

ABSTRACT
Endogenous retroviruses (ERVs) occupy extensive regions of the human genome. Although many of these retroviral elements have lost their ability to replicate, those whose insertion took place more recently, such as the HML-2 group of HERV-K elements, still retain intact open reading frames and the capacity to produce certain viral RNA and/or proteins. Transcription of these ERVs is, however, tightly regulated by dedicated epigenetic control mechanisms. Nonetheless, it has been reported that some pathological states, such as viral infections and certain cancers, coincide with ERV expression, suggesting that transcriptional reawakening is possible. HML-2 elements are reportedly induced during HIV-1 infection, but the conserved nature of these elements has, until recently, rendered their expression profiling problematic. Here, we provide comprehensive HERV-K HML-2 expression profiles specific for productively HIV-1-infected primary human CD4+ T cells. We combined enrichment of HIV-1 infected cells using a reporter virus expressing a surface reporter for gentle and efficient purification with long-read single-molecule real-time sequencing. We show that three HML-2 proviruses—6q25.1, 8q24.3, and 19q13.42—are upregulated on average between 3- and 5-fold in HIV-1-infected CD4+ T cells. One provirus, HML-2 12q24.33, in contrast, was repressed in the presence of active HIV replication. In conclusion, this report identifies the HERV-K HML-2 loci whose expression profiles differ upon HIV-1 infection in primary human CD4+ T cells. These data will help pave the way for further studies on the influence of endogenous retroviruses on HIV-1 replication.
IMPORTANCE Endogenous retroviruses inhabit big portions of our genome. Moreover, although they are mainly inert, some of the evolutionarily younger members maintain the ability to express both RNA and proteins. We have developed an approach using long-read single-molecule real-time (SMRT) sequencing that produces long reads that allow us to obtain detailed and accurate HERV-K HML-2 expression profiles. We applied this approach to study HERV-K expression in the presence or absence of productive HIV-1 infection of primary human CD4+ T cells. In addition to using SMRT sequencing, our strategy also includes the magnetic selection of the infected cells so that levels of background expression due to uninfected cells are kept at a minimum. The results presented here provide a blueprint for in-depth studies of the interactions of the authentic upregulated HERV-K HML-2 elements and HIV-1.
KEYWORDS: CD4+ T cells, HERV-K, HIV-1
INTRODUCTION
Interactions with the host cellular environment dictate fundamental properties of HIV-1 infectivity, immune recognition, and cytopathogenicity (1, 2). This cellular environment also includes less-appreciated, preexisting components deriving from endogenous retroviruses (ERVs). These proviruses, remnants of past retroviral germ line infections, account for more than 8% of the human genome (3), whose expression is tightly regulated by dedicated epigenetic control mechanisms (4, 5). Moreover, through evolutionary time, most ERVs have been subject to significant mutations, insertions, and deletions, which collectively have rendered them largely “inert.” Some ERVs, however, have retained open reading frames (ORFs) and, individually or in cooperation, can produce intact but noninfectious viral particles (6).
Many retroviruses have the flexibility to incorporate the products of other viruses they happen to be coexpressed with (7–9), and the interactions of HIV-1 with the ERVs harbored in the human genome are now starting to be investigated (10–13). The HERV-K HML-2 group, the most recent proviral insertions within the human genome, make up a large fraction of ERVs retaining ORFs and for this reason have been keenly studied (14, 15).
Previous reports present evidence that expression of HERV-K HML-2 proviruses is upregulated upon HIV-1 infection (12, 13, 16, 17) and that viral particle-associated RNAs within patient plasma samples are reportedly reduced by highly active antiretroviral therapy (10, 11). ERV-derived proteins and particles have also been shown to elicit immune responses (18–20).
Importantly, however, information on the specific proviruses expressed in the above-mentioned scenarios is currently very limited. Indeed, more than 90 HERV-K HML-2 elements are present within the human genome, with an increasing number now recognized to be insertionally polymorphic between individuals and human populations (21, 22). Understanding which members of this group are coexpressed with, and possibly upregulated by, HIV-1 would set the foundation to systematically study specific ERV-HIV interactions, with the ultimate goal of determining whether HERV-Ks influence HIV-1 replication and pathogenesis. Such data would also provide insight into the general physiology of HERV-Ks and help define candidates that might have potentially broader links to health and disease.
Studying individual proviruses therefore provides the advantage of observing expression from authentic loci, whose behaviors might considerably differ from those of the consensus sequence model systems often used to represent the whole HERV-K HML-2 group (23).
One of the main obstacles in characterizing the specific expression of any given ERV locus lies in their sequence similarity, which prevents their unambiguous discrimination by approaches such as PCR or conventional “next-generation” deep-sequencing technologies. Indeed, for the study of HERV-K HML-2 proviruses, this limitation represents one of the main factors confounding the literature, where various techniques and methodologies have often yielded inconsistent conclusions (17). To overcome this technical bottleneck, we have used long-read Pacific Biosciences single-molecule real-time (SMRT) sequencing to identify specific HERV-K HML-2 expression profiles of primary cells (9). Our approach provides the capability to unambiguously identify HERV-K HML-2 elements in the presence or absence of productive HIV-1 infection of primary human CD4+ T cells.
To obtain specific expression profiles, it is crucial that the RNA analyzed originates exclusively from infected cells, a nontrivial task when less than 0.2% of circulating CD4+ T cells are infected during chronic stages of HIV-1 infection in vivo (24). Even in vitro, infections of purified cells rarely reach beyond 10 to 25%, and interdonor variability often acts as a further confounding factor. Unless otherwise enriched, therefore, RNA deriving from uninfected cells dominates extractions, rendering subsequent analyses from bulk cell populations insensitive and their interpretation inaccurate. We have thus developed an HIV-1 clone harboring a surface reporter alongside nef that facilitates high-purity enrichment of infected cells. The resolution obtained by pairing this purification procedure with SMRT sequencing facilitates the effective determination of which HERV-K HML-2 elements contribute RNA during HIV-1 infection. Here, we provide the first detailed report of this transcriptional interaction at the locus level, paving the way for future studies.
RESULTS
Rapid selection strategy and HIV replication competent reporter construct.
Many reporter systems have been generated to monitor HIV-1 replication in infected cells, which harbor the required markers within either accessory (vpr and nef [25, 26]) or structural (27–29) genes. Examples of reporters include enzymes such as placental alkaline phosphatase and luciferase (25, 30, 31), small peptides such as myc (28) and FLAG (29), surface molecules such as murine heat-stable antigen (HSA) (26), and fluorescent molecules such as the enhanced green fluorescent protein (32).
We chose to develop a system that would facilitate both magnetic separation and flow cytometry-based analysis of the infected cells. Particularly, magnetic separation methods are practical and efficient, do not require highly specialized equipment, comply easily with biosafety requirements, and can be integrated into fast, high-throughput workflows that reduce cell stress associated with the selection procedures.
