

ABSTRACT
Chicken Na+/H+ exchanger type I (chNHE1), a multispan transmembrane protein, is a cellular receptor of the subgroup J avian leukosis virus (ALV-J). To identify the functional determinants of chNHE1 responsible for the ALV-J receptor activity, a series of chimeric receptors was created by exchanging the extracellular loops (ECL) of human NHE1 (huNHE1) and chNHE1 and by ECL replacement with a hemagglutinin (HA) tag. These chimeric receptors then were used in binding and entry assays to map the minimal ALV-J gp85-binding domain of chNHE1. We show that ECL1 of chNHE1 (chECL1) is the critical functional ECL that interacts directly with ALV-J gp85; ECL3 is also involved in ALV-J gp85 binding. Amino acid residues 28 to 39 of the N-terminal membrane-proximal region of chECL1 constitute the minimal domain required for chNHE1 binding of ALV-J gp85. These residues are sufficient to mediate viral entry into ALV-J nonpermissive cells. Point mutation analysis revealed that A30, V33, W38, and E39 of chECL1 are the key residues mediating the binding between chNHE1 and ALV-J gp85. Further, the replacement of residues 28 to 39 of huNHE1 with the corresponding chNHE1 residues converted the nonfunctional ALV-J receptor huNHE1 to a functional one. Importantly, soluble chECL1 and huECL1 harboring chNHE1 residues 28 to 39 both could effectively block ALV-J infection. Collectively, our findings indicate that residues 28 to 39 of chNHE1 constitute a domain that is critical for receptor function and mediate ALV-J entry.
IMPORTANCE chNHE1 is a cellular receptor of ALV-J, a retrovirus that causes infections in chickens and serious economic losses in the poultry industry. Until now, the domains determining the chNHE1 receptor function remained unknown. We demonstrate that chECL1 is critical for receptor function, with residues 28 to 39 constituting the minimal functional domain responsible for chNHE1 binding of ALV-J gp85 and efficiently mediating ALV-J cell entry. These residues are located in the membrane-proximal region of the N terminus of chECL1, suggesting that the binding site of ALV-J gp85 on chNHE1 is probably located on the apex of the molecule; the receptor-binding mode might be different from that of retroviruses. We also found that soluble chECL1, as well as huECL1 harboring chNHE1 residues 28 to 39, effectively blocked ALV-J infection. These findings contribute to a better understanding of the ALV-J infection mechanism and also provide new insights into the control strategies for ALV-J infection.
KEYWORDS: subgroup J avian leukosis virus, binding, chicken NHE1, receptors, virus entry
INTRODUCTION
Avian leukosis virus (ALV) is a retrovirus that is responsible for various tumor diseases in chicken. Depending on the host range, viral envelope interference, and cross-neutralization patterns, ALVs are divided into 10 subgroups, ALV-A to -J (1). Among these, ALV-A, ALV-B, and ALV-J commonly cause infections in poultry. Clinically, ALV-A and ALV-B mainly induce lymphocytoma, while ALV-J mainly induces myeloma (1–3). ALV-J is derived by recombination of an exogenous ALV with ancient endogenous avian viral sequences in the chicken genome and has a broader host range and higher transmission ability than the A to E subgroups (4, 5). ALV-J has been responsible for large economic losses in the last 2 decades, making it a research focus in the field of poultry viral disease (6–8). The epidemic strains of ALV-J are exhibiting not only an expanding host range but also increased pathogenicity (9, 10).
Retrovirus infection begins with the interactions between the viral envelope (Env) proteins and a specific cell surface protein that acts as a receptor. Env proteins are comprised of surface (SU) and transmembrane (TM) subunits. SU directly binds to the receptor and triggers conformational changes in the TM subunit, leading to the fusion of viral and cellular membranes (11, 12). The interaction of SU with its cell receptor is a very important and complex process involving many factors that affect receptor specificity and host range. ALVs, as simple retroviruses similar to murine leukemia viruses (13) and the equine infectious anemia virus (14), only require a single functional receptor to initiate an infection. Different ALV subgroups have evolved to utilize different cellular receptors to infect the host cells. The receptor of ALV-A is Tva, which is most related to the family of the low-density lipoprotein receptors (15, 16). The tvb gene encodes a tumor necrosis factor receptor-related protein that efficiently mediates ALV-B, -D, and -E infection (17, 18); a butyrophilin-related protein encoded by tvc serves as an ALV-C receptor (19, 20).
Chai et al. (21) identified the chicken Na+/H+ exchanger type I (chNHE1) as the cellular receptor for ALV-J. NHE1 is a ubiquitously expressed housekeeping protein that is involved in basic cellular functions, e.g., cell adhesion, manipulation of the extracellular pH, and Na+/H+ exchange (22, 23). Abnormal syntheses of NHE1 leads to various types of cell damage (24, 25). chNHE1 and human NHE1 (huNHE1) proteins of the NHE1 superfamily share high similarity (21). Based on the topology model of huNHE1, the putative structure of chNHE1 comprises 12 TM domains, six extracellular loops (ECLs), and a long intracellular C-terminal tail (21, 26, 27).
The receptors of most retroviruses, e.g., human immunodeficiency virus (HIV), feline immunodeficiency virus (FIV), equine infectious anemia virus (EIAV), and ALV-A to -E, are all type-I transmembrane proteins; most of them are immunoglobulin-like proteins (28–34). These immunoglobulin-like receptors are rod-shaped and protrude from the cell membrane. The tips of their N-terminal domains reach into the receptor-binding domain of a viral Env protein. In contrast, the ALV-J receptor chNHE1 is a multispan transmembrane protein. Its topology is completely different from those of type I transmembrane proteins (21). The virus-receptor interaction may follow a mechanism that is different from that of the other viral proteins and their receptors. Although W38 of chNHE1 was identified as a critical residue in ALV-J-resistant avian species (35), the functional determinants of chNHE1 responsible for its ALV-J receptor activity have not been identified. In the current study, we employed two complementary mapping strategies based on protein gain and loss of function to identify the critical domain responsible for the receptor function of chNHE1. Our analysis revealed that chECL1 is the critical functional ECL responsible for binding to gp85, the surface unit of ALV-J Env protein. Further, we demonstrated that residues 28 to 39 in the N terminus of chNHE1 form the minimal functional domain for chNHE1 gp85 binding, sufficient for mediating ALV-J entry.
RESULTS
chNHE1 ECL1 and ECL3 are involved in binding between the receptor and ALV-J gp85.
To evaluate the gp85-binding ability of the receptors, soluble forms of human IgG-Fc-tagged gp85, chNHE1, and huNHE1 were constructed and successfully produced in 293T cells, an ALV-J nonpermissive cell line (data not shown); the proteins then were used in a receptor binding assay that we developed. As shown in Fig. 1A, the gp85 binding to 293T cells expressing chNHE1 could be detected by fluorescence-activated cell sorting (FACS), with ca. 90% of cells binding gp85. In comparison, the binding percentages were less than 5% in the control reactions with human IgG-Fc protein and 293T cells expressing chNHE1, fluorescein isothiocyanate (FITC)-labeled anti-human IgG-Fc antibody and 293T cells expressing chNHE1, and gp85 protein and 293T cells expressing huNHE1 (Fig. 1A). These results indicated that gp85 specifically interacts with chNHE1 produced in 293T cells.
FIG 1.

ECL1 and ECL3 participate in the binding of chNHE1 and gp85 protein. (A) Validation of the receptor binding assay. The specific interaction of chNHE1 and gp85-Fc was evaluated using chNHE1-expressing 293T cells by fluorescence-activated cell sorting analysis (FACS). (a) chNHE1-expressing 293T cells incubated with human-Fc protein; (b) chNHE1-expressing 293T cells incubated with FITC-labeled anti-human Fc antibodies; (c) 293T cells expressing nonfunctional huNHE1 receptor incubated with gp85-Fc protein; (d) chNHE1-expressing 293T cells incubated with gp85-Fc protein. (B) Schematic representation of the strategy of constructing chimeric chNHE1 proteins with ECL1 to ECL6 replaced with the corresponding ECL domains of huNHE1 or hemagglutinin (HA) tag. Black, chNHE1 ECLs; white, huNHE1 ELCs; HA, HA tag. (C) The gp85-binding abilities of different chimeric chNHE1s expressed on the surface of transfected 293T cells, evaluated in a receptor binding assay. The binding capacity of wild-type chNHE1 was set to 100%, and the values for chimeric chNHE1 receptors were calculated as its proportion. (D) Entry of RCAS(J)-luciferase virus into 293T cells expressing different chimeric chNHE1s. Virus entry levels were determined using a luciferase reporter assay as described in Materials and Methods. The entry level of RCAS(J)-luciferase virus into 293T cells expressing wild-type chNHE1 was set to 1, and the values for chimeric chNHE1 receptors were calculated as its proportion. (E) Schematic representation of the strategy of constructing chimeric huNHE1s with ECL1 to ECL6 replaced with the corresponding ECLs of chNHE1. The color designations are as described for panel B. (F) The relative gp85-binding capacity of different chimeric huNHE1s expressed on the surface of transfected 293T cells. The binding ability was evaluated in a receptor binding assay. The binding capacity of wild-type chNHE1 was set as 100%, and the values for chimeric chNHE1 receptors were calculated as its proportion. (G) Entry of RCAS(J)-luciferase virus into 293T cells expressing different chimeric huNHE1s, as described for panel D. (B to D) Loss-of-function receptor analysis; (E to G) gain-of-function receptor analysis. (C, D, F, and G) Three independent experiments were performed, and data are shown as means ± standard deviations for triplicates from a representative experiment. **, P < 0.01.
