes of NRF2 and of CREB134. This analysis corroborates the contribution of ROS signaling after BP1 induction that was shown to contribute to the effect in functional studies with the antioxidant tempol27 (Figure 2E). FGF2 was also identified as one of the upstream regulators. The loss of the AngII contractile response in renal afferent arterioles from FGF2−/− mice (Figure 4C) and the inhibition of AngII-mediated vascular contractility by an FGFR kinase inhibitor (Figure 3B; 4B) supports a regulatory role of FGF2.
Figure 5.
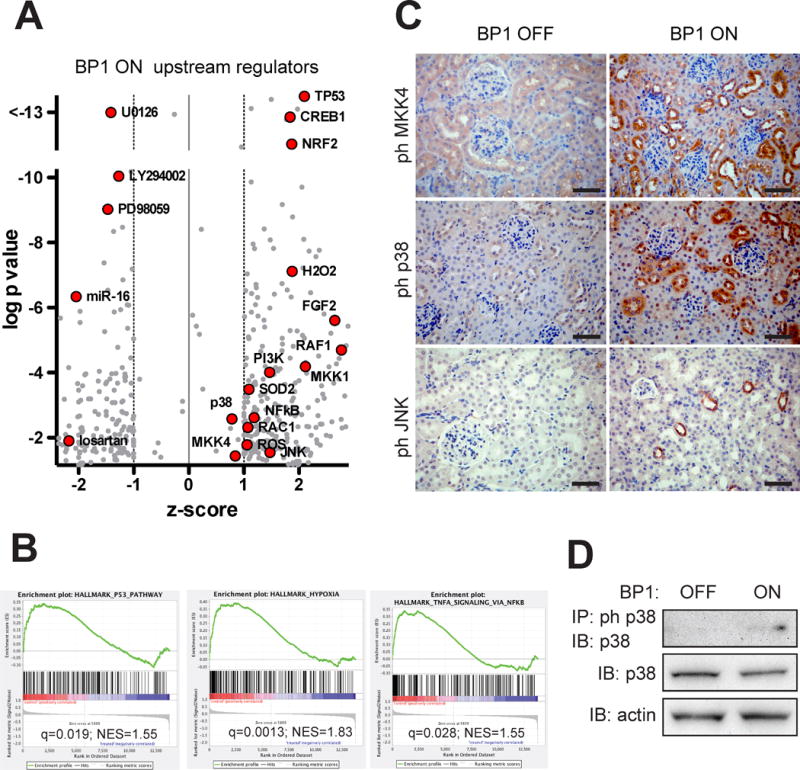
Signal transduction changes in kidneys after the induction of BP1. A, Gene expression data were subjected to an analysis for Upstream Regulators using the Ingenuity platform (see Supplemental Methods). z-scores and p-values (−log) are shown. Relevant data points are labeled (red symbols). A complete list (grey symbols) is provided in Table S1. B, Hallmark pathways from a Gene Set Enrichment Analysis of the gene expression data (see Supplemental Methods). NES, normalized enrichment score; q, false discovery adjusted p-value. A complete list is provided in Table S2. C, Detection of phospho-MKK4 or phospho-p38 or phospho-JNK by immunohistochemistry of kidneys from BP1 OFF and ON mice (total n=3). Size bar: 50 μm. Note: Positive immunoreactivity results in dark brown staining. D, Phospho-p38 Western blot analysis of kidney extracts from BP1 OFF and ON animals after immunoprecipitation for phospho-p38. Total p38 protein is shown for comparison.
Negative z-scores < −1 in the upstream regulator analysis were related to drugs that can inhibit effects of BP1 expression, i.e. AT1R (losartan), MKK (U0126; PD98059), PI3K (LY294002). Also, microRNA-16 (miR-16) was indicated as a significant (p<10−6) upstream regulator with a negative z-score < −2. miR-16 controls endothelial cell response to growth factors in vivo and thus provides a negative feed-back loop for growth factor signaling35.
The gene set enrichment analysis showed significantly impacted ‘hallmark’ pathways that are exemplified in Figure 5B (complete list in Table S2). ‘P53’ was identified as a hallmark pathway which matches with the identification of P53 as an upstream regulator. The ‘hypoxia hallmark’ pathway can be integrated with the NRF2 upstream regulator and ‘TNF-alpha pathway signaling via NFkB’ matches with NFkB as an upstream regulator. Thus, evaluation of gene expression with different platforms provides complementary insights into the regulation of tissue homeostasis after BP1 induction.