To build a reporter virus that we could utilize to select HIV-1-infected from uninfected cells by magnetic separation, we adopted a strategy similar to that described by Imbeault et al. (33). This approach takes advantage of the small surface molecule HSA as a means for selection. HSA was cloned upstream of an mCherry sequence to facilitate monitoring of ongoing infections by fluorescence microscopy or small-scale flow cytometry analysis. Both reporters have been extensively used alongside HIV-1 (33–38) without any described interference or detrimental consequence and therefore, although impossible to exclude, we believe that it is unlikely they would influence HERV-K expression. Following the stop codon of HSA, a Kozak sequence was inserted to drive the expression of mCherry, followed by an internal ribosome entry site (IRES) enabling the expression of nef (Fig. 1A). This layout provides stable expression of both reporters, as well as allowing the maintenance of nef expression (33, 39, 40), vital to attempts to recapitulate HIV-1 infection in its entirety. Finally, instead of utilizing the molecular chimeric strain NL4.3 (41) as the framework of our system, we opted for the molecular clone LAI, a direct derivative of the original HIV-1 isolate (42).
FIG 1.

(A) Schematic representation of the full-length HIV-1 LAI2 HSA-mCherry-IRES-Nef reporter virus used in this study and specifically built for the magnetic separation of infected cells. (B) HIV-1 LAI2 HSA-mCherry-IRES-Nef-infected CD4+ T cells express Gag and Nef. Western blots of cell lysates of infected CD4+ T cells and of uninfected controls assessed 4 days after infection are shown. (C) HIV-1 LAI2 HSA-mCherry-IRES-Nef infected CD4+ T cells express HSA and mCherry. At 4 days postinfection, the cells were stained with biotin-conjugated α-mCD24(HSA) antibody and counterstained with FITC-conjugated streptavidin. After formaldehyde fixation, the cells were analyzed by flow cytometry. Dot plots show the frequencies of cells positive for mCherry expression and anti-mCD24 staining. FSC, forward scatter.
Validating the approach, we observed that 4 days after infection with this reporter virus, primary human CD4+ T cells produced both Gag p24 and Nef (Fig. 1B). In addition, flow cytometry analysis confirmed the production and trafficking of HSA to the cell surface, as well as the production of mCherry (Fig. 1C). Furthermore, the majority of infected cells were mCherry-HSA double positive, ruling out expression interference between the neighboring ORFs. Thus, the HSA-mCherry-IRES-Nef cassette provided a stable tool for reporting infection in live cells while retaining expression of the viral accessory gene harboring the reporter sequences.
Efficient capture of productively infected CD4+ T cells.
We used primary CD4+ T cells isolated from buffy coat blood fractions obtained from eight anonymous healthy donors from the NY Blood Bank. Primary cells are naturally more representative of physiological conditions than CD4+ T cell lines, which have, over time, adapted to their less selective environmental constraints. Large numbers of host genes are involved in the control of repetitive elements, and the importance of accurate representations of these states between individuals is central to understanding the regulation of ERVs in pathological states (43).
Resting CD4+ T cells are largely resistant to HIV-1 infection due to blocks in the early stages of the viral life cycle, in part through SAMHD1 activity (44). To mitigate this, we activated CD4+ T cells using beads conjugated with α-CD3 and α-CD28 prior to infection. Infected cells where captured using biotinylated α-mCD24 (HSA) and streptavidin-conjugated magnetic beads (Fig. 2A outlines the stepwise procedure followed in this study). With this protocol, approximately 90% of the selected CD4+ T cells were either gp120 or mCherry positive and more than 94% positive when combining the two markers (Fig. 2B). These data confirmed that the strategy yielded infected CD4+ T cells at a sufficiently high purity to allow comprehensive downstream analyses.
FIG 2.

(A) Stepwise schematic representation of the procedure leading to the enrichment of infected CD4+ T cells necessary to achieve a low-background HIV-1-defined HERV-K expression profile. Virus production includes transfection of HEK293T and viral supernatant concentration/purification over density barrier layer (OptiPrep/odixanol). Activated CD4+ T cells are then infected overnight and incubated after a washing step for an additional 3 days. Separation of HSA-positive cells includes incubation with biotin-conjugated α-mCD24 (HSA), followed by the addition of streptavidin-coated magnetic nanoparticles, and magnetic separation. After washing, the cells are lysed in TRIzol, and the total RNA is extracted. The RNA is then used to generate HERV-K HML-2 expression cDNA libraries that are analyzed by long-read SMRT sequencing. (B) More than 90% of the HSA enriched are HIV-1 infected. At 4 days after infection with LAI2 HSA-mCherry-IRES-Nef, the cells magnetically separated for HSA were stained with α-gp120 human antibody (clone 2G12) and analyzed by flow cytometry. Dot plots show the frequencies of cells positive for mCherry expression and anti-gp120 staining. FSC, forward scatter.
Global HML-2 expression remains unchanged by productive HIV infection.
Total cellular RNA was extracted from selected HIV-1-positive CD4+ T cells and, separately, from uninfected controls. Preparations were twice treated with DNase to ensure purity of the RNA.
The levels of HIV-1 RNA transcription in the purified infected cells revealed no major differences between donors (Fig. 3A). We next compared global HERV-K HML-2 expression levels between donors and, within each individual donor, by comparing infected cells with the corresponding uninfected control. Real-time quantitative PCR (RT-qPCR) using primers specific to the TM region of the HERV-K HML-2 env (17) revealed no significant differences in average expression with or without HIV-1 infection, but there was marked variability between the individual donors (Fig. 3B). Indeed, in three donors, HML-2 expression increased ∼2-fold upon HIV-1 infection, while remaining stable (fold changes of between 1 and 1.39) or even decreasing (fold changes of 0.65 and 0.69) in the other five donors. In addition, no correlation between HIV-1 expression and overall changes in HERV-K levels was noted (Fig. 3C).
FIG 3.