In the NHE1 superfamily, huNHE1 shares the most similarity with chNHE1 but does not act as the receptor of ALV-J (21). To map the major chNHE1 gp85-binding domains, 12 chimeric receptors with ECL domains on the chNHE1 backbone swapped for the corresponding huNHE1 domains, and vice versa, were constructed, and their gp85-binding abilities were evaluated (Fig. 1B to G). The relative gp85-binding capacity of the chimeric chNHE1 harboring the huELC1 domain (ch-hu-ECL1) was less than 15%, while the binding capacities of chimeric chNHE1 harboring other huECLs were between 84% and 92% (Fig. 1C). At the same time, the relative gp85-binding capacity of huNHE1 harboring the chECL1 domain (hu-ch-ECL1) increased to ca. 80% of the wild-type chNHE1 value, while chimeric huNHE1s harboring other chECLs were unable to bind the gp85 protein (Fig. 1F). To evaluate the binding domains that were highly identical between chNHE1 and huNHE1, a strategy of replacement with a hemagglutinin (HA) tag was adopted. chECL2 was not replaced because it only contains five amino acids; chECL5 was also not evaluated because of its high similarity to huECL5 (96%) and the intricate structure of its intramembrane segment (26, 27). Ultimately, chimeric receptors with chECL3, chECL4, and chECL6 replaced with the HA tag were constructed and their receptor function was evaluated (Fig. 1B). We found that the relative gp85-binding capacity of chNHE1 harboring the HA tag instead of ECL3 (ECL3-HA) decreased to less than 33% (Fig. 1C); the relative gp85-binding capacities of chNHE1 receptor with chECL4 and chECL6 replaced with HA tags were 76% and 86%, respectively, showing no marked decrease (data not shown).
Viral entry assays then were performed to confirm the results of the receptor binding assays. An ALV-J enveloped replication-competent avian leucosis sarcoma (RCAS) virus harboring a luciferase reporter gene was constructed (36), with the env and gfp (encoding a green fluorescent protein) genes of RCAS(A)-GFP replaced with the ALV-J env gene and the firefly luciferase gene, respectively. The RCAS(J)-luciferase virus was produced in DF-1 cells and reached a titer of 105.21 50% tissue culture infective doses (TCID50)/ml. The subgroup specificity of the RCAS(J)-luciferase virus was confirmed by Western blotting with 4A3 anti-ALV-J gp85 monoclonal antibodies (MAb) (37 and data not shown).
The viral entry assay with 293T cells expressing various chimeric chNHE1s revealed that the ability to mediate viral entry was significantly decreased only in the case of the chimeric receptor ch-hu-ECL1 (ca. 87% decrease compared with wild-type chNHE1) (Fig. 1D). Conversely, the chimeric receptor hu-ch-ECL1 was able to efficiently mediate virus entry (ca. 95% increase compared with huNHE1, similar to the chNHE1-mediated virus entry level) (Fig. 1G). The ability of other chimeric receptors of chNHE1 harboring huECLs or huNHE1s harboring chECLs to mediate virus entry was almost completely abolished (Fig. 1D and G). In addition, the ability of ECL3-HA to mediate viral entry was decreased by ca. 66% (Fig. 1D). In contrast, chNHE1s with chECL4 and chECL6 replaced with HA tags showed the same capacity to mediate viral entry as wild-type chNHE1 (data not shown). Taken together, the results of the binding assay and viral entry assay demonstrated that chECL1 and chECL3 are involved in the binding of ALV-J envelope protein.
chECL1, as the major functional domain, directly interacts with gp85 protein and mediates virus entry.
To test whether chECL1 and/or chECL3 could directly interact with gp85, soluble chECL1 and chECL3 domains fused with the Fc region of a human IgG and the HA tag were constructed. Coimmunoprecipitation (co-IP) assays were performed by transiently coexpressing chECL1 or chECL3 and gp85 in 293T cells. HuECL1 and huECL3 were used as a negative control. The lysates of cells expressing ECL1 or ECL3 and gp85 were immunoprecipitated with anti-HA MAb, and gp85 was detected with the 4A3 anti-gp85 MAb (Fig. 2A). The results revealed that the coexpressed chECL1 and gp85 formed a specific complex; chECL3, huECL1, and huECL3 were not able to form a complex with gp85. A pulldown assay was performed next to further confirm the co-IP results. As shown in Fig. 2B, chECL1 pulled down gp85, while chECL3, huECL1, and huECL3 did not. This further demonstrated that chECL1 could directly interact with gp85.
FIG 2.

chECL1 directly interacts with gp85 protein and mediates viral entry. (A) Validation of the interaction of chECL1 and chECL3 with gp85 by a coimmunoprecipitation (co-IP) assay. 293T cells were transfected with gp85- and ECL1- or ECL3-expressing plasmids. HuECL1 and huECL3 were used as a negative control. Cell lysates were prepared 48 h posttransfection, and proteins were immunoprecipitated using anti-HA-agarose MAb; the proteins were immunoblotted with 4A3 anti-gp85 MAb or anti-HA antibodies. (B) Validation of the interaction of chECL1 and chECL3 with gp85 by a pulldown assay. Soluble chECL1 and chECL3 proteins were incubated with anti-HA-agarose MAb. huECL1, huECL3, and human IgG-Fc (huIgG-Fc) were used as a negative control. The bound complexes were mixed with raw lysates of gp85 expressed by 293T cells transfected with pCAF-gp85 plasmid. The proteins were separated by SDS-PAGE and probed by immunoblotting with 4A3 anti-gp85 MAb or anti-HA antibodies. (C) Confocal microscopy analysis of the cell surface expression of ECL1 constructs in 293T cells. 293T cells were transfected with chECL1 and huECL1 expression plasmids for 24 h and fixed and stained with anti-FLAG M2 MAb, followed by incubation with Alexa Fluor 488 donkey anti-mouse IgG(H+L) (green). Nuclei were stained with DAPI. chNHE1 was used as a positive control. (D) The gp85-binding capacity of chECL1 and huECL1 expressed on the surface of transfected 293T cells, evaluated in a receptor binding assay. (E) Entry of RCAS(J)-luciferase virus into 293T cells expressing chECL1 or huECL1, assayed as described in the legend to Fig. 1. (F) Modeling of the chNHE1 transmembrane (TM) domain. For clarity, only TM domains of ECL1 (TM1 and TM2) and ECL3 (TM5 and TM6) are shown. Cell membranes are indicated by dotted lines. The distance between C-terminal portions of TM1 and TM5 is shown to indicate the size of the molecule. The structural modeling of the receptors was generated using SWISS-MODEL and was based on homology molecules found in the PDB database. (D and E) Three independent experiments were performed, and data are shown as means ± standard deviations for triplicates from a representative experiment.
To evaluate whether chECL1 could mediate viral entry, TM versions of chECL1 and huECL1 were constructed and produced. As showed in Fig. 2C, the TM protein versions were well expressed on the cell membrane. The gp85-binding assay indicated that gp85 specifically bound to the 293T cells expressing TM chELC1 (relative binding capacity of over 83%); it did not bind to 293T cells expressing TM huELC1 (Fig. 2D). The viral entry assay demonstrated that TM chECL1 effectively mediated RCAS(J)-luciferase virus entry into 293T cells (Fig. 2E). These observations further demonstrated that chECL1 directly interacts with gp85 protein and mediates viral entry.
To further evaluate the corresponding roles of ECL1 and ECL3 of chNHE1 in the receptor-gp85 interaction, chNHE1 TM domains were modeled based on the structure of the Escherichia coli Na+/H+ antiporter NhaA (38). As shown in Fig. 2F, the N terminus of chECL1 is ca. 35 Å away from chECL3. If we assume that the size of ALV-J gp85 is similar to that of HIV-1 gp120 (with a diameter of ca. 40 Å), and because the binding domain is very close to the cell membrane, it is not likely that a gp85 molecule would bind the two domains at same time. Taken together, these results indicated that chECL1 directly interacts with gp85 protein and mediates virus entry.
N-terminal chECL1 residues 28 to 39 form the minimal binding domain directly involved in receptor-gp85 binding.
chECL1 contains 72 amino acids and is the largest ECL of chNHE1 (21). To map the chECL1 segments responsible for receptor binding in greater detail, a series of sequential domain-swapping strategies were employed. First, chECL1 was split into three segments: ECL1-1, amino acids 28 to 63; ECL1-2, amino acids 64 to 99; and ECL1-3, amino acids 46 to 81. They were replaced with the corresponding amino acids of huECL1 (Fig. 3A). The gp85-binding assay indicated that the relative binding capacity of the ECL1-1 chimeric receptor was significantly decreased (to under 15%), which was the same as that of the chimeric receptor ch-hu-ECL1; the relative gp85-binding capacities of ECL1-2 and ECL1-3 were 104% and 94%, respectively (Fig. 3B). The ECL1-1 fragment then was split into four equal-length segments (ECL1-1A, ECL1-1B, ECL1-1C, and ECL1-1D; Fig. 3C), replaced as in the first round described above. The gp85-binding assay indicated that the relative binding capacity of both ECL1-1A and ECL1-1B decreased by 50%, with the binding capacities of ECL1-1C and ECL1-1D at 96% and 101%, respectively (Fig. 3D). Finally, in the third round, ECL1-1A and ECL1-1B sections were further split into six equal-length segments (ECL1-1A-1/2/3 and ECL1-1B-1/2/3; Fig. 3E) and replaced as described above. Evaluation of the gp85-binding ability revealed that chimeric receptors ECL1-1A-1/2/3 and ECL1-1B-1 (collectively covering amino acids 28 to 39 in the N terminus of chECL1) exhibited relatively low binding capacities (5 to 52%; Fig. 3F). These findings indicated that the amino acids 28 to 39 in the N terminus of chECL1 comprise the minimal binding domain directly involved in receptor-gp85 binding.