Signal transduction proteins
We hypothesized that an analysis of changes in protein phosphorylation and formation of signaling complexes could provide additional insight into altered pathways in BP1 ON versus OFF mice. Thus, kidney proteins were extracted with mild detergent to maintain protein/protein interactions, and protein complexes captured with an immobilized anti-phospho-tyrosine (pY) monoclonal antibody. As established previously, changes in signal complexes can thus be revealed if one of the protein partners in the complexes is tyrosine phosphorylated (pY)36. 2D gel electrophoresis was used to separate the pY containing protein complexes. Imaging analysis of Coomassie-stained gels quantitated distinct spots that were identified by mass spectrometry36.
Four proteins that integrate into the known signaling pathway were covered by at least 8 distinct peptides (Figure S4 and Figure 6). Two of these pY-complexed proteins identified were increased by ~4-fold after BP1 induction and are known downstream effectors of the Rho family of small G-proteins37 that regulate a multitude of cellular functions, PAK2 (=p21 CDC42/RAC1-activated kinase 2)38 and CIT/STK21 (citron or RHO-interacting, serine/threonine kinase 21)39. In earlier studies40 Gutkind et al. have reported that Ras-related small GTP-binding proteins such as RAC1 function as cellular regulators of ROS, an effect that was mimicked by exogenously added growth factors. A third protein, MKK4 (=MAP2K4) was found 3.6-fold upregulated in a pY complex after BP1 expression. MKK4 integrates mitogenic and stress signaling and targets the MAP kinases JNK and p38. A fourth protein, PTPN12 (Protein Tyrosine Phosphatase, Non-Receptor Type 12) was found reduced by 2.8-fold after BP1 induction. In general, PTPs are thought to function as negative feed-back controls of tyrosine kinase activities that can be inactivated by ROS signaling41. PTPN12 has been linked to altered oxidative stress and its loss to enhanced oncogenic signaling42.
Figure 6.
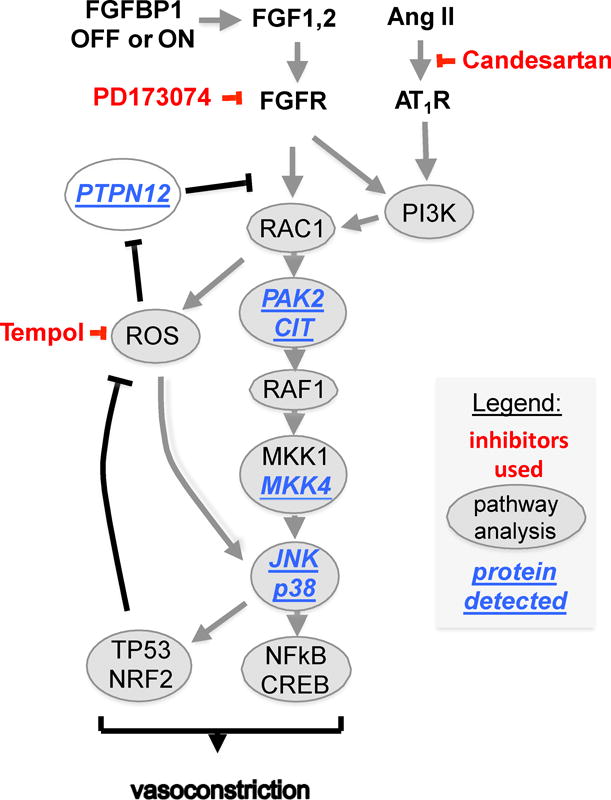
Integration of signaling changes after the induction of BP1. The crosstalk between FGFR and AngII/GPCR signaling based on the Upstream Regulator, pathway and protein analyses. Signaling proteins identified by tissue staining (pMKK4, pJNK, pp38; Figure 5B,C) or by mass spectrometry analysis of phospho-tyrosine protein complexes isolated from tissues (CIT, MKK4, PAK2, PTPN12; Figure S4) are underlined. Inhibitors used in the experiments are shown in red.
