

ABSTRACT
Conserved across the family Herpesviridae, glycoprotein B (gB) is responsible for driving fusion of the viral envelope with the host cell membrane for entry upon receptor binding and activation by the viral gH/gL complex. Although crystal structures of the gB ectodomains of several herpesviruses have been reported, the membrane fusion mechanism has remained elusive. Here, we report the X-ray structure of the pseudorabies virus (PrV) gB ectodomain, revealing a typical class III postfusion trimer that binds membranes via its fusion loops (FLs) in a cholesterol-dependent manner. Mutagenesis of FL residues allowed us to dissect those interacting with distinct subregions of the lipid bilayer and their roles in membrane interactions. We tested 15 gB variants for the ability to bind to liposomes and further investigated a subset of them in functional assays. We found that PrV gB FL residues Trp187, Tyr192, Phe275, and Tyr276, which were essential for liposome binding and for fusion in cellular and viral contexts, form a continuous hydrophobic patch at the gB trimer surface. Together with results reported for other alphaherpesvirus gBs, our data suggest a model in which Phe275 from the tip of FL2 protrudes deeper into the hydrocarbon core of the lipid bilayer, while the side chains of Trp187, Tyr192, and Tyr276 form a rim that inserts into the more superficial interfacial region of the membrane to catalyze the fusion process. Comparative analysis with gBs from beta- and gamma-herpesviruses suggests that this membrane interaction model is valid for gBs from all herpesviruses.
IMPORTANCE Herpesviruses are common human and animal pathogens that infect cells by entering via fusion of viral and cellular membranes. Central to the membrane fusion event is glycoprotein B (gB), which is the most conserved envelope protein across the herpesvirus family. Like other viral fusion proteins, gB anchors itself in the target membrane via two polypeptide segments called fusion loops (FLs). The molecular details of how gB FLs insert into the lipid bilayer have not been described. Here, we provide structural and functional data regarding key FL residues of gB from pseudorabies virus, a porcine herpesvirus of veterinary concern, which allows us to propose, for the first time, a molecular model to understand how the initial interactions by gBs from all herpesviruses with target membranes are established.
KEYWORDS: alphaherpesvirus, class III fusion protein, entry, fusion loops, fusion protein, glycoprotein B, herpesvirus, membrane fusion, pseudorabies virus
INTRODUCTION
The family Herpesviridae contains a large number of enveloped, double-stranded DNA viruses, which are classified into alpha-, beta-, and gammaherpesvirus subfamilies based on their evolutionary relationships and biological properties (1). Pseudorabies virus (PrV) (suid alphaherpesvirus 1), the etiological agent of Aujeszky's disease in swine (2), belongs to the Alphaherpesvirinae, which also includes human pathogens, such as herpes simplex virus 1 (HSV-1) and HSV-2 and varicella-zoster virus (VZV). PrV has become a useful model for studying the biology of alphaherpesviruses in general (3).
Herpesviruses enter cells by fusion of their envelope with the host cell plasma membrane or with the membrane of an endocytic vesicle, depending on the virus and the cell type (4, 5). While many enveloped viruses require only one or two proteins to mediate receptor binding and entry, herpesviruses rely on the concerted actions of at least four glycoproteins. Distinct viral proteins first engage specific cellular receptors in an interaction that provides a trigger for membrane fusion (e.g., glycoprotein D [gD] in HSV-1/2 and PrV, gO in the betaherpesvirus human cytomegalovirus [HCMV], and gp42 in the gammaherpesvirus Epstein-Barr virus [EBV]) (6–9). The merger of the viral and cellular membranes is then catalyzed by fusion machinery that is conserved across the herpesvirus family and consists of gB and a heterodimeric complex of membrane-bound gH, in association with the anchorless gL (gH/gL) (reviewed in references 10 and 11).