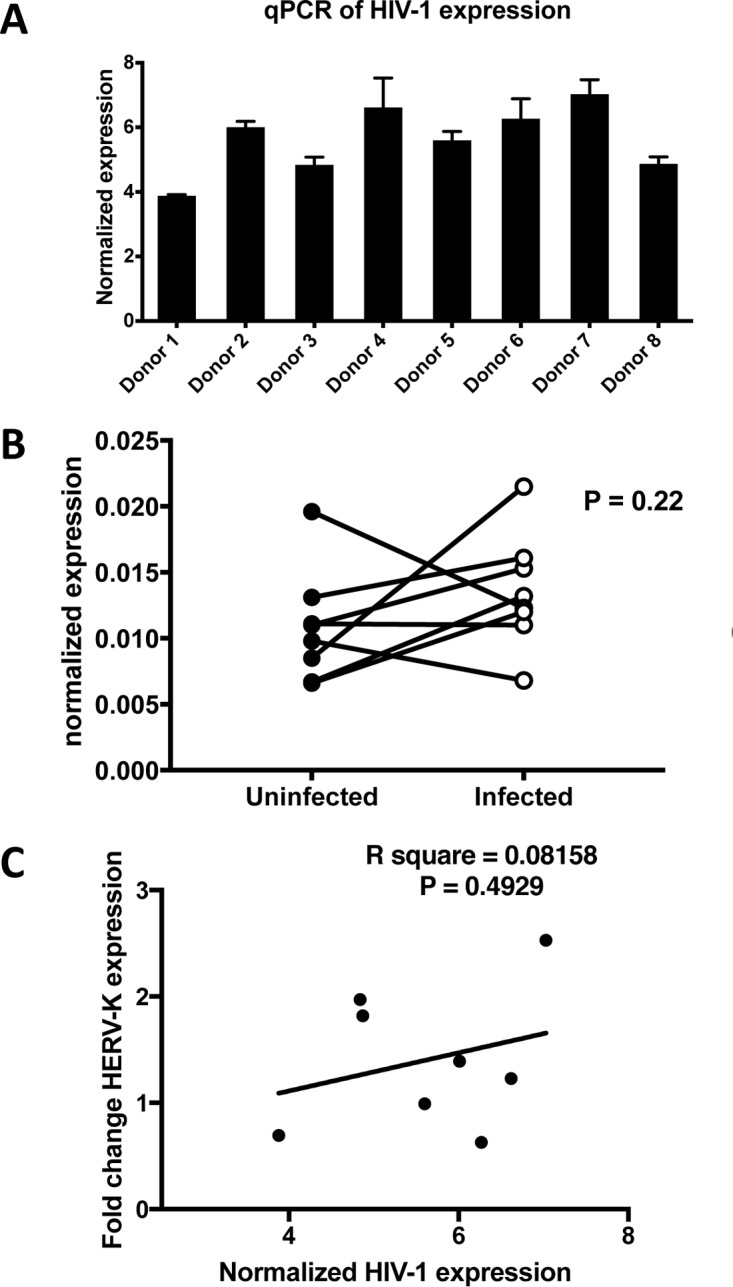
(A) Cells from all donors support HIV-1 infection. HIV-1-specific RT-qPCR was used to assess HIV-1 expression levels at the end of the infection step of each experiment. (B) No significant difference in the overall HERV-K HML-2 expression and marked donor variability is observed in HIV-1 infection of CD4+ T cells. HERV-K HML-2-specific RT-qPCR was used to assess overall expression levels at the end of the infection step of each experiment. (C) No correlation between HIV-1 expression levels and a change in HERV-K HML-2 expression was detected.
These data indicate that, when mimicking physiologic conditions, rather than forcing high infection rates by spinoculation; for example, HIV-1 infection was insufficient to significantly increase overall HERV-K HML-2 expression levels in primary CD4+ T cells. This further suggests that previously reported links between the two are likely dependent on additional factors not present within our primary human CD4+ T cell model system. Indeed, the increased HERV-K detection reported in HIV-1 patients (12, 17) might occur by secondary means, for example, through immune activation (45), the effects of microbial translocation (46), or mechanisms influencing the cellular epigenetic environment (47).
HML-2 identification using SMRT sequencing.
The high sequence similarity of HERV-K HML-2 group members hinders their individual expression assessment by PCR-based methods and prevents the unambiguous mapping of short reads from common high-throughput sequencing platforms. We instead used long-read SMRT sequencing, a technology able to make multiple read passes around circularized templates and, at once, produce long reads with unparalleled consensus accuracy derived via the compression and error correction of those single molecule passes. This approach is ideal for the assessment of complex libraries with high levels of sequence similarity among its individual components.
As previously described (9), libraries of 700-bp amplicons of the HERV-K HML-2 env from specifically primed reverse transcription products were prepared. Unique 9-mer molecular tags were incorporated into individual cDNA molecules to allow the downstream identification of individual amplicons and, thus, to account for and overcome PCR amplification resampling biases (48). Incorporation of uniquely identifying tags in this manner allows for up to 262,144 individual cDNAs to be monitored within each library.
From each HIV-1-infected or uninfected control sample library, we obtained an average of 2,766 high-quality reads (standard deviation [SD], ±670) passing filters to eliminate potential duplicates and chimeras (9). A total of 44,265 reads, where each read denotes an expressed RNA molecule, were mapped against a custom-made library of HERV-K HML-2 elements derived from the latest human reference genome, GRCh38). We found that a total of 47 distinct proviruses were expressed in at least two donors (Table 1).
TABLE 1.
Detailed HERV-K HML-2 expression in HIV-1-infected cells and uninfected controls from eight donorsa

HERV-K HML-2 proviruses are identified by the locus they occupy in the human genome. The expression of each provirus is represented as the percentage of the total HERV-K HML-2 expression of each individual sample. On the vertical side of the table are the chromosomal loci that specify the different HERV-K HML-2 proviruses (21). INF, HIV-1 infected; CNTL, uninfected control.
In good agreement with our previous report (9), we found that the expression of four loci (HML-2 1q22, 1q23.3, 3q12.3, and 6q25.1 [GenBank accession numbers JN675014, JN675013, JN675019, and JN675042, respectively]) largely defined the overall expression profile of primary human CD4+ T cells in the absence of exogenous lentiviral infection. As commonly observed when profiling HERV-K HML-2 expression (49–51), a considerable number of elements were expressed with very low frequency (Table 1). To streamline the comparative data analysis, we excluded those HML-2 loci that contributed <0.2% of the total HERV-K HML-2 expression across both infected and uninfected samples for each donor (Fig. 4A).
FIG 4.

(A) Heat map representation of HERV-K HML-2 expression in HIV-1-infected cells and uninfected controls from eight different donors. Elements contributing <0.2% of the total HERV-K HML-2 expression (in both samples of each infected/uninfected pair) are excluded. Arrows in the uninfected controls indicate elements with higher levels of expression, while in the infected samples, in addition to the higher expressers, the arrows also indicate the elements whose level of expression consistently changes upon infection. On the vertical side of the map are the chromosomal loci that specify the different HERV-K HML-2 proviruses (21); on the horizontal side of the map are the donors (1 to 8). (B) Heat map representing the change in HERV-K HML-2 expression upon HIV-1 infection. Values are provided as the natural logarithm (LN) of the ratio between the expression levels in infected and uninfected cells. Arrows indicate the elements whose expression level consistently changes upon infection. On the vertical side of the map are the chromosomal loci that specify the different HERV-K HML-2 proviruses (21); on the horizontal side of the map are the donors (1 to 8).
When comparing infected cells with the uninfected controls from the same donor, we observed little change in expression of the highly expressed HML-2 1q22 and 3q12.3 loci (Fig. 4A). In contrast, the expression of HML-2 6q25.1, 8q24.3, and 19q13.42 increased 3.0- to 5.7-fold upon HIV-1 infection (Fig. 4A and B). HML-2 1q23.3 expression also increased substantially in five of the eight donors, underlining a different regulation from the other two highly expressed elements, HML-2 1q22 and 3q12.3. HML-2 19q13.41 expression also increased upon infection (Fig. 4B) but never exceeded 0.35% of the total HML-2 expression. Finally, across all loci, we observed only a single example of a decrease in expression upon HIV-1 infection: HML-2 12q24.33 expression decreased 1.3- to 5.0-fold across individual donors.