Validation of signaling proteins
For an independent validation of the nodes of signaling uncovered in the above studies, we analyzed kidney tissues by immunohistochemical staining. Staining of parallel tissue sections for phospho-proteins in the MAP kinase pathway downstream of the FGF receptor1 showed an increase in phospho-MKK4, phospho-p38 and phospho-JNK after BP1 expression (Figure 5C). For p38, the induction of phosphorylation was confirmed by Western blotting of protein extracts (Figure 5D). No differences in phospho-ERK staining of kidneys were seen (not shown).
FGF and AngII pathway crosstalk in cultured human cells
Cultured kidney cells (HEK293) were used to expand the above findings in an animal model to human cells. Combinations of increasing concentrations of FGF2 and/or AngII were studied for their induction of phosphorylation of MKK4 and the MAP kinases ERK, JNK and p38. An increase in phospho-MKK4, phospho-JNK and phospho-p38 was found in the co-stimulation with FGF2 and AngII (Figure S5). In contrast, co-stimulation with FGF and AngII did not induce phospho-ERK. A parallel analysis in human endothelial and smooth muscle cells (Figure S6) corroborates this analysis. After co-stimulation with FGF2 and AngII phospho-p38 was induced in contrast to phospho-JNK and phospho-ERK.
Discussion
Activation of G-protein coupled receptors (GPCRs) and receptor tyrosine kinases such as the FGFR initiate converging signaling cascades in cells to elicit a phenotypic response. Earlier studies in rat aortic smooth muscle cells showed an increase of AngII-stimulated Ca2+ release after treatment with FGF2 and a blockade of the increase after inhibition of the MAP kinase pathway suggesting a crosstalk that could be relevant for the initiation of hypertension43. Also, FGF2 was found to be essential for mediating AngII-induced cardiomyopathy that utilized the JNK and p38 MAP kinase pathways44. Moreover, endogenous FGF2 has been implicated in pulmonary hypertension45 and cardiac hypertrophy46, conditions associated with increased vascular resistance.
Some mechanistic insight into the crosstalk between pathways that control vascular tone and angiogenic signals emerged from inhibitors of VEGF-driven tumor angiogenesis used to treat cancers. VEGF pathway activity is restricted to endothelial cells and the major side effect of systemic therapy with VEGF pathway inhibitors is hypertension due to the reduction of constitutive eNOS47. FGFs also act on endothelial cells and their effects overlap with VEGF. Indeed, intravascular administration of exogenous FGF1 or FGF2 lowers the blood pressure in experimental animals and can correct hypertension due to preferential targeting of endothelial signaling due to this route of administration48, 49. Because endogenous FGFs also act on vascular smooth muscle cells, this balance of their endothelial activity explains the apparent paradox that FGF2-deficient mice are hypotensive31, 32 (see Figure S2) despite the hypotensive effect of intravenously administered FGF2. It is conceivable that the lower MAP observed after induction of BP1 in tempol treated animals (Figure 2E, grey vs. black bar) is also due to a shift to the vasodilatory FGF signaling under inhibition of ROS by tempol. Interestingly AngII also shows distinct activity on blood pressure that depends on the cell type stimulated. Typically, AngII will cause vasoconstriction and a rise in blood pressure (see Figure 2 and S2). However, AngII reduced blood pressure in animals that only expressed endothelial AngII receptors due to the vasodilatory effects of the endothelial stimulus50.
Here we show that the predominant effect of FGF pathway activation by the induction of BP1 expression is to increase blood pressure by sensitizing resistance vessels to AngII (Figure 3 and 4). This sensitization to AngII vasoconstriction was reversed by an FGFR kinase inhibitor, supporting the notion that increased BP1 expression activates the FGF pathway and thus increases vessel sensitivity for AngII. The increased baseline sensitivity of resistance vessels was reflected in increased blood pressure that is dependent on AngII receptor signaling as evidenced by the inhibitory effects of the receptor antagonist Candesartan (Figure 2D). We do not know, if other antihypertensives may also be effective.
When evaluating the GPCR/FGFR crosstalk further, we found a qualitative difference between AngII and alpha-adrenoceptor sensitization of arteriolar contractility after FGF pathway activation (Figure 3B vs. C and Figure 4C vs. D). We propose that this difference is due to the distinct downstream effectors of these GPCRs. Previous studies showed that chronic AngII infusion increased vascular superoxide which enhanced the pressor response26 and increased arteriole constriction by AngII but not by norepinephrine51. Thus, distinct intracellular effectors can modulate the crosstalk of FGFR and AngII.