The molecular basis of the entry mechanism has been best described for alphaherpesviruses. In HSV-1, the cascade begins with gD binding to cellular receptors, such as herpesvirus entry mediator (HVEM), nectin-1, or 3-O-sulfonated heparin sulfate (reviewed in reference 12). This interaction leads to a conformational change in gD (13, 14) that is believed to enable its interaction with the gH/gL complex, which in turn triggers gB to carry out membrane fusion (15, 16). It should be noted that unlike HSV-1, PrV does not require gD for direct viral cell-cell spread and gB-induced cell-cell fusion (17–19). Moreover, PrV virions can acquire the ability to infect cells in the absence of gD, gL, or the N-terminal domain of gH by compensatory mutations in other envelope glycoproteins (20, 21).
gB was proposed to be the bona fide fusion protein of herpesviruses due to its structural homology with the fusogenic G protein of the otherwise unrelated vesicular stomatitis virus (VSV) (22). Together with VSV G and the baculovirus fusion protein gp64 (23), gB was classified as a class III fusion protein. Viral fusion proteins are in general presented in a metastable prefusion state on the viral membrane. Upon an activation signal, they undergo conformational rearrangements unmasking the initially buried hydrophobic regions (fusion peptide [FP] or fusion loops [FLs]) for interactions with the target membrane, resulting in simultaneous anchorage of the fusion protein in the viral and cellular membrane at opposite ends of the protein. This extended intermediate is unstable and rapidly folds back into a “hairpin,” an energetically more favorable postfusion conformation (24, 25).
gB is the most conserved envelope glycoprotein of herpesviruses, although only ∼5% of its residues are identical across the entire herpesvirus family. The level of conservation is higher within each subfamily, and PrV and HSV-1 gBs share 50% amino acid sequence identity. The X-ray structures determined for the postfusion gB ectodomains from HSV-1 (26), EBV (27), and HCMV (28, 29) demonstrate conservation of the three-dimensional structural organization. Although the crystal structure of gB in a prefusion state has not yet been determined, a low-resolution structure of full-length HSV-1 gB in a state different from the postfusion conformation was obtained by cryo-electron tomography (30). VSV G protein has been crystallized in both pre- and postfusion conformations. The former revealed a trimeric molecule with the FLs pointing toward the viral membrane and with a different spatial organization of domains than the postfusion, low-pH form (31). Hypothetical models of the prefusion gB using the VSV G conformation as a template have been proposed (27, 32), but supporting structural data are still lacking. The high structural conservation observed for gB also applies to the gH/gL complex, as illustrated by the X-ray structures of the gH/gL ectodomains from HSV-2, EBV, VZV, and a core domain of PrV gH (33–36), which revealed a common fold despite low sequence conservation (37). gH/gL has no structural resemblance to any known fusion protein but was shown to play a role in regulation of the fusion activity of gB (16, 37).
Membrane-interacting regions of fusion proteins are typically well conserved within the virus family and are rich in hydrophobic and aromatic residues that insert into the outer leaflet of a cell membrane (38). In class I fusion proteins, they are called FPs, which can be N-terminal peptides or an internal loop near the N terminus, as in the Ebola virus envelope glycoprotein GP2, for example (39) (to avoid confusion, we reserve the term FL for the loops of class II and III fusion proteins, and we refer to all class I membrane-interacting regions as fusion peptides, even when they correspond to an internal loop). FPs were shown to adopt a different structure upon insertion into target membranes (reviewed in reference 40). The FLs of class II and class III fusion proteins, in contrast, are conformationally constrained by being part of a larger structured β-barrel domain and appear to largely maintain their structure upon membrane insertion. In class II fusion proteins, the membrane-interacting surface can be composed of residues from one (flaviviruses and alphaviruses [41, 42]), two (rubella virus and phleboviruses [43, 44]), or three (hantaviruses [45]) FLs, while the known class III fusion proteins use two FLs to interact with membranes. FLs of herpesviruses are unusual because their amino acid sequences are poorly conserved even within subfamilies. Therefore, their identification was impossible until structural data became available. Residues exposed at the tips of the recombinant crystallized HSV-1 gB ectodomain (26) were proposed to form the FLs based on structural homology with the well-defined FLs of the VSV G protein (46, 47). A similar organization of the FLs was observed in the reported gB structures, with the difference that the aromatic and apolar residues are predominantly presented at the sides of the HSV-1 FLs while they protrude from the tips of the EBV and HCMV FLs. These exposed hydrophobic residues in EBV and HCMV gBs caused the recombinant ectodomains to aggregate, forming rosette-like structures (48, 49). Mutagenesis of the hydrophobic residues within the FL, while abolishing their fusion function, was essential for solubility of these proteins for structure determination (49, 50). In contrast, HSV-1 gB (51) and PrV gB (this study) exhibit less hydrophobic tips, form soluble trimers, and can be studied and crystallized as functional wild-type (WT) proteins.