These results provide the most accurate assessment to-date of HERV-K HML-2 expression in the context of active HIV-1 replication in untransformed primary human lymphocytes. Of note, these data reveal that the lack of observable global expression change (Fig. 3B) is likely due to the stability of HML-2 1q22 and 3q12.3 expression. These elements contribute 82 and 66% of total HML-2 expression within uninfected and infected CD4+ T cells, respectively, effectively masking the HIV-1-induced changes that become apparent using SMRT sequencing. The improved resolution granted by our combined approach reveals a proportional increase in env reads derived from four HML-2 loci (1q23.3, 6q25.1, 8q24.3b, and 19q13.42) from 9.4 to 28.4%. Taken together, productive HIV-1 replication in primary CD4+ T cells results in a clear HML-2 expression signature.
DISCUSSION
The HERV-K HML-2 group includes the youngest retroviral elements known within the human genome (21). Indeed, several integrations are sufficiently recent to have retained insertional polymorphism between human populations (52). In addition, a potentially unique example of an unfixed HERV element retaining all retroviral ORFs has been described (22). Although there is no evidence that any HML-2 proviruses found in significant proportions of the human population are individually infectious, two recombinants have been shown to be moderately infectious in vitro (53, 54).
The study of HERV-K regulation in health and disease was initiated by the discovery of viral particles within germ cell tumors (55–60). Subsequently, both HERV RNA expression and protein products have been noted in a variety of autoimmune conditions (61, 62) and in malignancy (51, 63, 64). In spite of the increasing number of these reports, no causal associations have been identified, and any such suggestions should require diligent investigation prior to their acceptance (65). Regardless of potential roles in pathogenesis, HERV-K HML-2 elements have also been suggested to have utility as biomarkers of disease (66), and their repeated observation in HIV-1 infection, for example, has led to the suggestion that their products might constitute targets for novel vaccination strategies (13).
To date, studies have been somewhat limited by the absence of unambiguous data detailing which HERV-K HML-2 loci contribute the expression seen in homeostasis and in conditions of dysregulation. As a consequence, many reports describing interactions with HERV-K HML-2 viruses have made use of the two aforementioned recombinants, Phoenix (53) and HERV-KCON (54). The extent to which either of these accurately recapitulates potential interactions with the individual HERV-K HML-2 proviruses of the human genome is unknown. Accurate data profiling which loci contribute expression and may be differentially regulated in the context of HIV-1 infection is therefore very important and will facilitate their specific incorporation into future work.
In this study, we combined long-read SMRT sequencing with enrichment of infected cells through magnetic separation, a method achieving high levels of target purity while avoiding the cellular stress caused by flow cytometry sorting. Using primary CD4+ T cell populations, we note no significant differences in global HERV-K HML-2 expression upon HIV-1 infection (Fig. 3B), and no correlation with HIV-1 transcript expression levels (Fig. 3C). The unaltered overall expression was likely due to the unchanging levels of the two most highly expressed loci in uninfected cells, HML-2 1q22 and 3q12.3 (Fig. 4A and B), the same two loci previously found to dominate expression across human peripheral blood mononuclear cells (PBMCs) (9). This strong baseline expression likely mask the changes visible in other elements and may, in part, explain discrepancies between previous reports if their methodologies differed in the capacity to report the expression of these specific elements.
Underneath this baseline expression, stable across all donors tested in this study, at least, three loci—HML-2 6q25.1, 8q24.3, and 19q13.42 (and, in part, 1q23.3)—were found to be upregulated in comparison to uninfected controls. Interdonor expression variability was greater within this group, but, interestingly, any lack of upregulation of these proviruses upon HIV-1 infection (e.g., 1q23.3) was concomitant with greater levels of their initial expression (Fig. 4A).
Interestingly, two of the upregulated proviruses, HML-2 6q25.1 and 19q13.42, are members of the LTR5A and LTR5B subgroups (21), which include the older members of the HERV-K HML-2 family. Indeed, as might be expected from their greater age, both lack their 5′ long terminal repeats (LTRs) and thus likely achieve their expression and responsiveness through other means.
The presented data reveal that the majority of the HERV-K HML-2 elements capable of expression within either whole PBMCs (9) or within purified CD4+ T cells are unaffected by HIV-1 infection. Although global expression is unaltered, we nonetheless detail noticeable expression increases for a subset of HML-2 proviruses in HIV-1 infection. Our results describe the effects of HIV-1 infection on HERV-K expression using a single strain, and therefore it would be interesting to further assess different HIV strains, especially transmitter-founder ones, and test the conservation level of the HERV-K expression profiles observed.
The mechanism by which HIV-1 infection and replication could induce HERV-K expression is still poorly understood, although there are studies suggesting an active role for Tat and Vif (13, 67). Alternatively, one could envision HERV-K upregulation to be due to broader consequences of HIV-1 infection, such as the remodeling of host cell chromatin, as suggested by others (68). Further studies are needed in order to better comprehend the underlying mechanisms of HIV-1 regulation of HERV-K expression; however, the methodologies described here provide a robust platform against which different hypotheses can be tested.
MATERIALS AND METHODS
Cell culture.
HEK293T cells were cultured in Dulbecco modified Eagle medium in the presence of 10% fetal calf serum (FCS; GemCell), 100 IU/ml penicillin-streptomycin, and 2 mM l-glutamine at 37°C and 5% CO2.
CD4+ T cells from anonymous donors were obtained from buffy coats (New York Blood Bank) by Ficoll (Sigma) density centrifugation and negative selection using magnetic beads (CD4+ T cell isolation kit II; Miltenyi Biotec) according to the manufacturer's instructions. CD4+ T cells were cultured in RPMI 1640 supplemented with 10% FCS (Gibco/Thermo Fisher), 100 IU/ml penicillin, 100 μg/ml streptomycin, 0.1 M HEPES, 2 mM l-glutamine, and 20 IU/ml IL-2 (NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH).
Construction of pLAI2 HSA-mCherry-IRES-Nef HIV-1 molecular clone and virus stock production.
The HSA-mCherry-IRES cassette was cloned by overlap PCR fusing HSA, obtained from pNL4-3.HSA.R+E− (34), to the mCherry-IRES cassette of reporter vector NLCI (35, 36). The resulting HSA-mCherry-IRES cassette was then fused between the env and nef junction, in the nef frame, and finally cloned into full-length pLAI2 (69) using BamHI and XhoI restriction sites and In-Fusion technology (Clontech). The construct was verified by sequencing. The NLCI was a gift from Benjamin Chen, Mount Sinai Icahn School of Medicine. pNL4-3.HSA.R+E− and pLAI2 were obtained from the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH.
Virus stocks were produced by transfecting 293T cells (6 × 106 per 75-cm2 tissue culture flask) with 15 μg of pLAI2-HSA-mCherry-IRES-Nef using 3 μg/ml polyethyleneimine (Polysciences). After approximately 20 h, the transfection medium was changed, and viral supernatants were collected 48 and 72 h after transfection. Viral supernatants were first filtered (0.45-μm pore size) and then concentrated over a 6% iodixanol (OptiPrep; Invitrogen/Thermo Fisher) cushion at a 15,000 relative centrifugal force at 4°C for 6 h. Aliquots of the resuspended viral stocks were maintained at −80°C until use. Viral stocks were quantified by measuring p24 with HIV-1 p24 enzyme-linked immunosorbent assay kit (XpressBio) according to the manufacturer's instructions.