FIG 3.

Residues 28 to 39 in the N-terminal part of chECL1 constitute the minimal gp85-binding domain. (A) Schematic representation of the strategy for constructing chimeric chNHE1s with three equal-length chECL1 fragments replaced with the corresponding fragments of huECL1. (B) The gp85-binding capacity of 293T cells expressing ECL1-1, ECL1-2, and ECL1-3 chimeric receptors evaluated in a receptor binding assay. (C) Schematic representation of the strategy of constructing chimeric chNHE1s with four equal-length fragments of ECL1-1 replaced with the corresponding fragments of huECL1. (D) The relative gp85-binding capacity of 293T cells expressing ECL1-1A, ECL1-1B, ECL1-1C, and ECL1-1D chimeric receptors, evaluated in a receptor binding assay. (E) Schematic representation of the strategy of constructing chimeric chNHE1s with six equal-length segments of ECL1-1A and ECL1-1B replaced with the corresponding fragments of huECL1. (F) The relative gp85-binding capacity of 293T cells expressing ECL1-1A-1/2/3 and ECL1-1B-1/2/3 chimeric receptors, evaluated in a receptor binding assay. In panels A, C, and E, the black bars represent chECL1 and the white bars represent huECL1. (B, D, and F) Three independent experiments were performed, and data are shown as means ± standard deviations for triplicates from a representative experiment. **, P < 0.01.
chECL1 residues A30, V33, W38, and E39 are the key residues for chNHE1 binding of gp85.
Among the residues 28 to 39 in the N terminus of chECL1, 11 are different between chECL1 and huECL1. To pinpoint the key amino acids responsible for gp85 binding, chimeric chNHE1s were constructed where these 11 amino acids were replaced (one at a time) with the corresponding residues from huNHE1 (Fig. 4A). The relative gp85-binding capacity of A30Q, V33P, W38I, and E39R variants decreased to 12 to 56%; the remaining seven variants exhibited a relative binding capacity above 80%, with values similar to that of wild-type chNHE1 (Fig. 4B). The V33P substitution had the greatest impact on receptor binding, with the relative gp85-binding capacity of the respective variant decreasing to 12%. Viral entry assay further confirmed that substitution of the four suspect residues (A30Q, V33P, W38I, and E39R) significantly decreased viral entry by 41% to 87% (Fig. 4C). These observations clearly indicated that A30, V33, W38, and E39 of chECL1 are the key residues involved in receptor binding of gp85.
FIG 4.
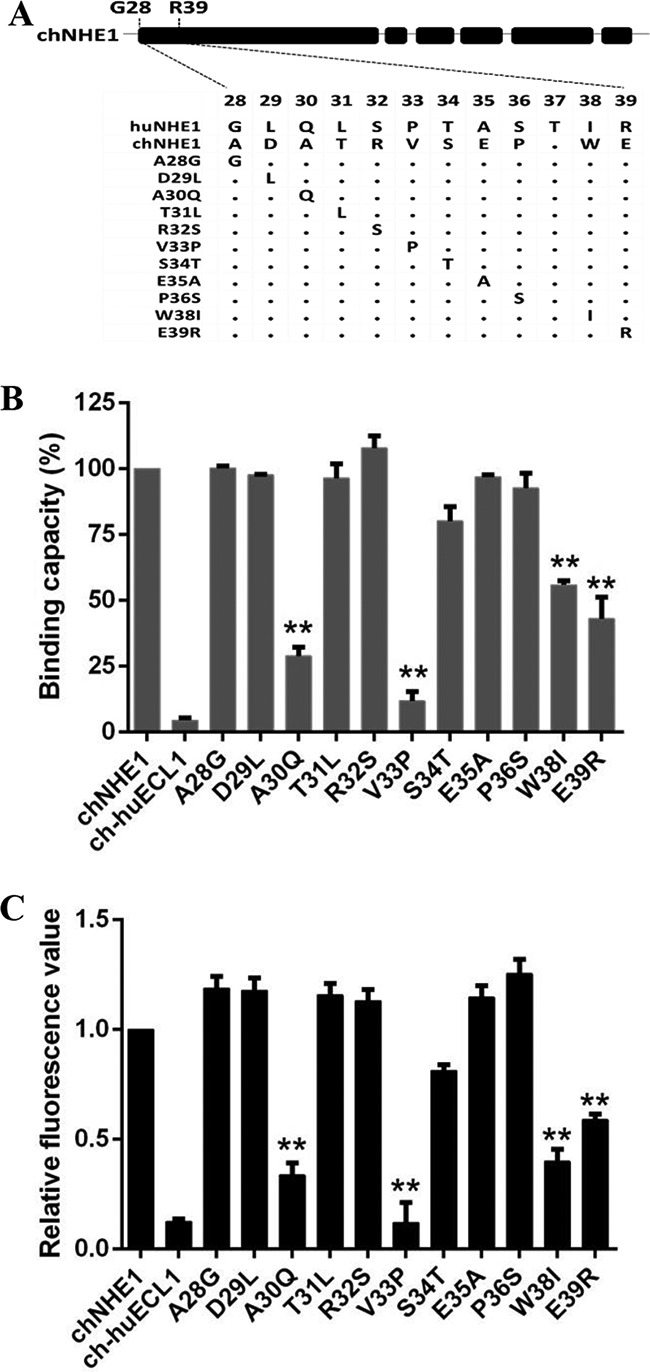
Residues A30, V33, W38, and E39 of ECL1 are key residues for the gp85-binding activity of chNHE1. Eleven amino acids from among residues 28 to 39 of chNHE1 were changed by site-directed mutagenesis to the corresponding amino acids of huNHE1, and the gp85-binding and viral entry-mediating activities of the chimeric receptors were evaluated. (A) Schematic representation of the strategy of constructing chimeric chNHE1s with 11 different single-residue substitutions. (B) The relative gp85-binding capacity of 293T cells expressing the wild type or different single-residue variants of chNHE1. The binding capacity of wild-type chNHE1 was set at 100%, and the values for the other receptors were calculated as proportions of the wild-type value. (C) Entry of RCAS(J)-luciferase virus into 293T cells expressing the wild type or different single-residue variants of chNHE1, assayed as described in the legend to Fig. 1. (B and C) Three independent experiments were performed, and data are shown as means ± standard deviations for triplicates from a representative experiment. **, P < 0.01.
Residues 28 to 39 of chNHE1 convert huNHE1 into a functional ALV-J receptor and mediate viral entry.
The preceding experiments indicated that A30, V33, W38, and E39 and amino acids 28 to 39 of chNHE1 were critical for gp85 binding. To further explore the roles of these amino acids in receptor function, a series of chimeric huNHE1s, four with the corresponding single amino acids replaced and one with residues 28 to 39 exchanged for the corresponding amino acids of chECL1, were constructed (Fig. 5A). The gp85-binding assay indicated that the four single-residue variants (Q30A, P33V, I38W, and R39E) failed to bind gp85 (binding capacity decreased by 8%). In contrast, replacement of residues 28 to 39 in huNHE1 enabled the naturally nonfunctional ALV-J receptor to efficiently bind gp85, with the relative binding capacity being over 90% (Fig. 5B). Consistent with these results, the replacement of residues 28 to 39 converted huNHE1 to a functional ALV-J receptor, enabling it to mediate RCAS(J)-luciferase virus entry into 293T cells, while the four single-residue variants failed to mediate virus entry (Fig. 5C). These observations confirmed that residues 28 to 39 in the N-terminal region of chECL1 comprise the minimal functional domain for efficient viral entry.
FIG 5.