Extensive mutagenesis studies have been performed on HSV-1 gB, demonstrating that the hydrophobic residues Trp174, Phe175, and Tyr179 in FL1 and Ala261 in FL2, as well as polar and charged residues, such as His263 and Arg264, presented at the sides of FL2, play an important role in fusion (51–54). The hydrophobic residues were proposed to form a patch that inserts into the membrane, while the His and Arg side chains were speculated to interact with phospholipid head groups without penetrating deeper into the membrane. In support of this model, the low-resolution structure of HSV-1 gB ectodomains bound to liposomes and obtained by cryo-electron tomography showed that the interactions of the HSV-1 gB ectodomains are limited to the outer membrane leaflet. This model predicted that the FLs would insert into the bilayer at an oblique angle, indicating that the residues from β-strands leading to the FLs may be involved in the interactions with lipids (55). HSV-1 gB FL residues Trp174, Tyr179, His263, and Arg264, furthermore, were shown to be the contact sites for interactions of the ectodomain with liposomes (54).
Cholesterol (CH) is a lipid that is ubiquitously present in mammalian cell membranes, where it plays an important role in the entry of many viruses (56), including PrV and HSV-1 (reviewed in references 57 to 59). The HSV-1 gB ectodomain was shown to bind to liposomes only in the presence of cholesterol (54), and full-length HSV-1 gB expressed in cells was found to associate with lipid rafts (60).
While the current HSV-1 model has provided an important step forward in understanding how gB binds to membranes, it has been unclear whether a similar model of insertion would hold true for other gB proteins due to the poor conservation of the FLs, highlighting the need to study each gB individually and to perform comparative analyses. With this in mind, we sought to enlarge the structural and functional repertoire available for alphaherpesviruses by carrying out studies on PrV gB. We report here the X-ray crystal structure of the PrV gB ectodomain at 2.7-Å resolution and demonstrate that the recombinant ectodomains bind to liposomes in a cholesterol-dependent fashion. The association with liposomes occurs via the trimer tip that contains the FLs, resembling the shallow insertion observed for HSV-1 gB. We designed the PrV gB mutagenesis to complement what had already been reported for HSV-1 gB and to target all aromatic and hydrophobic residues within the FLs, some that are unique to PrV and some that are conserved in HSV-1 gB, as well. The resulting pattern of active and inactive FL mutants matches in part what had been observed previously for HSV-1 gB, but with some interesting differences that shine new light on the way the FLs insert into membranes. Based on our results, we propose a model in which the tip of FL2 penetrates deeper into the lipid bilayer, reaching into the hydrophobic core, while residues on the sides of the FL1 and FL2 tips position their side chains within the same plane, inserting into the amphipathic, interfacial region of the membrane. Comparative analysis with the FLs of HCMV and EBV gBs further suggests that, in spite of considerable sequence divergence, this model could be used to describe in general the way herpesvirus gBs interact with membranes.
RESULTS
PrV gB expression, crystallization, and structure determination.