Infection and selection of HIV-1-infected cells.
CD4+ T cells were activated using α-CD3-CD28-coated beads (Gibco/Thermo Fisher) for 3 days. Approximately 1 × 107 to 1.5 × 107 cells per donor were infected overnight with 250-ng p24 equivalents of LAI2-HSA-mCherry-IRES-Nef virus, an amount empirically chosen to reach 10 to 20% infection in primary CD4+ T cells after 4 days of infection in the presence of 2 μg/ml Polybrene (Sigma). Uninfected controls were treated with 2 μg/ml Polybrene, but no virus.
Selection of infected cells was carried as described by Imbeault et al. (33) using EasySep magnetic separation kits (Stem Cell). Briefly, the cells were incubated with biotinylated α-mCD24(HSA) antibody (M1/69; BioLegend) for 15 min, then for an additional 15 min with an EasySep biotin selection cocktail, and finally for 10 min with EasySep magnetic nanoparticles. Labeled cells were then selected with three to four rounds of washing and magnetic selection.
Western blotting.
Infected primary CD4+ T cells and their uninfected controls were lysed in radioimmunoprecipitation assay buffer (10 mM Tris-Cl [pH 8.0], 1 mM EDTA, 1% Triton X-100, 0.1% sodium dodecyl sulfate, 140 mM NaCl) in the presence of Complete protease inhibitor cocktail (Roche). Proteins from the lysates were separated on 10% polyacrylamide gels (Invitrogen/Thermo Fisher) and transferred to polyvinylidene difluoride membranes (Pierce/Thermo Fisher). Nef was detected using α-HIV-1 Nef polyclonal antibody (70) (catalog no. 2949; NIH AIDS Reagent Program Division of AIDS, NIAID, NIH). HIV-1 Gag was probed with α-HIV-1 p24 monoclonal antibody 183-H12-5C (71) (catalog no. 3537; NIH AIDS Reagent Program Division of AIDS, NIAID, NIH). We used the horseradish peroxidase-conjugated secondary anti-rabbit antibody from Sigma (catalog no. F2555). SuperSignal West Femto (Pierce/Thermo Fisher) was used for development of the membranes, and imaging was carried out using the FluorChem E imaging system (ProteinSimple).
Flow cytometry.
HSA was probed with a 1:1,000 dilution of fluorescein isothiocyanate (FITC)-conjugated α-mCD24 (M1/69; BioLegend). HIV-1 Env was detected with α-gp120 antibody 2G12 (72) (NIH AIDS Reagent Program) at a 1:300 dilution and Alexa Fluor 488-conjugated α-human antibody (Invitrogen/Thermo Fisher) at a 1:1,000 dilution. Gates were set by comparison with uninfected cells stained as described above. mCherry gates were set based on uninfected cells. An average of 5 × 104 cells were collected on a Guava easyCyte HT-BGR system (Millipore) flow cytometer and analyzed using FlowJo.
RNA purification and RT-qPCR.
RNA extraction was performed using TRIzol (Invitrogen/Thermo Fisher) according to the manufacturer's directions. Each sample was DNase treated twice using the DNA-free kit (Ambion). First-strand cDNA for qPCR analysis was obtained using iScript cDNA synthesis kit (Bio-Rad). The global expression of HERV-K HML-2 and HIV-1 was assessed by qPCR using iQ SYBR green Supermix (Bio-Rad).
The primers used to assess HIV-1 (forward, TGTGTGCCCGTCTGTTGTGT; reverse, GAGTCCTGCGTCGAGAGATC) covered 143 nucleotides (nt) of the first exon, from nt 102 to 244 (73). Global HERV-K HML-2 expression was determined for env (forward, CTAACCATGTCCCAGTGATG; reverse, GGAGACAGACTCATGAGCTTAGAA) (17). Both primers sets were normalized to ribosomal protein S11 (RPS11) expression (forward, GCCGAGACTATCTGCACTAC; reverse, ATGTCCAGCCTCAGAACTTC) (74).
SMRT sequencing.
SMRT sequencing and analysis was performed as described previously (9). Briefly, 650 ng of double DNase-treated total RNA from each sample were reverse transcribed using ThermoScript III (Invitrogen/Thermo Fisher), purified, and amplified with CloneAmp HiFi PCR Premix (Clontech/TaKaRa). Purified PCR-derived libraries were converted to SMRTbell templates using the PacBio RS DNA template preparation kit and sequenced. Fluorescent phospho-linked nucleotide incorporation was recorded in 45-min movies, and automated data processing was performed.
Sequence analysis.
Filtered CCS subreads from the initial sequencing were processed with a dedicated Python3 pipeline as previously described (9) to separate sequences by barcode and remove PCR duplicates. Reads were matched to an HERV-K HML-2 database (21) by local BLASTn (BLAST 2.2.28+), and chimeric sequences were removed using UCHIME (USEARCH 7.0.1001) (75). Raw counts for each HERV-K were converted to the percentage of the total reads for each donor, allowing standardization of the read counts between donor samples.
Statistical analysis.
Statistical analysis was performed using GraphPad Prism 7 software. P values are two sided, and values of <0.05 were considered significant.
ACKNOWLEDGMENTS
We thank Jonathan Stoye and Ines Chen for critically reading the manuscript and Benjamin Chen for the NLCI plasmid. The following reagents were obtained through the National Institutes of Health (NIH) AIDS Reagent Program (Division of AIDS, NIAID, NIH): α-HIV-1 p24 monoclonal antibody (183-H12-5C; catalog number 3537) from Bruce Chesebro and Kathy Wehrly, α-HIV-1 gp120 monoclonal (2G12; catalog number 1476) from Polymun Scientific, and α-HIV-1 Nef polyclonal antibody (catalog number 2949) from Ronald Swanstrom. pLAI2 (catalog number 2532) was obtained from Keith Peden, courtesy of the MRC AIDS Directed Program, and HIV-1 pNL4-3.HSA.R+E- (catalog number 3420) was obtained from Nathaniel Landau.
L.C.F.M. is funded by NIH grants R01 GM113886, GM113886-01S1, and GM113886-03S1. V.S. is funded by NIH grants R01 AI064001 and R01 AI120998. G.R.Y. was supported by the Francis Crick Institute under award FC001162 (Jonathan P. Stoye). The Crick receives its core funding from Cancer Research UK, the UK Medical Research Council, and the Wellcome Trust. A.F.-S. is funded by NIH/NIAID grants R01 AI07345, R21 AI116022, and U19 AI118610.