Supplementary Material
Perspectives.
The modulation of FGF signaling by BP1 can regulate vascular sensitivity to endogenous AngII and hence control steady state blood pressure. Together with the finding of genomic polymorphisms of the human FGFBP1 gene resulting in increased expression of BP1 in tissues, our studies establish BP1 as a potential driver gene of hypertension.
Novelty and Significance.
What is new: We report on a secreted FGF binding protein that enhances FGF signaling, modulates AngII vascoconstriction and blood pressure.
What is relevant: Genomic polymorphisms of genes in the FGF pathway have been found associated with an increased risk of hypertension.
Summary. We show the crosstalk between FGF and AngII pathway signaling in regulating blood pressure.
Acknowledgments
We thank the Shared Resources for cDNA array and protein mass spectrometry analysis.
Sources of Funding: Supported by NIH P01 HL068686 (CSW, WJW, AW), R01s CA71508 (AW), DK36079, DK49870 (CSW), HL102497 (PER), P30 CA51008 (AW), George E Schreiner Chair of Nephrology (CSW), National Nature Science Foundation of China (EYL; 31471100).
Footnotes
Disclosure of Conflict of Interest
There are no known conflicts of interest to report.
Author contributions
ET, EYL performed experiments, analysed data; AW developed concept, analyzed data and wrote manuscript. PER, ATR, WJW, CSW provided advice. LL, GS, YFC performed experiments. WK analyzed data. All authors reviewed and edited the manuscript.
References
- 1.Goetz R, Mohammadi M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nature Reviews Mol Cell Biol. 2013;14:166–180. doi: 10.1038/nrm3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Padmanabhan S, Newton-Cheh C, Dominiczak AF. Genetic basis of blood pressure and hypertension. Trends Genetics. 2012;28:397–408. doi: 10.1016/j.tig.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Levy D, Ehret GB, Rice K, et al. Genome-wide association study of blood pressure and hypertension. Nature Genetics. 2009;41:677–687. doi: 10.1038/ng.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Newton-Cheh C, Johnson T, Gateva V, et al. Genome-wide association study identifies eight loci associated with blood pressure. Nature Genetics. 2009;41:666–676. doi: 10.1038/ng.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takeuchi F, Isono M, Katsuya T, et al. Blood pressure and hypertension are associated with 7 loci in the Japanese population. Circulation. 2010;121:2302–2309. doi: 10.1161/CIRCULATIONAHA.109.904664. [DOI] [PubMed] [Google Scholar]
- 6.Tomaszewski M, Charchar FJ, Lynch MD, Padmanabhan S, Wang WYS, Miller WH, Grzeszczak W, Maric C, Zukowska-Szczechowska E, Dominiczak AF. Fibroblast growth factor 1 gene and hypertension: from the quantitative trait locus to positional analysis. Circulation. 2007;116:1915–1924. doi: 10.1161/CIRCULATIONAHA.107.710293. [DOI] [PubMed] [Google Scholar]
- 7.Tomaszewski M, Eales J, Denniff M, et al. Renal Mechanisms of Association between Fibroblast Growth Factor 1 and Blood Pressure. J Am Soc Nephrol. 2015;26:3151–3160. doi: 10.1681/ASN.2014121211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tomaszewski M, Charchar FJ, Nelson CP, Barnes T, Denniff M, Kaiser M, Debiec R, Christofidou P, Rafelt S, Van Der Harst P, Wang WYS, Maric C, Zukowska-Szczechowska E, Samani NJ. Pathway Analysis Shows Association between FGFBP1 and Hypertension. J Am Soc Nephrol. 2011;22:947–955. doi: 10.1681/ASN.2010080829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braun MC, Herring SM, Gokul N, Monita M, Bell R, Hicks MJ, Wenderfer SE, Doris PA. Hypertensive renal disease. J Hypertension. 2013;31:2050–2059. doi: 10.1097/HJH.0b013e328362f9a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coffman TM. Under pressure: the search for the essential mechanisms of hypertension. Nat Med. 2011;17:1402–1409. doi: 10.1038/nm.2541. [DOI] [PubMed] [Google Scholar]