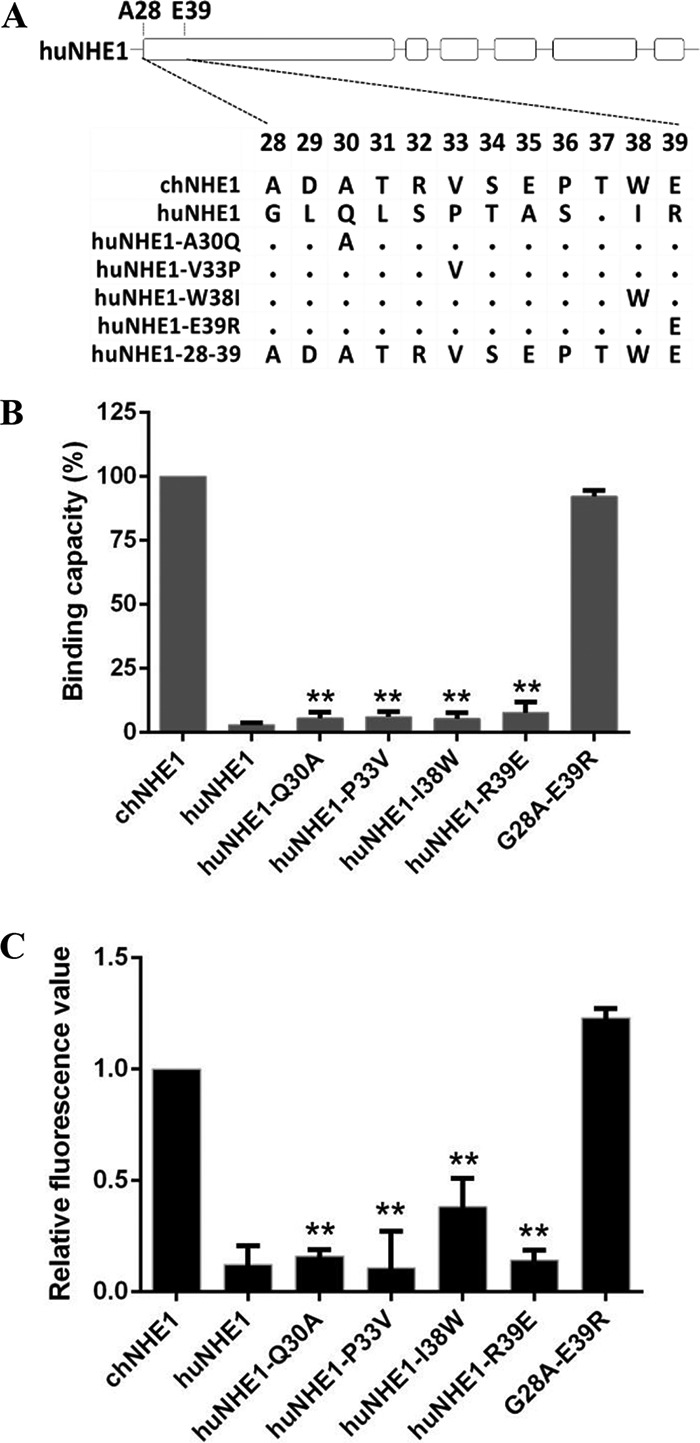
Residues 28 to 39 of chNHE1 are sufficient to convert huNHE1 into a functional ALV-J receptor and mediate virus entry. (A) Schematic representation of the strategy of constructing chimeric huNHE1s with single-residue or 12-amino-acid substitutions within ECL1. The huNHE1 residues were replaced with the corresponding chNHE1 residues. (B) The relative gp85-binding capacity of 293T cells expressing different chimeric huNHE1s. The binding capacity of wild-type chNHE1 was set at 100%, and the values for the chimeric huNHE1 receptors were calculated as its proportion. (C) Entry of RCAS(J)-luciferase virus into 293T cells expressing different chimeric huNHE1s, assayed as described in the legend to Fig. 1. (B and C) Three independent experiments were performed, and data are shown as means ± standard deviations for triplicates from a representative experiment. **, P < 0.01.
Soluble huECL1 with residues 28 to 39 replaced by the corresponding residues of chNHE1 efficiently blocks viral infection.
To verify the role of chECL1 residues 28 to 39 in the soluble form of the receptor and to evaluate their potential application for viral infection blocking, soluble chimeric huECL1 proteins, four with different single residues and one with a 12-amino-acid segment replaced with the corresponding chECL1 residues, were constructed and successfully expressed. First, the interaction of soluble chimeric huECL1s with gp85 was evaluated in a co-IP assay. Similar to chELC1, the coexpressed chimeric huECL1 with 12 amino acids replaced formed a complex with gp85, in contrast to chimeric huECL1s with single-residue substitutions and huECL1 alone (Fig. 6A). This indicated that residues 28 to 39 of chNHE1 enable huECL1 binding to ALV-J gp85.
FIG 6.

Soluble huECL1 harboring the corresponding residues 28 to 39 of chNHE1 efficiently blocks ALV-J infection. (A) Coimmunoprecipitation (co-IP) validation of the interaction of gp85 and soluble chimeric huECL1s described in the legend to Fig. 5A. 293T cells were transfected with gp85- and the appropriate soluble chimeric huECL1-expressing plasmids. Cell lysates were prepared 48 h posttransfection, and the proteins were immunoprecipitated by anti-HA-agarose MAb; the proteins were immunoblotted with 4A3 anti-gp85 MAb or anti-HA antibodies. (B) Soluble chECL1 blocks RCAS(J)-luciferase virus entry into DF-1 cells. RCAS(J)-luciferase virus was incubated with different concentrations of soluble chECL1 or bovine serum albumin (BSA) for 1 h at 4°C. This was followed by infection of DF-1 cells. To detect viral entry, the cells infected with RCAS(J)-luciferase virus were lysed 24 h postinfection and luciferase activity was detected in a luciferase reporter assay. BSA was used as a negative control. (C and D) chECL1 and huECL1 harboring the corresponding residues 28 to 39 of chNHE1 block viral entry (C) and reduce the degree of viral replication (D). DF-1 cells were infected with RCAS(J)-luciferase virus or HPRS-103 preincubated with soluble chimeric huECL1s or chECL1. To detect viral entry, the cell culture infected with RCAS(J)-luciferase virus was lysed 24 h postinfection and the luciferase activity was determined by a luciferase reporter assay. To detect viral replication, the cell culture infected with HPRS-103 was harvested 24, 48, and 72 h postinfection, and RT activity was detected using an RT assay. Uninfected DF-1 cells were used as a negative control. RCAS(J)-luciferase virus or HPRS-103 that had not been preincubated with soluble chimeric receptors was used as a positive control. (B, C, and D) Three independent experiments were performed, and data are shown as means ± standard deviations for triplicates from a representative experiment. **, P < 0.01.
To accurately test the ability of soluble chECL1 and chimeric huECL1s to block viral infection, the optimum concentrations of soluble chECL1 for blocking RCAS(J)-luciferase virus were first determined. RCAS(J)-luciferase virus was preincubated with various concentrations of soluble chECL1 before infecting DF-1 cells. The infected cells were harvested 24 h postinfection and the viral levels determined. The infective ability of RCAS(J)-luciferase virus incubated with soluble chECL1 was reduced; this correlated with increasing amounts of soluble chECL1 in a dose-dependent manner (Fig. 6B). RCAS(J)-luciferase virus infection was significantly blocked with 200 μg/ml of soluble chECL1, with no such effect observed in the case of huECL1. Hence, that concentration was used in subsequent assays.
RCAS(J)-luciferase virus and HPRS-103 were incubated with 200 μg/ml of different soluble chimeric huECL1s and then were used to infect DF-1 cells. huECL1 with residues 28 to 39 replaced by the corresponding chNHE1 residues abolished the cell entry of RCAS(J)-luciferase virus (Fig. 6C) and decreased the degree of ALV-J replication (Fig. 6D). In contrast, chimeric huECL1s with single-residue substitutions (A30Q, V33P, I38W, and E39R) and huECL1 alone had no effect on RCAS(J)-luciferase virus entry and ALV-J replication (Fig. 6C and D). These findings demonstrated that soluble chECL1 and chimeric huECL1 with residues 28 to 39 replaced with the corresponding chNHE1 sequence efficiently blocked viral infection.
DISCUSSION
In the current study, using the ECL of chNHE1 and huNHE1 domains and HA tag replacement strategies, we demonstrated that chECL1 is critical for gp85 binding, with chECL3 also being involved in gp85 binding. More importantly, the data presented here indicate that residues 28 to 39 in the N terminus of chNHE1 comprise the minimal critical functional domain of chNHE1. These residues were responsible for converting the nonfunctional ALV-J receptor huNHE1 into a functional receptor and efficiently mediated ALV-J entry. Furthermore, soluble forms of chECL1 and huECL1 harboring residues 28 to 39 of chNHE1 both significantly blocked viral entry. These systematic analyses enabled us to identify, for the first time, the critical domains and amino acids of chNHE1 involved in binding of gp85. The efficient blocking of ALV-J infection by soluble ECL1 informs a new strategy for antiviral drug design. In addition, utilization of soluble SU-Fc expression in receptor binding ability studies also may constitute a convenient approach for related studies of other retroviruses.
The putative structure of chNHE1 contains six ECLs and shares the most similarities with huNHE1 (21). Our findings indicated that chECL1 and chECL3 are involved in gp85 binding. This is consistent with receptor binding and entry of the amphotropic murine leukemia virus, which requires both loop 2 and loop 4 of its cellular receptor Pit2 for infection (39). As mentioned, to evaluate the corresponding roles of ECL1 and ECL3 of chNHE1 in the receptor-gp85 interaction, chNHE1 TM domains were modeled based on the structure of E. coli Na+/H+ antiporter NhaA (38). According to the model (Fig. 2F), it is not likely that a gp85 molecule would bind chECL1 and chECL3 at the same time. In addition, the co-IP and pulldown experiments indicated that chECL1 could interact directly with gp85, whereas chECL3 could not. Therefore, we speculate that chECL3 plays a role in maintaining the correct chNHE1-binding conformation rather than in directly contacting the gp85 molecule.