REFERENCES
- 1.Iwasaki A. 2012. Innate immune recognition of HIV-1. Immunity 37:389–398. doi: 10.1016/j.immuni.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simon V, Bloch N, Landau NR. 2015. Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat Immunol 16:546–553. doi: 10.1038/ni.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, et al. . 2001. Initial sequencing and analysis of the human genome. Nature 409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 4.Rowe HM, Jakobsson J, Mesnard D, Rougemont J, Reynard S, Aktas T, Maillard PV, Layard-Liesching H, Verp S, Marquis J, Spitz F, Constam DB, Trono D. 2010. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 463:237–240. doi: 10.1038/nature08674. [DOI] [PubMed] [Google Scholar]
- 5.Turelli P, Castro-Diaz N, Marzetta F, Kapopoulou A, Raclot C, Duc J, Tieng V, Quenneville S, Trono D. 2014. Interplay of TRIM28 and DNA methylation in controlling human endogenous retroelements. Genome Res 24:1260–1270. doi: 10.1101/gr.172833.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boller K, Schoenfeld K, Lischer S, Fischer N, Hoffmann A, Kurth R, Tonjes RR. 2008. Human endogenous retrovirus HERV-K113 is capable of producing intact viral particles. J Gen Virol 89:567–572. doi: 10.1099/vir.0.83534-0. [DOI] [PubMed] [Google Scholar]
- 7.Landau NR, Page KA, Littman DR. 1991. Pseudotyping with human T-cell leukemia-virus type-I broadens the human immunodeficiency virus host range. J Virol 65:162–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM, McKeating JA. 2003. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc Natl Acad Sci U S A 100:7271–7276. doi: 10.1073/pnas.0832180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brinzevich D, Young GR, Sebra R, Ayllon J, Maio SM, Deikus G, Chen BK, Fernandez-Sesma A, Simon V, Mulder LC. 2014. HIV-1 interacts with human endogenous retrovirus K (HML-2) envelopes derived from human primary lymphocytes. J Virol 88:6213–6223. doi: 10.1128/JVI.00669-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Contreras-Galindo R, Almodovar-Camacho S, Gonzalez-Ramirez S, Lorenzo E, Yamamura Y. 2007. Comparative longitudinal studies of HERV-K and HIV-1 RNA titers in HIV-1-infected patients receiving successful versus unsuccessful highly active antiretroviral therapy. AIDS Res Hum Retroviruses 23:1083–1086. doi: 10.1089/aid.2007.0054. [DOI] [PubMed] [Google Scholar]
- 11.Contreras-Galindo R, Kaplan MH, Contreras-Galindo AC, Gonzalez-Hernandez MJ, Ferlenghi I, Giusti F, Lorenzo E, Gitlin SD, Dosik MH, Yamamura Y, Markovitz DM. 2012. Characterization of human endogenous retroviral elements in the blood of HIV-1-infected individuals. J Virol 86:262–276. doi: 10.1128/JVI.00602-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Contreras-Galindo R, Kaplan MH, Markovitz DM, Lorenzo E, Yamamura Y. 2006. Detection of HERV-K(HML-2) viral RNA in plasma of HIV type 1-infected individuals. AIDS Res Hum Retroviruses 22:979–984. doi: 10.1089/aid.2006.22.979. [DOI] [PubMed] [Google Scholar]
- 13.Jones RB, Garrison KE, Mujib S, Mihajlovic V, Aidarus N, Hunter DV, Martin E, John VM, Zhan W, Faruk NF, Gyenes G, Sheppard NC, Priumboom-Brees IM, Goodwin DA, Chen L, Rieger M, Muscat-King S, Loudon PT, Stanley C, Holditch SJ, Wong JC, Clayton K, Duan E, Song H, Xu Y, Sengupta D, Tandon R, Sacha JB, Brockman MA, Benko E, Kovacs C, Nixon DF, Ostrowski MA. 2012. HERV-K-specific T cells eliminate diverse HIV-1/2 and SIV primary isolates. J Clin Invest 122:4473–4489. doi: 10.1172/JCI64560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van der Kuyl AC. 2012. HIV infection and HERV expression: a review. Retrovirology 9:6. doi: 10.1186/1742-4690-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanke K, Hohn O, Bannert N. 2016. HERV-K(HML-2), a seemingly silent subtenant, but still waters run deep. APMIS 124:67–87. doi: 10.1111/apm.12475. [DOI] [PubMed] [Google Scholar]
- 16.Contreras-Galindo R, Lopez P, Velez R, Yamamura Y. 2007. HIV-1 infection increases the expression of human endogenous retroviruses type K (HERV-K) in vitro. AIDS Res Hum Retroviruses 23:116–122. doi: 10.1089/aid.2006.0117. [DOI] [PubMed] [Google Scholar]
- 17.Bhardwaj N, Maldarelli F, Mellors J, Coffin JM. 2014. HIV-1 infection leads to increased transcription of human endogenous retrovirus HERV-K (HML-2) proviruses in vivo but not to increased virion production. J Virol 88:11108–11120. doi: 10.1128/JVI.01623-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garrison KE, Jones RB, Meiklejohn DA, Anwar N, Ndhlovu LC, Chapman JM, Erickson AL, Agrawal A, Spotts G, Hecht FM, Rakoff-Nahoum S, Lenz J, Ostrowski MA, Nixon DF. 2007. T cell responses to human endogenous retroviruses in HIV-1 infection. PLoS Pathog 3:e165. doi: 10.1371/journal.ppat.0030165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.SenGupta D, Tandon R, Vieira RG, Ndhlovu LC, Lown-Hecht R, Ormsby CE, Loh L, Jones RB, Garrison KE, Martin JN, York VA, Spotts G, Reyes-Teran G, Ostrowski MA, Hecht FM, Deeks SG, Nixon DF. 2011. Strong human endogenous retrovirus-specific T cell responses are associated with control of HIV-1 in chronic infection. J Virol 85:6977–6985. doi: 10.1128/JVI.00179-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tandon R, SenGupta D, Ndhlovu LC, Vieira RG, Jones RB, York VA, Vieira VA, Sharp ER, Wiznia AA, Ostrowski MA, Rosenberg MG, Nixon DF. 2011. Identification of human endogenous retrovirus-specific T cell responses in vertically HIV-1-infected subjects. J Virol 85:11526–11531. doi: 10.1128/JVI.05418-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Subramanian RP, Wildschutte JH, Russo C, Coffin JM. 2011. Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology 8:90. doi: 10.1186/1742-4690-8-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wildschutte JH, Williams ZH, Montesion M, Subramanian RP, Kidd JM, Coffin JM. 2016. Discovery of unfixed endogenous retrovirus insertions in diverse human populations. Proc Natl Acad Sci U S A 113:e2326-34. doi: 10.1073/pnas.1602336113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Terry SN, Manganaro L, Cuesta-Dominguez A, Brinzevich D, Simon V, Mulder LCF. 2017. Expression of HERV-K108 envelope interferes with HIV-1 production. Virology 509:52–59. doi: 10.1016/j.virol.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Josefsson L, King MS, Makitalo B, Brannstrom J, Shao W, Maldarelli F, Kearney MF, Hu WS, Chen J, Gaines H, Mellors JW, Albert J, Coffin JM, Palmer SE.. 2011. Majority of CD4+ T cells from peripheral blood of HIV-1-infected individuals contain only one HIV DNA molecule. Proc Natl Acad Sci U S A 108:11199–11204. doi: 10.1073/pnas.1107729108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Connor RI, Chen BK, Choe S, Landau NR. 1995. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206:935–944. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- 26.Jamieson BD, Zack JA. 1998. In vivo pathogenesis of a human immunodeficiency virus type 1 reporter virus. J Virol 72:6520–6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hubner W, Chen P, Del Portillo A, Liu Y, Gordon RE, Chen BK. 2007. Sequence of human immunodeficiency virus type 1 (HIV-1) Gag localization and oligomerization monitored with live confocal imaging of a replication-competent, fluorescently tagged HIV-1. J Virol 81:12596–12607. doi: 10.1128/JVI.01088-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muller B, Daecke J, Fackler OT, Dittmar MT, Zentgraf H, Krausslich HG. 2004. Construction and characterization of a fluorescently labeled infectious human immunodeficiency virus type 1 derivative. J Virol 78:10803–10813. doi: 10.1128/JVI.78.19.10803-10813.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petit C, Schwartz O, Mammano F. 1999. Oligomerization within virions and subcellular localization of human immunodeficiency virus type 1 integrase. J Virol 73:5079–5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen BK, Gandhi RT, Baltimore D. 1996. CD4 down-modulation during infection of human T cells with human immunodeficiency virus type 1 involves independent activities of vpu, env, and nef. J Virol 70:6044–6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen BK, Saksela K, Andino R, Baltimore D. 1994. Distinct modes of human immunodeficiency virus type 1 proviral latency revealed by superinfection of nonproductively infected cell lines with recombinant luciferase-encoding viruses. J Virol 68:654–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen BK, Feinberg MB, Baltimore D. 1997. The κB sites in the human immunodeficiency virus type 1 long terminal repeat enhance virus replication yet are not absolutely required for viral growth. J Virol 71:5495–5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Imbeault M, Lodge R, Ouellet M, Tremblay MJ. 2009. Efficient magnetic bead-based separation of HIV-1-infected cells using an improved reporter virus system reveals that p53 upregulation occurs exclusively in the virus-expressing cell population. Virology 393:160–167. doi: 10.1016/j.virol.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 34.He J, Choe S, Walker R, Di Marzio P, Morgan DO, Landau NR.. 1995. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J Virol 69:6705–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cohen GB, Gandhi RT, Davis DM, Mandelboim O, Chen BK, Strominger JL, Baltimore D. 1999. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 10:661–671. doi: 10.1016/S1074-7613(00)80065-5. [DOI] [PubMed] [Google Scholar]
- 36.Li H, Zony C, Chen P, Chen BK. 2017. Reduced potency and incomplete neutralization of broadly neutralizing antibodies against cell-to-cell transmission of HIV-1 with transmitted founder Envs. J Virol 91:e02425-16. doi: 10.1128/JVI.02425-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cavrois M, Banerjee T, Mukherjee G, Raman N, Hussien R, Rodriguez BA, Vasquez J, Spitzer MH, Lazarus NH, Jones JJ, Ochsenbauer C, McCune JM, Butcher EC, Arvin AM, Sen N, Greene WC, Roan NR.. 2017. Mass cytometric analysis of HIV entry, replication, and remodeling in tissue CD4+ T cells. Cell Rep 20:984–998. doi: 10.1016/j.celrep.2017.06.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ito Y, Remion A, Tauzin A, Ejima K, Nakaoka S, Iwasa Y, Iwami S, Mammano F. 2017. Number of infection events per cell during HIV-1 cell-free infection. Sci Rep 7:6559. doi: 10.1038/s41598-017-03954-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown A, Gartner S, Kawano T, Benoit N, Cheng-Mayer C. 2005. HLA-A2 down-regulation on primary human macrophages infected with an M-tropic EGFP-tagged HIV-1 reporter virus. J Leukoc Biol 78:675–685. doi: 10.1189/jlb.0505237. [DOI] [PubMed] [Google Scholar]
- 40.Levy DN, Aldrovandi GM, Kutsch O, Shaw GM. 2004. Dynamics of HIV-1 recombination in its natural target cells. Proc Natl Acad Sci U S A 101:4204–4209. doi: 10.1073/pnas.0306764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 59:284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wain-Hobson S, Vartanian JP, Henry M, Chenciner N, Cheynier R, Delassus S, Martins LP, Sala M, Nugeyre MT, Guetard D, et al. . 1991. LAV revisited: origins of the early HIV-1 isolates from Institut Pasteur. Science 252:961–965. doi: 10.1126/science.2035026. [DOI] [PubMed] [Google Scholar]
- 43.Maksakova IA, Mager DL, Reiss D. 2008. Keeping active endogenous retroviral-like elements in check: the epigenetic perspective. Cell Mol Life Sci 65:3329–3347. doi: 10.1007/s00018-008-8494-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruffin N, Brezar V, Ayinde D, Lefebvre C, Schulze Zur Wiesch J, van Lunzen J, Bockhorn M, Schwartz O, Hocini H, Lelievre JD, Banchereau J, Levy Y, Seddiki N.. 2015. Low SAMHD1 expression following T-cell activation and proliferation renders CD4+ T cells susceptible to HIV-1. AIDS 29:519–530. doi: 10.1097/QAD.0000000000000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paiardini M, Muller-Trutwin M. 2013. HIV-associated chronic immune activation. Immunol Rev 254:78–101. doi: 10.1111/imr.12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, Blazar BR, Rodriguez B, Teixeira-Johnson L, Landay A, Martin JN, Hecht FM, Picker LJ, Lederman MM, Deeks SG, Douek DC. 2006. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med 12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 47.Hurst TP, Magiorkinis G. 2017. Epigenetic control of human endogenous retrovirus expression: focus on regulation of long-terminal repeats (LTRs). Viruses 9:E130. doi: 10.3390/v9060130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jabara CB, Jones CD, Roach J, Anderson JA, Swanstrom R. 2011. Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc Natl Acad Sci U S A 108:20166–20171. doi: 10.1073/pnas.1110064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhardwaj N, Montesion M, Roy F, Coffin JM. 2015. Differential expression of HERV-K (HML-2) proviruses in cells and virions of the teratocarcinoma cell line Tera-1. Viruses 7:939–968. doi: 10.3390/v7030939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fuchs NV, Loewer S, Daley GQ, Izsvak Z, Lower J, Lower R. 2013. Human endogenous retrovirus K (HML-2) RNA and protein expression is a marker for human embryonic and induced pluripotent stem cells. Retrovirology 10:115. doi: 10.1186/1742-4690-10-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmitt K, Reichrath J, Roesch A, Meese E, Mayer J. 2013. Transcriptional profiling of human endogenous retrovirus group HERV-K(HML-2) loci in melanoma. Genome Biol Evol 5:307–328. doi: 10.1093/gbe/evt010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Turner G, Barbulescu M, Su M, Jensen-Seaman MI, Kidd KK, Lenz J. 2001. Insertional polymorphisms of full-length endogenous retroviruses in humans. Curr Biol 11:1531–1535. doi: 10.1016/S0960-9822(01)00455-9. [DOI] [PubMed] [Google Scholar]