- 11.Tassi E, Al-Attar A, Aigner A, Swift MR, McDonnell K, Karavanov A, Wellstein A. Enhancement of fibroblast growth factor (FGF) activity by an FGF-binding protein. J Biol Chem. 2001;276:40247–40253. doi: 10.1074/jbc.M104933200. [DOI] [PubMed] [Google Scholar]
- 12.Werner S. A novel enhancer of the wound healing process the fibroblast growth factor-binding protein. Am J Pathol. 2011;179:2144–2147. doi: 10.1016/j.ajpath.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu DQ, Kan MK, Sato GH, Okamoto T, Sato JD. Characterization and molecular cloning of a putative binding protein for heparin-binding growth factors. J Biol Chem. 1991;266:16778–16785. [PubMed] [Google Scholar]
- 14.Zhang W, Chen Y, Swift MR, Tassi E, Stylianou DC, Gibby KA, Riegel AT, Wellstein A. Effect of FGF-binding protein 3 on vascular permeability. J Biol Chem. 2008;283:28329–28337. doi: 10.1074/jbc.M802144200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie B, Tassi E, Swift MR, McDonnell K, Bowden ET, Wang S, Ueda Y, Tomita Y, Riegel AT, Wellstein A. Identification of the fibroblast growth factor (FGF)-interacting domain in a secreted FGF-binding protein by phage display. J Biol Chem. 2006;281:1137–1144. doi: 10.1074/jbc.M510754200. [DOI] [PubMed] [Google Scholar]
- 16.Beer H-D, Bittner M, Niklaus G, Munding C, Max N, Goppelt A, Werner S. The fibroblast growth factor binding protein is a novel interaction partner of FGF-7, FGF-10 and FGF-22 and regulates FGF activity: implications for epithelial repair. Oncogene. 2005;24:5269–5277. doi: 10.1038/sj.onc.1208560. [DOI] [PubMed] [Google Scholar]
- 17.Czubayko F, Smith RV, Chung HC, Wellstein A. Tumor growth and angiogenesis induced by a secreted binding protein for fibroblast growth factors. J Biol Chem. 1994;269:28243–28248. [PubMed] [Google Scholar]
- 18.Mongiat M, Otto J, Oldershaw R, Ferrer F, Sato JD, Iozzo RV. Fibroblast growth factor-binding protein is a novel partner for perlecan protein core. J Biol Chem. 2001;276:10263–10271. doi: 10.1074/jbc.M011493200. [DOI] [PubMed] [Google Scholar]
- 19.Czubayko F, Liaudet-Coopman ED, Aigner A, Tuveson AT, Berchem GJ, Wellstein A. A secreted FGF-binding protein can serve as the angiogenic switch in human cancer. Nat Med. 1997;3:1137–1140. doi: 10.1038/nm1097-1137. [DOI] [PubMed] [Google Scholar]
- 20.Gibby KA, McDonnell K, Schmidt MO, Wellstein A. A distinct role for secreted fibroblast growth factor-binding proteins in development. Proc Natl Acad Sci U S A. 2009;106:8585–8590. doi: 10.1073/pnas.0810952106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ray PE, Al-Attar A, Liu X, Das J, Tassi E, Wellstein A. Expression of a Secreted Fibroblast Growth Factor Binding Protein-1 (FGFBP1) in Angioproliferative Kaposi Sarcoma. J AIDS Clin Res. 2014;5:1–8. doi: 10.4172/2155-6113.1000309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu H-y, Bai W-d, Liu J-q, Zheng Z, Guan H, Zhou Q, Su L-l, Xie S-t, Wang Y-c, Li J, Li N, Zhang Y-j, Wang H-t, Hu D-h. Up-regulation of FGFBP1 signaling contributes to miR-146a-induced angiogenesis in human umbilical vein endothelial cells. Scientific Reports. 2016:1–11. doi: 10.1038/srep25272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tassi E, Mcdonnell K, Gibby KA, Tilan JU, Kim SE, Kodack DP, Schmidt MO, Sharif GM, Wilcox CS, Welch WJ, Gallicano GI, Johnson MD, Riegel AT, Wellstein A. Impact of fibroblast growth factor-binding protein-1 expression on angiogenesis and wound healing. Am J Pathol. 2011;179:2220–2232. doi: 10.1016/j.ajpath.2011.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmidt MO, Garman KA, Lee YG, et al. The Role of Fibroblast Growth Factor Binding Protein 1 in Skin Carcinogenesis and Inflammation. J Invest Dermatol. 2017 doi: 10.1016/j.jid.2017.1007.1847. epub Aug 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McDonnell K, Bowden ET, Cabal-Manzano R, Hoxter B, Riegel AT, Wellstein A. Vascular leakage in chick embryos after expression of a secreted binding protein for fibroblast growth factors. Lab Invest. 2005;85:747–755. doi: 10.1038/labinvest.3700269. [DOI] [PubMed] [Google Scholar]