The N terminus of chECL1 shows low sequence identity with huNHE1; four key residues in the region (A30, V33, W38, and E39) were identified as ones that significantly affect receptor function. However, the gain-of-function assays demonstrated that only replacement of the entire 12 amino acids (residues 28 to 39 of chNHE1) enabled huNHE1 binding to gp85 and blocked virus entry; no such effects were apparent after the individual replacement of the four residues. A number of studies demonstrated that the binding interfaces between proteins rely on multiple intermolecular contacts involving 10 to 30 side chains from each protein (40, 41). The free energy of protein-protein binding could be attained through only a few strong interactions or by an accumulation of many weaker contacts at the entire protein-protein interface. For example, only a few functionally important residues of the human growth hormone (hGH) and the extracellular domain of its first bound receptor (hGHbp) confer their tight binding affinity (42); the hGH-hGHbp interaction belongs to the former few strong interactions. In the current study, we identified at least four residues that constitute the binding surface between the receptor and gp85. It appears that no individual substitution provided enough energy for receptor binding. To better understand the binding, the role of these amino acids during the interaction between gp85 and chNHE1 should be investigated further.
A previous study reported that W38 of chHNE1 is a determining site for different ALV-J susceptibilities of the resistant avian species (35). In the current study, the relative binding capacity of the chNHE1 W38I variant was significantly reduced (to 56%), while the complementary gain-of-function assay demonstrated that huNHE1 harboring I38W substitution did not bind gp85 protein and failed to mediate virus entry. Furthermore, the soluble huECL1 I38W variant could not directly interact with gp85 or block viral entry. These results indicate that although W38 plays an important role in defining the ALV-J host range in avian species (35), it is only one of the key residues required for chNHE1 binding.
As verified by both loss- and gain-of-function assays, residues 28 to 39 in the N terminus of chNHE1 constitute the minimal domain required for full receptor function. The chNHE1 and huNHE1 sequences differ the most from each other in this region, which might explain their different gp85-binding abilities. Furthermore, a soluble huECL1 harboring chNHE1 residues 28 to 39 produced in a eukaryotic cell line was able to block viral entry. This further confirmed the crucial role of the residue 28 to 39 domain of chNHE1 in receptor function. Previous studies demonstrated that HIV-1 replication is effectively inhibited in human T-cell lines and in primary peripheral blood lymphocytes overexpressing soluble forms of CD4, an HIV-1 receptor (43). Similarly, soluble forms of Tva protein also significantly inhibited ALV-A infection in vitro and in vivo (32). In the present study, soluble forms of chECL1 and huECL1 variants harboring residues 28 to 39 of chNHE1 also exhibited a remarkable antiviral effect against ALV-J-enveloped RCAS virus as well as ALV-J. These observations suggested that small soluble receptor functional domains could serve as antiviral drug candidates. Because ALV-J has caused serious economic losses in China in recent years (2, 10), the identification of the minimal functional domain provides a basis for the development of antiviral therapeutics and provides us a new ALV-J control strategy.
ALVs utilize a two-step fusion mechanism, namely, that receptor binding primes the second trigger of low pH for cell entry (11, 44–46). The initial contact between SU and its cell receptor is a crucial step for ALV infection. chNHE1 is a multispan TM protein that is structurally significantly different from the receptors of ALV-A to -E viruses and other retroviruses (e.g., HIV, EIAV, and FIV, etc.). To explore the possible mechanism of interaction between ALV-J gp85 and chNHE1, we compared modeled structures of the identified retrovirus receptors (Fig. 7) (14, 33, 47–51). Tva, Tvb, Tvc, and other retrovirus receptors have rod-like configuration, protruding high from the cell membrane, which allows them to reach the binding sites at various geometric locations on the ligand surface. For example, the CD4-binding site of HIV gp120 is a depression formed at the interface of the outer domain, the inner domain, and the bridging sheet of gp120 (47). The identified binding sites at membrane-distal regions of Tva, Tvb, Tvc, CD4 (52), tumor necrosis factor receptors, and immunoglobulin superfamily molecules (14, 20, 29, 33, 52, 53) can reach the binding sites on the virus surface without any steric hindrance. A remarkable observation made in the current study was that the identified 12 amino acids of the gp85-binding domain mapped to the membrane-proximal region of the N-terminal part of chECL1 (Fig. 7B). Such geometric location apparently would not allow the receptor to reach a binding pocket at the side of the envelope molecule. This location suggests that the chNHE1-binding site on ALV-J gp85 is located on the apex of the molecule. Recent studies showed that lentiviral vectors carrying Nipah virus glycoproteins enter the cell much more efficiently by binding to the membrane-proximal receptors than to membrane-distal receptors (54). ALV-J has a wider host range and greater horizontal transmission capacity than ALV-A to -E (4, 55, 56), which might be associated with its membrane-proximal receptor binding. This interesting aspect requires further research.
FIG 7.

Comparison of chNHE1, ECL1, and the receptors of other ALV subgroups and retroviruses. Putative topology of ECL1 and the connected TM domains of chNHE1 are shown. The ALV-J gp85 contour is shown in blue in the size of an HIV-1 gp120 molecule. The identified binding sites on receptors are colored red. The receptors shown are the following: ALV-A, Tva; ALV-J, chNHE1; HIV-1, CD4; ALV-B, -D, and -E, Tvb; ALV-C, Tvc; EIAV, ELR1; and FIV, CD134. CD4 is shown in complex with HIV-1 gp120 (PDB entry 1GC1). The structural modeling of the receptors was generated using SWISS-MODEL and was based on homology molecules found in the PDB database.
In conclusion, we identified chECL1 as the critical functional ECL, and residues 28 to 39 of chNHE1 as the minimal functional domain, required for chNHE1 binding to gp85 and efficient mediation of ALV-J entry. In addition, we demonstrated that soluble chECL1 and huECL1 variants harboring residues 28 to 39 of chNHE1 both effectively blocked ALV-J infection. These observations not only contribute to a better understanding of the molecular basis of the interaction between the ALV-J and its receptor but also provide potential targets for development of antiviral drugs and transgenic chicken resistant to ALV-J.
MATERIALS AND METHODS
Cells and viruses.
293T cells and DF-1 cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Thermo Scientific, Rockford, IL) containing 10% fetal bovine serum (FBS) (Sigma-Aldrich, St. Louis, MO), 100 U/ml penicillin, and 100 μg/ml streptomycin (Summus, Beijing, China). Chicken embryo fibroblast cells (CEFs) were prepared from 9-day-old embryos of specific-pathogen-free chicken. All cells were maintained in a humidified incubator containing 5% CO2 at 37°C. The ALV prototype strain HPRS-103 was kindly provided by Venugopal Nair (the Pirbright Institute, Pirbright, United Kingdom) and was propagated in DF-1 cells (9).
Expression of soluble gp85 protein.
The gp85 gene (GenBank accession number HQ634809.1) was amplified by PCR from proviral DNA of ALV-J HPRS-103. The gene encoding soluble gp85 (sgp85) was obtained by fusing the gp85 sequence with a fragment encoding a signal peptide sequence (for N-terminal protein fusion) and a fragment encoding the constant region fragment (Fc) of human IgG (sgp85-hIgG) or a FLAG tag (sgp85-Flag; for C-terminal protein fusion) by overlapping PCR. The sgp85-hIgG gene was cloned into the CAG vector (Addgene, Cambridge, MA) to generate CAG-sgp85-Fc plasmid. The sgp85-Flag construct was cloned into the pCAGGS vector to generate pCAF-gp85 plasmid. To evaluate the gp85-binding ability of chimeric receptors, sgp85-hIgG protein was prepared. Briefly, 293T cells were transfected with the CAG-sgp85-Fc plasmid using an X-treme GENE-HP-DNA transfection reagent (Roche, Indianapolis, IN); 48 h posttransfection, the cell culture medium was collected and the recombinant protein purified on a protein A column (GenScript, Jiangsu, China) by following the manufacturer's instructions. The concentration of sgp85-hIgG was determined using the bicinchoninic acid (BCA) protein assay kit (Thermo Scientific, Rockford, IL) by following the manufacturer's instructions.
RCAS(J)-luciferase reporter virus.
To detect viral entry into host cells, an ALV-J-enveloped RCAS virus expressing a luciferase reporter gene was constructed and rescued. RCAS(A)-GFP vector (36, 57) was digested by KpnI and StuI (Thermo Scientific); the 10,150-bp RCAS backbone was retained. A 2,120-bp fragment containing part of the pol gene, the entire env gene, and the redundant TM region (rTM) was amplified by PCR from the proviral DNA of HPRS-103. It was then inserted into the RCAS backbone to generate the RCAS(J)-GFP plasmid. The constructed plasmid then was digested by EagI and SalI (Thermo Scientific) to remove the gfp gene; the 11,339-bp RCAS(J) backbone was retained. The luciferase-coding gene (GenBank accession number U47295.2) was amplified by PCR from pGL3 Luciferase plasmid (Promega, Madison, WI) using primers with incorporated EagI and SalI enzyme restrictions sites; the digested amplification product was subsequently inserted into the RCAS(J) backbone to generate RCAS(J)-luciferase plasmid. The plasmid was used to transfect DF-1 monolayer cells at 80% density; 7 days posttransfection, cellular supernatant containing the RCAS(J)-luciferase reporter virus was harvested by centrifugation (2,000 × g for 10 min) and stored at −80°C. The titer of the rescued reporter virus was determined on the basis of the TCID50 value using the Reed and Muench method (9).