- 53.Dewannieux M, Harper F, Richaud A, Letzelter C, Ribet D, Pierron G, Heidmann T. 2006. Identification of an infectious progenitor for the multiple-copy HERV-K human endogenous retroelements. Genome Res 16:1548–1556. doi: 10.1101/gr.5565706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee YN, Bieniasz PD. 2007. Reconstitution of an infectious human endogenous retrovirus. PLoS Pathog 3:e10. doi: 10.1371/journal.ppat.0030010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bronson DL, Fraley EE, Fogh J, Kalter SS. 1979. Induction of retrovirus particles in human testicular tumor (Tera-1) cell cultures: an electron microscopic study. J Natl Cancer Inst 63:337–339. [PubMed] [Google Scholar]
- 56.Herbst H, Sauter M, Kuhler-Obbarius C, Loning T, Mueller-Lantzsch N. 1998. Human endogenous retrovirus (HERV)-K transcripts in germ cell and trophoblastic tumours. APMIS 106:216–220. doi: 10.1111/j.1699-0463.1998.tb01338.x. [DOI] [PubMed] [Google Scholar]
- 57.Herbst H, Sauter M, Mueller-Lantzsch N. 1996. Expression of human endogenous retrovirus K elements in germ cell and trophoblastic tumors. Am J Pathol 149:1727–1735. [PMC free article] [PubMed] [Google Scholar]
- 58.Lower R, Lower J, Frank H, Harzmann R, Kurth R. 1984. Human teratocarcinomas cultured in vitro produce unique retrovirus-like viruses. J Gen Virol 65(Pt 5):887–898. doi: 10.1099/0022-1317-65-5-887. [DOI] [PubMed] [Google Scholar]
- 59.Sauter M, Roemer K, Best B, Afting M, Schommer S, Seitz G, Hartmann M, MuellerLantzsch N. 1996. Specificity of antibodies directed against Env protein of human endogenous retroviruses in patients with germ cell tumors. Cancer Res 56:4362–4365. [PubMed] [Google Scholar]
- 60.Sauter M, Schommer S, Kremmer E, Remberger K, Dolken G, Lemm I, Buck M, Best B, Neumann-Haefelin D, Mueller-Lantzsch N. 1995. Human endogenous retrovirus K10: expression of Gag protein and detection of antibodies in patients with seminomas. J Virol 69:414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de la Hera B, Varade J, Garcia-Montojo M, Lamas JR, de la Encarnacion A, Arroyo R, Fernandez-Gutierrez B, Alvarez-Lafuente R, Urcelay E. 2013. Role of the human endogenous retrovirus HERV-K18 in autoimmune disease susceptibility: study in the Spanish population and meta-analysis. PLoS One 8:e62090. doi: 10.1371/journal.pone.0062090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tai AK, O'Reilly EJ, Alroy KA, Simon KC, Munger KL, Huber BT, Ascherio A. 2008. Human endogenous retrovirus-K18 Env as a risk factor in multiple sclerosis. Mult Scler 14:1175–1180. doi: 10.1177/1352458508094641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Agoni L, Guha C, Lenz J. 2013. Detection of human endogenous retrovirus K (HERV-K) transcripts in human prostate cancer cell lines. Front Oncol 3:180. doi: 10.3389/fonc.2013.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Contreras-Galindo R, Kaplan MH, Leissner P, Verjat T, Ferlenghi I, Bagnoli F, Giusti F, Dosik MH, Hayes DF, Gitlin SD, Markovitz DM. 2008. Human endogenous retrovirus K (HML-2) elements in the plasma of people with lymphoma and breast cancer. J Virol 82:9329–9336. doi: 10.1128/JVI.00646-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Young GR, Stoye JP, Kassiotis G. 2013. Are human endogenous retroviruses pathogenic? An approach to testing the hypothesis. Bioessays 35:794–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhao J, Rycaj K, Geng S, Li M, Plummer JB, Yin B, Liu H, Xu X, Zhang Y, Yan Y, Glynn SA, Dorsey TH, Ambs S, Johanning GL, Gu L, Wang-Johanning F. 2011. Expression of human endogenous retrovirus type K envelope protein is a novel candidate prognostic marker for human breast cancer genes. Cancer 2:914–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gonzalez-Hernandez MJ, Swanson MD, Contreras-Galindo R, Cookinham S, King SR, Noel RJ Jr, Kaplan MH, Markovitz DM. 2012. Expression of human endogenous retrovirus type K (HML-2) is activated by the Tat protein of HIV-1. J Virol 86:7790–7805. doi: 10.1128/JVI.07215-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Deshiere A, Joly-Beauparlant C, Breton Y, Ouellet M, Raymond F, Lodge R, Barat C, Roy MA, Corbeil J, Tremblay MJ. 2017. Global mapping of the macrophage-HIV-1 transcriptome reveals that productive infection induces remodeling of host cell DNA and chromatin. Sci Rep 7:5238. doi: 10.1038/s41598-017-05566-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Peden K, Emerman M, Montagnier L. 1991. Changes in growth properties on passage in tissue culture of viruses derived from infectious molecular clones of HIV-1LAI, HIV-1MAL, and HIV-1ELI. Virology 185:661–672. doi: 10.1016/0042-6822(91)90537-L. [DOI] [PubMed] [Google Scholar]
- 70.Shugars DC, Smith MS, Glueck DH, Nantermet PV, Seillier-Moiseiwitsch F, Swanstrom R. 1993. Analysis of human immunodeficiency virus type 1 nef gene sequences present in vivo. J Virol 67:4639–4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chesebro B, Wehrly K, Nishio J, Perryman S. 1992. Macrophage-tropic human immunodeficiency virus isolates from different patients exhibit unusual V3 envelope sequence homogeneity in comparison with T-cell-tropic isolates: definition of critical amino acids involved in cell tropism. J Virol 66:6547–6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Buchacher A, Predl R, Strutzenberger K, Steinfellner W, Trkola A, Purtscher M, Gruber G, Tauer C, Steindl F, Jungbauer A, et al. . 1994. Generation of human monoclonal antibodies against HIV-1 proteins: electrofusion and Epstein-Barr virus transformation for peripheral blood lymphocyte immortalization. AIDS Res Hum Retroviruses 10:359–369. doi: 10.1089/aid.1994.10.359. [DOI] [PubMed] [Google Scholar]
- 73.Butler SL, Hansen MS, Bushman FD. 2001. A quantitative assay for HIV DNA integration in vivo. Nat Med 7:631–634. doi: 10.1038/87979. [DOI] [PubMed] [Google Scholar]
- 74.Manganaro L, Pache L, Herrmann T, Marlett J, Hwang Y, Murry J, Miorin L, Ting AT, Konig R, Garcia-Sastre A, Bushman FD, Chanda SK, Young JA, Fernandez-Sesma A, Simon V. 2014. Tumor suppressor cylindromatosis (CYLD) controls HIV transcription in an NF-κB-dependent manner. J Virol 88:7528–7540. doi: 10.1128/JVI.00239-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]