- 26.Modlinger P, Chabrashvili T, Gill PS, Mendonca M, Harrison DG, Griendling KK, Li M, Raggio J, Wellstein A, Chen Y, Welch WJ, Wilcox CS. RNA silencing in vivo reveals role of p22phox in rat angiotensin slow pressor response. Hypertension. 2006;47:238–244. doi: 10.1161/01.HYP.0000200023.02195.73. [DOI] [PubMed] [Google Scholar]
- 27.Wilcox CS, Pearlman A. Chemistry and antihypertensive effects of tempol and other nitroxides. Pharmacol Rev. 2008;60:418–469. doi: 10.1124/pr.108.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang D, Chabrashvili T, Wilcox CS. Enhanced contractility of renal afferent arterioles from angiotensin-infused rabbits: roles of oxidative stress, thromboxane prostanoid receptors, and endothelium. Circulation Research. 2004;94:1436–1442. doi: 10.1161/01.RES.0000129578.76799.75. [DOI] [PubMed] [Google Scholar]
- 29.Lai EY, Wellstein A, Welch WJ, Wilcox CS. Superoxide Modulates Myogenic Contractions of Mouse Afferent Arterioles. Hypertension. 2011;50:650–656. doi: 10.1161/HYPERTENSIONAHA.111.170472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ray PE, Tassi E, Liu X-H, Wellstein A. Role of fibroblast growth factor-binding protein in the pathogenesis of HIV-associated hemolytic uremic syndrome. Am J Physiol. 2006;290:R105–113. doi: 10.1152/ajpregu.00492.2005. [DOI] [PubMed] [Google Scholar]
- 31.Dono R, Texido G, Dussel R, Ehmke H, Zeller R. Impaired cerebral cortex development and blood pressure regulation in FGF-2-deficient mice. EMBO J. 1998;17:4213–4225. doi: 10.1093/emboj/17.15.4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou M, Sutliff RL, Paul RJ, Lorenz JN, Hoying JB, Haudenschild CC, Yin M, Coffin JD, Kong L, Kranias EG, Luo W, Boivin GP, Duffy JJ, Pawlowski SA, Doetschman T. Fibroblast growth factor 2 control of vascular tone. Nat Med. 1998;4:201–207. doi: 10.1038/nm0298-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gambino V, De Michele G, Venezia O, et al. Oxidative stress activates a specific p53 transcriptional response that regulates cellular senescence and aging. Aging cell. 2013;12:435–445. doi: 10.1111/acel.12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malhotra D, Portales-Casamar E, Singh A, Srivastava S, Arenillas D, Happel C, Shyr C, Wakabayashi N, Kensler TW, Wasserman WW, Biswal S. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Research. 2010;38:5718–5734. doi: 10.1093/nar/gkq212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chamorro-Jorganes A, Araldi E, Penalva LOF, Sandhu D, Fernández-Hernando C, Suárez Y. MicroRNA-16 and microRNA-424 regulate cell-autonomous angiogenic functions in endothelial cells via targeting vascular endothelial growth factor receptor-2 and fibroblast growth factor receptor-1. ATVB. 2011;31:2595–2606. doi: 10.1161/ATVBAHA.111.236521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu Z-Z, Kagan BL, Ariazi EA, Rosenthal DS, Zhang L, Li JV, Huang H, Wu C, Jordan VC, Riegel AT, Wellstein A. Proteomic analysis of pathways involved in estrogen-induced growth and apoptosis of breast cancer cells. PLoS ONE. 2011;6:e20410. doi: 10.1371/journal.pone.0020410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vega FM, Ridley AJ. SnapShot: Rho family GTPases. Cell. 2007;129:1430. doi: 10.1016/j.cell.2007.06.021. [DOI] [PubMed] [Google Scholar]