The ectodomain expression construct of PrV gB (suid alphaherpesvirus 1, strain Kaplan [PrV-Ka]; GenBank accession number AEM64049.1) encodes gB residues 59 to 756 (Fig. 1A). The construct was designed to exclude the gB signal sequence (residues 1 to 58), which was replaced by the Drosophila Bip signal peptide, present in the expression vector and known to drive efficient expression of heterologous proteins (61). The expression construct ends with double Strep-tag II, as described in Materials and Methods, added just before the hydrophobic membrane-proximal region (MPR) (residues 757 to 800), which is followed by the transmembrane region (residues 801 to 821) and the cytoplasmic domain (residues 822 to 916) in the intact protein.
FIG 1.

PrV gB ectodomain structure. (A) Schematic representation of the PrV gB expression construct. The PrV gB signal peptide (residues 1 to 58) was replaced by the Drosophila BiP secretion signal, which was cleaved off and not part of the secreted protein. The secreted PrV gB ectodomain used for crystallization contains residues 59 to 756, followed by a double Strep-tag II (DST). Regions forming the five gB domains are labeled with Roman numbers I to V below the bar and are colored as follows: domain I, blue; domain II, green; domain III, yellow; domain IV, orange; and domain V, red. Two linker regions (residues 147 to 154 and 493 to 528) are shown in gray. The dashed lines mark the regions that were unresolved in the gB structure. The location of the furin cleavage site (RRARR; residues 501 to 505) is indicated. (B) SDS-PAGE analysis of the recombinant PrV gB. The Coomassie blue-stained 4 to 20% SDS-PAGE gel shows the purified PrV gB ectodomain under nonreducing (lanes 1 and 2) and reducing (lanes 3 and 4) conditions. The samples in lanes 2 and 4 were treated with Endo D. The N-terminal and C-terminal fragments generated by furin cleavage are labeled gBb and gBc, and the uncleaved gB is marked gBa. DTT, dithiothreitol. (C) Structure of the PrV gB monomer. The molecule is colored from blue (N terminus) to red (C terminus). The locations of the N and C termini are indicated, and domains are labeled with Roman numbers I to V. The C terminus is followed by the 50-residue-long MPR, not present in the expression construct, leading to the transmembrane anchor; the anticipated location of the membrane is indicated by the arrow. Fusion loops presented by domain I are marked by asterisks. The extra N-terminal residues that were resolved for the first time in this structure form a strand labeled β1. The linker connecting domains II and III, which is not visible in our structure, is plotted as a yellow dotted line to indicate the putative location of the furin cleavage site (orange star). The location of the glycosylation site Asn264, to which a single NAG residue is attached, is indicated by the yellow star. (D) Structure of the PrV gB trimer. The colors of the protomers are the same as in panel C. Ribbon and molecular-surface representations are shown. The N and C termini of the same protomer represented in panel B are labeled. Strand β29, which runs antiparallel to strand β1, is indicated (domain IV). PyMOL (103) was used to create the structures shown in panels C and D.
The PrV gB ectodomain was expressed using the stably transfected Drosophila Schneider 2 (S2) cell line, as described previously (62). After affinity and size exclusion chromatography (SEC), we obtained 8 to 12 mg of pure protein from 1 liter of cell culture. The protein eluted as a single peak from a SEC column and exhibited no signs of aggregation. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis of the purified ectodomain showed a single band migrating just below the 100-kDa marker under nonreducing conditions (Fig. 1B, lane 1). PrV gB contains a furin cleavage site, 501RRARR505, and the recombinant protein is cleaved in S2 cells, as demonstrated by the presence of two protein bands of lower molecular mass under reducing conditions (∼60-kDa and ∼40-kDa fragments labeled, respectively, gBb and gBc) (Fig. 1B, lane 3).