Construction of various chimeric receptors.
The chNHE1-encoding gene (GenBank accession number DQ256198) was amplified from the cDNA of CEF cells by reverse transcription-PCR using specific primers (21). The huNHE1-encoding gene (GenBank accession number S68616) was obtained from Sino Biological Inc. (Beijing, China). Both genes were cloned into the eukaryotic expression vector pCAGGS and were fused with a 3′ Flag tag sequence (pCAGGS-Flag); the resulting constructs were designated pCAF-chNHE1 and pCAF-huNHE1, accordingly.
The strategy for constructing chimeric NHE1s with various ECL domains exchanged is shown in Fig. 1B. Six different domain-exchange chimeric receptors based on the chNHE1 backbone were constructed by replacing domains chECL1 to chECL6 with the corresponding ECL1 to ECL6 domains of huNHE1 by overlapping PCR; the constructs were named ch-huECL1 to ch-huECL6. Similarly, six different domain exchange chimeric receptors based on the huNHE1 backbone were constructed by replacing domains huECL1 to huECL6 with the corresponding ECL1 to ECL6 domains of chNHE1; the constructs were named hu-chECL1 to hu-chECL6 (Fig. 1E). Furthermore, overlapping PCR was used to replace chECL3, chECL4, and chECL6 of chNHE1 with the 9-amino-acid HA; the constructs were named chECL3-HA, chECL4-HA, and chECL6-HA, respectively.
To map the chECL1 segments and key amino acid residues responsible for its receptor activity, a series of ECL1 segment substitutions and 12-amino-acid or point variants of chNHE1 or huNHE1 were generated by replacing the corresponding segment or amino acid coding sequence by overlapping PCR. All of the mutated NHE1 sequences then were cloned into the pCAGGS-Flag vector.
For TM expression of chECL1 and huECL1, the TM1-ECL1-TM2-TM3 sequence-encoding fragments were amplified by PCR from the pCAF-chNHE1 or pCAF-huNHE1 gene and inserted into the pCAGGS-Flag vector. For soluble expression of chECL1, chECL3, huECL1, huECL3, point variants, and chimeric ECL1, the respective gene sequences were amplified by PCR or overlapping PCR from the pCAF-chNHE1 or pCAF-huNHE1 gene. The recombinant soluble ECL polypeptides subsequently were obtained by fusing the coding sequences with a signal peptide-encoding sequence (for an N-terminal fusion with ECL) and HA tag and a sequence encoding an Fc region of a human IgG (for a C-terminal fusion). Finally, the constructs were inserted into the CAG vector (Addgene). The primer sequences of all oligonucleotides used in the study are available upon request.
Receptor binding assay.
The receptor binding assay was performed to evaluate receptor gp85-binding ability. Plasmids encoding wild-type chNHE1 or the different chimeric receptors were used to transfect 293T cells; 24 h posttransfection, 293T cells were detached with a solution of phosphate-buffered saline (PBS) and 5 mM EDTA, pelleted (1,000 × g) for 10 min, and washed three times with ice-cold PBS containing 5% (wt/vol) FBS. Approximately 2 × 106 transfected 293T cells first were incubated with 500 μl of sgp85 (100 ng/μl) on ice for 1 h; this was followed by staining with a 1:200 dilution of anti-human IgG (Fc specific)-FITC antibody produced in goat (F9512; Sigma, St. Louis, MO). After fixing in 4% (vol/vol) paraformaldehyde (Solarbio, Beijing, China) for 15 min at room temperature, the cells were incubated in a 1:200 dilution of anti-FLAG M2 MAb produced in mouse (F1804; Sigma) and then in a 1:200 dilution of anti-mouse IgG (whole-molecule)-tetramethyl rhodamine isothiocyanate (TRITC) antibodies produced in rabbit (T2402; Sigma). The stained cells were analyzed by FACS using a FACS Aria II flow cytometer (BD Biosciences, Franklin Lakes, NJ).
Viral entry assay.
The ability of chNHE1 and different chimeric receptors to mediate viral entry was evaluated in a luciferase reporter assay with the RCAS(J)-luciferase reporter virus. 293T cells were transfected with plasmids encoding wild-type chNHE1 or different chimeric receptors as described above; 24 h posttransfection, the transfected 293T cells were infected with RCAS(J)-luciferase virus at a multiplicity of infection of 0.1. After 2 h at 37°C and 5% CO2 (i.e., conditions allowing efficient viral entry), the cells were washed three times with room temperature PBS and maintained in DMEM containing 5% (wt/vol) FBS at 37°C and 5% CO2. The virus entry levels were determined 24 h postinfection by detecting luciferase activity of the cell lysates using a Steady-Glo luciferase assay system (Promega) by following the manufacturer's instructions. 293T cells infected with the RCAS(J)-luciferase virus but not transfected with the chNHE1-encoding plasmids were used as a negative control. The entry level of RCAS(J)-luciferase into 293T cells expressing wild-type chNHE1 was set as 1, and the values for chimeric chNHE1s receptors were calculated as its proportion.
Co-IP experiments.
293T cells cultured in 6-well plates were transfected with the respective plasmids (2 μg of each) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. At 48 h posttransfection, the transfected 293T cells were washed three times with ice-cold PBS and then lysed in 200 μl of IP lysis buffer (Beyotime, Beijing, China) supplemented with 1 mM phenylmethanesulfonyl fluoride (Beyotime). After centrifugation, 40 μl of the supernatant was removed as the input fraction; the remaining supernatant (160 μl) was incubated with anti-HA-agarose MAb (A2095; Sigma) overnight at 4°C. After five washes with ice-cold PBS containing a complete protease inhibitor cocktail (1 tablet/50 ml) (Roche Diagnostics GmbH, Mannheim, Germany), the immunoprecipitated proteins were separated by SDS-PAGE and detected by Western blotting.
Pulldown assay.
For the in vitro binding assay, human IgG-Fc fusions of huNHE1 or chNHE1 ECL1 or ECL3 were expressed in 293T cells. The cell culture medium was collected and the proteins purified on a protein A column (GenScript). Eighty microliters of anti-HA-agarose MAb (A2905; Sigma) was incubated with 20 μg of chECL1, huECL1, chECL3, huECL3, or human IgG-Fc for 2 h at 4°C with gentle agitation. After washing five times with ice-cold PBS, the agarose was incubated with a lysate of 293T cells transfected with pCAF-gp85 and expressing gp85 for 2 h at 4°C with gentle agitation. After five washes with ice-cold PBS, the bound proteins were separated by SDS-PAGE and detected by Western blotting.
Western blotting.
Proteins were separated on 12.5% SDS-PAGE gels and transferred onto a nitrocellulose membrane (Hybond-C Super; GE Healthcare, Piscataway, NJ). After blocking in 5% (wt/vol) skim milk at 4°C overnight, the membrane was incubated with 4A3 anti-gp85 MAb (37) or anti-HA MAb (H9658; Sigma) for 1 h at room temperature. After washing three times with PBS, the membrane was incubated with IRDye 680RD donkey anti-mouse IgG(H+L) antibodies (LI-COR Biosciences, Lincoln, NE) for 1 h at room temperature. Finally, the membrane blots were scanned using an Odyssey infrared imaging system (LI-COR Biosciences).
Confocal microscopy.
To visualize the expression of NHE1 and ECL1 proteins on the cell surface, 293T cells were transfected with plasmids encoding chNHE1, huECL1, and chECL1; 24 h posttransfection, cells were washed with PBS, fixed in 4% (vol/vol) paraformaldehyde for 30 min at room temperature, and blocked with 5% (wt/vol) bovine serum albumin in PBS for 1 h at room temperature. The cells were incubated with anti-FLAG M2 MAb produced in mouse (F1804; Sigma) for 2 h at room temperature. After washing three times with PBS, the cells were incubated with Alexa Fluor 488 donkey anti-mouse IgG (H+L) (A21202; Life Technologies, Grand Island, NY) for 1 h at room temperature. The cells were then washed three times with PBS and permeabilized with 0.1% (vol/vol) Triton X-100 for 10 min. After staining with 4′,6-diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific, Waltham, MA) for 15 min, the cells were examined using a Leica SP2 confocal system (Leica Microsystems, Wetzlar, Germany).
Blocking assay of viral entry.
The blocking assay was performed to evaluate the blocking effect of ECL1 and its variants on ALV-J entry. Plasmids encoding chNHE1 and huNHE1 ECL1 and different chimeric chNHE1s were used to transfect 293T cells; 48 h posttransfection, the cell culture medium containing soluble ECL1s was collected and the proteins purified on protein A columns (GenScript) by following the manufacturer's instructions.