- 38.Yi C, Maksimoska J, Marmorstein R, Kissil JL. Development of small-molecule inhibitors of the group I p21-activated kinases, emerging therapeutic targets in cancer. Biochem Pharmacol. 2010;80:683–689. doi: 10.1016/j.bcp.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Di Cunto F, Calautti E, Hsiao J, Ong L, Topley G, Turco E, Dotto GP. Citron rho-interacting kinase, a novel tissue-specific ser/thr kinase encompassing the Rho-Rac-binding protein Citron. J Biol Chem. 1998;273:29706–29711. doi: 10.1074/jbc.273.45.29706. [DOI] [PubMed] [Google Scholar]
- 40.Sundaresan M, Yu ZX, Ferrans VJ, Sulciner DJ, Gutkind JS, Irani K, Goldschmidt-Clermont PJ, Finkel T. Regulation of reactive-oxygen-species generation in fibroblasts by Rac1. Biochem J. 1996;318(Pt 2):379–382. doi: 10.1042/bj3180379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 42.Harris IS, Blaser H, Moreno J, et al. PTPN12 promotes resistance to oxidative stress and supports tumorigenesis by regulating FOXO signaling. Oncogene. 2014;33:1047–1054. doi: 10.1038/onc.2013.24. [DOI] [PubMed] [Google Scholar]
- 43.Samain E, Bouillier H, Miserey S, Perret C, Renaud JF, Safar M, Dagher G. Extracellular signal-regulated kinase pathway is involved in basic fibroblast growth factor effect on angiotensin II-induced Ca(2+) transient in vascular smooth muscle cell from Wistar-Kyoto and spontaneously hypertensive rats. Hypertension. 2000;35:61–67. doi: 10.1161/01.hyp.35.1.61. [DOI] [PubMed] [Google Scholar]
- 44.Pellieux C, Foletti A, Peduto G, Aubert JF, Nussberger J, Beermann F, Brunner HR, Pedrazzini T. Dilated cardiomyopathy and impaired cardiac hypertrophic response to angiotensin II in mice lacking FGF-2. J Clin Invest. 2001;108:1843–1851. doi: 10.1172/JCI13627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Izikki M, Guignabert C, Fadel E, Humbert M, Tu L, Zadigue P, Dartevelle P, Simonneau G, Adnot S, Maitre B, Raffestin B, Eddahibi S. Endothelial-derived FGF2 contributes to the progression of pulmonary hypertension in humans and rodents. J Clin Invest. 2009;119:512–523. doi: 10.1172/JCI35070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schultz JE, Witt SA, Nieman ML, Reiser PJ, Engle SJ, Zhou M, Pawlowski SA, Lorenz JN, Kimball TR, Doetschman T. Fibroblast growth factor-2 mediates pressure-induced hypertrophic response. J Clin Invest. 1999;104:709–719. doi: 10.1172/JCI7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Humphreys BD, Atkins MB. Rapid development of hypertension by sorafenib: toxicity or target? Clin Cancer Res. 2009;15:5947–5949. doi: 10.1158/1078-0432.CCR-09-1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cuevas P, Carceller F, Ortega S, Zazo M, Nieto I, Giménez-Gallego G. Hypotensive activity of fibroblast growth factor. Science. 1991;254:1208–1210. doi: 10.1126/science.1957172. [DOI] [PubMed] [Google Scholar]
- 49.Cuevas P, García-Calvo M, Carceller F, Reimers D, Zazo M, Cuevas B, Muñoz-Willery I, Martínez-Coso V, Lamas S, Giménez-Gallego G. Correction of hypertension by normalization of endothelial levels of fibroblast growth factor and nitric oxide synthase in spontaneously hypertensive rats. Proc Natl Acad Sci USA. 1996;93:11996–12001. doi: 10.1073/pnas.93.21.11996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramchandran R, Takezako T, Saad Y, Stull L, Fink B, Yamada H, Dikalov S, Harrison DG, Moravec C, Karnik SS. Angiotensinergic stimulation of vascular endothelium in mice causes hypotension, bradycardia, and attenuated angiotensin response. Proc Natl Acad Sci USA. 2006;103:19087–19092. doi: 10.1073/pnas.0602715103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang D, Jose P, Wilcox CS. Beta(1) receptors protect the renal afferent arteriole of angiotensin-infused rabbits from norepinephrine-induced oxidative stress. J Am Soc Neprol. 2006;17:3347–3354. doi: 10.1681/ASN.2006030212. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.