The RCAS(J)-luciferase virus (5,000 TCID50) or HPRS-103 (5,000 TCID50) was incubated with various concentrations of soluble ECL1s for 1 h at 4°C. The viral suspension then was added to DF-1 cells in 12-well plates, and the plates were incubated at 4°C for 2 h. Unbound viruses then were removed by washing three times with PBS. The cells were maintained in DMEM containing 5% (wt/vol) FBS at 37°C and 5% CO2 for a further 24 h. The infected cells then were harvested. Viral levels in the RCAS(J)-luciferase virus infection group were determined by detecting luciferase activity in the cell lysates using the Steady-Glo luciferase assay system (Promega) by following the manufacturer's instructions. Viral levels in the HPRS-103 infection group were determined by a reverse transcriptase (RT) assay as described below.
RT assay.
RT activity was used to evaluate the viral proliferation (58). Cells infected with HPRS-103 were harvested and lysed by freezing and thawing three times. The lysates were centrifuged (5,000 × g for 10 min); RT activity was detected in the supernatants using an RT assay kit (Roche) by following the manufacturer's instructions.
Structure modeling.
Structures of the receptors were modeled using SWISS-MODEL based on the homologous molecules found in the Protein Data Bank (PDB; www.wwpdb.org). PDB entries for receptor homology modeling were the following: 4ZCD (chNHE1), 1GC1 (CD4-gp120), 4I9X (Tvb), 1K7B (TVa), 4F80 (TVc), 2HEY (CD134), and 1JMA (ELR1). Figures were created using the PyMOL program (www.pymol.org).
Statistical analysis.
Statistical significance of the differences between the obtained values was established using the Student t test. An unadjusted P value below 0.05 was considered to indicate statistical significance.
ACKNOWLEDGMENTS
We thank Stephen H. Hughes (National Cancer Institute, Frederick, MD) for the gift of the RCAS(A)-GFP vector.
This work was supported by The Heilongjiang Science Fund for Distinguished Young Scholars (JC2015007), by the State Key Laboratory of Veterinary Biotechnology Foundation (SKLVBF201504), by the National Natural Science Foundation of China (31372437), and by the Intramural Research Program of the Vaccine Research Center, NIAID, NIH.
X.G., Y.Z., M.Y., and C.R. performed the experiments and interpreted the results. Y.G., B.Y., Y.L., Y.W., X.Q., C.L., H.C., Y.Z., L.G., K.L., and Q.P. were involved in the interpretation of the results and critically read the manuscript. B.Z., X.W., and Y.G. were responsible for study design, results analysis, manuscript preparation, and editing.
REFERENCES
- 1.Payne LN, Brown SR, Bumstead N, Howes K, Frazier JA, Thouless ME. 1991. A novel subgroup of exogenous avian leukosis virus in chickens. J Gen Virol 72:801–807. doi: 10.1099/0022-1317-72-4-801. [DOI] [PubMed] [Google Scholar]
- 2.Gao Y, Yun B, Qin L, Pan W, Qu Y, Liu Z, Wang Y, Qi X, Gao H, Wang X. 2012. Molecular epidemiology of avian leukosis virus subgroup J in layer flocks in China. J Clin Microbiol 50:953–960. doi: 10.1128/JCM.06179-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pan W, Gao Y, Sun F, Qin L, Liu Z, Yun B, Wang Y, Qi L, Gao H, Wang X. 2011. Novel sequences of subgroup J avian leukosis viruses associated with hemangioma in Chinese layer hens. Virology J 8:1–9. doi: 10.1186/1743-422X-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bai J, Payne LN, Skinner MA. 1995. HPRS-103 (exogenous avian leukosis virus, subgroup J) has an env gene related to those of endogenous elements EAV-0 and E51 and an E element found previously only in sarcoma viruses. J Virol 69:779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benson SJ, Ruis BL, Fadly AM, Conklin KF. 1998. The unique envelope gene of the subgroup J avian leukosis virus derives from ev/J proviruses, a novel family of avian endogenous viruses. J Virol 72:10157–10164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng Z, Liu J, Cui Z, Zhang L. 2010. Tumors associated with avian leukosis virus subgroup J in layer hens during 2007 to 2009 in China. J Vet Med Sci 72:1027–1033. doi: 10.1292/jvms.09-0564. [DOI] [PubMed] [Google Scholar]
- 7.Lai H, Zhang H, Ning Z, Chen R, Zhang W, Qing A, Xin C, Yu K, Cao W, Liao M. 2011. Isolation and characterization of emerging subgroup J avian leukosis virus associated with hemangioma in egg-type chickens. Vet Microbiol 151:275–283. doi: 10.1016/j.vetmic.2011.03.037. [DOI] [PubMed] [Google Scholar]
- 8.Wu X, Qian K, Qin A, Shen H, Wang P, Jin W, Eltahir YM. 2010. Recombinant avian leukosis viruses of subgroup J isolated from field infected commercial layer chickens with hemangioma and myeloid leukosis possess an insertion in the E element. Vet Res Commun 34:619–632. doi: 10.1007/s11259-010-9436-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Q, Gao Y, Wang Y, Qin L, Qi X, Qu Y, Gao H, Wang X. 2012. A 205-nucleotide deletion in the 3′ untranslated region of avian leukosis virus subgroup J, currently emergent in China, contributes to its pathogenicity. J Virol 86:12849–12860. doi: 10.1128/JVI.01113-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao YL, Qin LT, Pan W, Wand YQ, Le Qi X, Gao HL, Wang XM. 2010. Avian leukosis virus subgroup J in layer chickens, China. Emerg Infect Dis 16:1637–1638. doi: 10.3201/eid1610.100780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mothes W, Boerger AL, Narayan S, Cunningham JM, Young JA. 2000. Retroviral entry mediated by receptor priming and low pH triggering of an envelope glycoprotein. Cell 103:679–689. doi: 10.1016/S0092-8674(00)00170-7. [DOI] [PubMed] [Google Scholar]
- 12.Barnard RJ, Elleder D, Young JA. 2006. Avian sarcoma and leukosis virus-receptor interactions: from classical genetics to novel insights into virus-cell membrane fusion. Virology 344:25–29. doi: 10.1016/j.virol.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 13.Battini JL, Danos O, Heard JM. 1998. Definition of a 14-amino-acid peptide essential for the interaction between the murine leukemia virus amphotropic envelope glycoprotein and its receptor. J Virol 72:428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang B, Sun C, Jin S, Cascio M, Montelaro RC. 2008. Mapping of equine lentivirus receptor 1 residues critical for equine infectious anemia virus envelope binding. J Virol 82:1204–1213. doi: 10.1128/JVI.01393-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bates P, Young JA, Varmus HE. 1993. A receptor for subgroup A Rous sarcoma virus is related to the low density lipoprotein receptor. Cell 74:1043–1051. doi: 10.1016/0092-8674(93)90726-7. [DOI] [PubMed] [Google Scholar]
- 16.Young JA, Bates P, Varmus HE. 1993. Isolation of a chicken gene that confers susceptibility to infection by subgroup A avian leukosis and sarcoma viruses. J Virol 67:1811–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adkins HB, Brojatsch J, Naughton J, Rolls MM, Pesola JM, Young JA. 1997. Identification of a cellular receptor for subgroup E avian leukosis virus. Proc Natl Acad Sci U S A 94:11617–11622. doi: 10.1073/pnas.94.21.11617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adkins HB, Brojatsch J, Young JA. 2000. Identification and characterization of a shared TNFR-related receptor for subgroup B, D, and E avian leukosis viruses reveal cysteine residues required specifically for subgroup E viral entry. J Virol 74:3572–3578. doi: 10.1128/JVI.74.8.3572-3578.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elleder D, Stepanets V, Melder DC, Senigl F, Geryk J, Pajer P, Plachý J, Hejnar J, Svoboda J, Federspiel MJ. 2005. The receptor for the subgroup C avian sarcoma and leukosis viruses, Tvc, is related to mammalian butyrophilins, members of the immunoglobulin superfamily. J Virol 79:10408–10419. doi: 10.1128/JVI.79.16.10408-10419.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Munguia A, Federspiel MJ. 2008. Efficient subgroup C avian sarcoma and leukosis virus receptor activity requires the IgV domain of the Tvc receptor and proper display on the cell membrane. J Virol 82:11419–11428. doi: 10.1128/JVI.01408-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chai N, Bates P. 2006. Na+/H+ exchanger type 1 is a receptor for pathogenic subgroup J avian leukosis virus. Proc Natl Acad Sci U S A 103:5531–5536. doi: 10.1073/pnas.0509785103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhartur SG, Ballarin LJ, Musch MW, Bookstein C, Chang EB, Rao MC. 1999. A unique Na+/H+ exchanger, analogous to NHE1, in the chicken embryonic fibroblast. Am J Physiol 276:838–846. [DOI] [PubMed] [Google Scholar]
- 23.Gupta A, Edwards JC, Hruska KA. 1996. Cellular distribution and regulation of NHE-1 isoform of the Na-H exchanger in the avian osteoclast. Bone 18:87–95. doi: 10.1016/8756-3282(95)00455-6. [DOI] [PubMed] [Google Scholar]
- 24.Denker SP, Barber DL. 2002. Cell migration requires both ion translocation and cytoskeletal anchoring by the Na-H exchanger NHE1. J Cell Biol 159:1087–1096. doi: 10.1083/jcb.200208050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cardone RA, Casavola A, Reshkin SJ. 2005. The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat Rev Cancer 5:786–795. doi: 10.1038/nrc1713. [DOI] [PubMed] [Google Scholar]
- 26.Hendusaltenburger R, Kragelund BB, Pedersen SF.. 2014. Structural dynamics and regulation of the mammalian SLC9A family of Na+/H+ exchangers. Curr Topics Membr 73:69–148. doi: 10.1016/B978-0-12-800223-0.00002-5. [DOI] [PubMed] [Google Scholar]
- 27.Kemp G, Young H, Fliegel L. 2008. Structure and function of the human Na/Hexchanger isoform 1. Channels 2:329–336. doi: 10.4161/chan.2.5.6898. [DOI] [PubMed] [Google Scholar]
- 28.Liu Q, Acharya P, Dolan MA, Zhang P, Guzzo C, Lu J, Kwon A, Gururani D, Miao H, Bylund T, Chuang GY, Druz A, Zhou T, Rice WJ, Wigge C, Carragher B, Potter CS, Kwong PD, Lusso P. 2017. Quaternary contact in the initial interaction of CD4 with the HIV-1 envelope trimer. Nat Struct Mol Biol 24:370–378. doi: 10.1038/nsmb.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Parseval A, Chatterji U, Morris G, Sun P, Olson AJ, Elder JH. 2005. Structural mapping of CD134 residues critical for interaction with feline immunodeficiency virus. Nat Struct Mol Biol 12:60–66. doi: 10.1038/nsmb872. [DOI] [PubMed] [Google Scholar]
- 30.Connolly L, Zingler K, Young JA. 1994. A soluble form of a receptor for subgroup A avian leukosis and sarcoma viruses (ALSV-A) blocks infection and binds directly to ALSV-A. J Virol 68:2760–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rong L, Bates P. 1995. Analysis of the subgroup A avian sarcoma and leukosis virus receptor: the 40-residue, cysteine-rich, low-density lipoprotein receptor repeat motif of Tva is sufficient to mediate viral entry. J Virol 69:4847–4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holmen SL, Salter DW, Payne WS, Dodgson JB, Hughes SH, Federspiel MJ. 1999. Soluble forms of the subgroup A avian leukosis virus [ALV(A)] receptor Tva significantly inhibit ALV(A) infection in vitro and in vivo. J Virol 73:10051–10060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knauss DJ, Young JA. 2002. A fifteen-amino-acid TVB peptide serves as a minimal soluble receptor for subgroup B avian leukosis and sarcoma viruses. J Virol 76:5404–5410. doi: 10.1128/JVI.76.11.5404-5410.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klucking S, Young JA. 2004. Amino acid residues Tyr-67, Asn-72, and Asp-73 of the TVB receptor are important for subgroup E avian sarcoma and leukosis virus interaction. Virology 318:371–380. doi: 10.1016/j.virol.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 35.Kučerová D, Plachy J, Reinisová M, Šenigl F, Trejbalová K, Geryk J, Hejnar J. 2013. Nonconserved tryptophan 38 of the cell surface receptor for subgroup J avian leukosis virus discriminates sensitive from resistant avian species. J Virol 87:8399–8407. doi: 10.1128/JVI.03180-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hughes SH. 2004. The RCAS vector system. Folia Biol 50:107–119. [PubMed] [Google Scholar]
- 37.Li X, Zhu H, Wang Q, Sun J, Gao Y, Qi X, Wang Y, Gao H, Gao Y, Wang X. 2015. Identification of a novel B-cell epitope specific for avian leukosis virus subgroup J gp85 protein. Arch Virol 160:995–1004. doi: 10.1007/s00705-014-2318-6. [DOI] [PubMed] [Google Scholar]
- 38.Slepkov ER, Rainey JK, Sykes BD, Fliegel L. 2007. Structural and functional analysis of the Na+/H+ exchanger. Biochem J 401:623–633. doi: 10.1042/BJ20061062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lundorf MD, Pedersen FS, O'Hara B, Pedersen L. 1999. Amphotropic murine leukemia virus entry is determined by specific combinations of residues from receptor loops 2 and 4. J Virol 73:3169–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lo Conte L, Chothia C, Janin J. 1999. The atomic structure of protein-protein recognition sites. J Mol Biol 285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 41.Clark AJ, Gindin T, Zhang B, Wang L, Abel R, Murret CS, Xu F, Bao A, Lu NJ, Zhou T, Kwong PD, Shapiro L, Honig B, Friesner RA. 2017. Free energy perturbation calculation of relative binding free energy between broadly neutralizing antibodies and the gp120 glycoprotein of HIV-1. J Mol Biol 429:930–947. doi: 10.1016/j.jmb.2016.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clackson T, Wells JA. 1995. A hot spot of binding energy in a hormone-receptor interface. Science 267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 43.Morgan RA, Baler-Bitterlich G, Ragheb JA, Wong-Staal F, Gallo RC, Anderson WF. 1994. Further evaluation of soluble CD4 as an anti-HIV type 1 gene therapy: demonstration of protection of primary human peripheral blood lymphocytes from infection by HIV type 1. AIDS Res Hum Retrovir 10:1507–1515. doi: 10.1089/aid.1994.10.1507. [DOI] [PubMed] [Google Scholar]
- 44.Smith JG, Cunningham JM. 2007. Receptor-induced thiolate couples Env activation to retrovirus fusion and infection. PLoS Pathog 3:e198. doi: 10.1371/journal.ppat.0030198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Justice JIV, Beemon KL. 2013. Avian retroviral replication. Curr Opin Virol 3:664–669. doi: 10.1016/j.coviro.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Côté M, Zheng YM, Liu SL. 2009. Receptor binding and low pH coactivate oncogenic retrovirus envelope-mediated fusion. J Virol 83:11447–11455. doi: 10.1128/JVI.00748-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu H, Kwong PD, Hendrickson WA. 1997. Dimeric association and segmental variability in the structure of human CD4. Nature 387:527–530. doi: 10.1038/387527a0. [DOI] [PubMed] [Google Scholar]
- 48.Tonelli M, Peters RJ, James TL, Agard DA. 2001. The solution structure of the viral binding domain of Tva, the cellular receptor for subgroup A avian leukosis and sarcoma virus. FEBS Lett 509:161–168. doi: 10.1016/S0014-5793(01)03086-1. [DOI] [PubMed] [Google Scholar]
- 49.Hoag MR, Roman J, Beaupre BA, Silvaggi NR, Moran GR. 2015. Bacterial renalase: structure and kinetics of an enzyme with 2- and 6-dihydro-β-NAD(P) oxidase activity from Pseudomonas phaseolicola. Biochemistry 54:3791–3802. doi: 10.1021/acs.biochem.5b00451. [DOI] [PubMed] [Google Scholar]
- 50.Nemčovičová I, Benedict CA, Zajonc DM. 2013. Structure of human cytomegalovirus UL141 binding to TRAIL-R2 reveals novel, non-canonical death receptor interactions. PLoS Pathog 9:e1003224. doi: 10.1371/journal.ppat.1003224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Palakodeti A, Sandstrom A, Sundaresan L, Harly C, Nedellec S, Olive D, Scotet E, Bonneville M, Adams EJ. 2012. The molecular basis for modulation of human Vγ9Vδ2 T cell responses by CD277/butyrophilin-3 (BTN3A)-specific antibodies. J Biol Chem 287:32780–32790. doi: 10.1074/jbc.M112.384354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rong L, Gendron K, Bates P. 1998. Conversion of a human low-density lipoprotein receptor ligand binding repeat to a virus receptor: identification of residues important for ligand specificity. Proc Natl Acad Sci U S A 95:8467–8472. doi: 10.1073/pnas.95.15.8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moebius U, Clayton LK, Abraham S, Harrison SC, Reinherz EL. 1992. The human immunodeficiency virus gp120 binding site on CD4: delineation by quantitative equilibrium and kinetic binding studies of mutants in conjunction with a high-resolution CD4 atomic structure. J Exp Med 176:507–517. doi: 10.1084/jem.176.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bender RR, Muth A, Schneider IC, Friedel T, Hartmann J, Plückthun A, Maisner A, Buchholz CJ. 2016. Receptor-targeted Nipah virus glycoproteins improve cell-type selective gene delivery and reveal a preference for membrane-proximal cell attachment. PLoS Pathog 12:e1005641. doi: 10.1371/journal.ppat.1005641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Payne LN, Howes K, Gillespie AM, Smith LM. 1992. Host range of Rous sarcoma virus pseudotype RSV(HPRS-103) in 12 avian species: support for a new avian retrovirus envelope subgroup, designated J. J Gen Virol 73:2995–2997. doi: 10.1099/0022-1317-73-11-2995. [DOI] [PubMed] [Google Scholar]
- 56.Payne LN, Gillespie AM, Howes K. 1992. Myeloid leukaemogenicity and transmission of the HPRS-103 strain of avian leukosis virus. Leukemia 6:1167–1176. [PubMed] [Google Scholar]
- 57.Federspiel MJ, Hughes SH. 1997. Retroviral gene delivery. Methods Cell Biol 52:179–214. doi: 10.1016/S0091-679X(08)60379-9. [DOI] [PubMed] [Google Scholar]
- 58.Lichtenstein DL, Rushlow KE, Cook RF, Raabe ML, Swardson CJ, Kociba GJ, Issel CJ, Montelaro RC. 1995. Replication in vitro and in vivo of an equine infectious anemia virus mutant deficient in dUTPase activity. J Virol 69:2881–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]