Compared to its 100-kDa apparent molecular mass, the polypeptide chain of the gB ectodomain has a calculated mass of 82 kDa, corresponding to the 49-kDa N-terminal and 33-kDa C-terminal furin cleavage products, which indicates the presence of posttranslational modifications in the mature protein. There are six predicted N-glycosylation sites (four in the N-terminal and two in the C-terminal fragments), and a shift to a lower molecular mass is indeed observed for both fragments upon treatment with a deglycosidase (Fig. 1B, lanes 2 and 4). Drosophila S2 cells add 1- to 2-kDa simple mannose core structures to form N-linked sugars (63), accounting for the presence of several N-linked sugars in the expressed protein.
The purified gB ectodomains crystallized easily under numerous conditions, but the crystals were fragile and of poor diffraction quality. Upon enzymatic deglycosylation with endo-β-N-acetylglucosaminidase (Endo D), we obtained diffraction quality crystals in 0.1 M Tris, pH 8.5, 7% polyethylene glycol (PEG) 4000, 0.6 M LiCl. Diffraction data were collected and processed as described in Materials and Methods. The crystals belong to the H3 space group (a = 99.9 Å, b = 99.9 Å, c = 272.9 Å; α = β = 90°; γ = 120°), with one gB protomer per asymmetric unit. Crystallographic statistics of data processing and structure refinement are given in Table 1.
TABLE 1.
X-ray diffraction data collection and refinement for PrV gB

Outer-shell values are given in parentheses.
The Rfree test set was composed of 5% randomly chosen reflections.
Number of protein/water atoms.
Overall/protein/water B factors.
Preferred/allowed/outliers as calculated by Coot (109).
PrV gB forms a typical class III postfusion trimer.
The PrV gB ectodomain folds into an ∼16-nm by ∼8-nm trimeric spike, resembling the structures reported for the postulated postfusion conformation of the HSV-1 (26), EBV (27), and HCMV (28, 29) gB ectodomains. Briefly, the N terminus of the protein (amino acid 113) is located at the top end of the trimer, with the polypeptide chain running down along the entire length of the spike, folding into domain I, which carries the FLs exposed at the base of the molecule (Fig. 1C). Domain II is positioned laterally, adopting a fold reminiscent of the pleckstrin homology domains. The furin cleavage site is located in the flexible linker that connects domains II and III and could not be resolved in our structure. Domain III contains the prominent, centrally located helix that extends to the top of the molecule, followed by domain IV, also known as the “top” or “crown” domain. Residues 697 to 750 form a stretched chain that packs tightly into the crevice formed by the other two protomers. This so-called “domain V” has the appearance of a zipper that seals the trimer (Fig. 1D), resulting in the C terminus of the ectodomain (amino acid 750) being brought into close proximity to the FLs.
Although the expression construct encodes gB residues 59 to 756, the first residue resolved in the crystal structure was Arg113, consistent with the highly flexible N-terminal end of the protein, which is rich in glycine and proline residues. The same applies to HSV-1, EBV, and HCMV gBs, in which the N-terminal region of the crystallized construct also was not resolved. The other PrV gB regions that were not built into the structure are the above-mentioned unstructured segment that contains the furin cleavage site and connects domains II and III (residues 478 to 521) and the C-terminal 6 residues of the ectodomain (residues 750 to 756), which are followed by the double Strep-tag II in the expression construct. The PrV gB structure, however, does reveal 3 residues at the very N terminus (residues 113 to 116), which had not been observed in the other gB structures. This segment forms a short β-strand (β1) in domain IV that packs against β29 in an antiparallel fashion (Fig. 1C and D).
Of six potential N-glycosylation sites, we observed densities for the sugar moieties attached to Asn264 (domain I), Asn444 (domain II), and Asn636 (domain IV). The electron density was of sufficient quality to allow building of one N-acetylglucosamine (NAG) residue only to Asn264 (Fig. 1C and D).
As anticipated, PrV gB showed higher structural conservation with the HSV-1 homolog than with the more distant gBs of EBV or HCMV. The root mean square deviation (RMSD) for the superposition of the PrV and HSV-1 structures is 1.04 Å for C-α atoms (Table 2). The main difference between the PrV and HSV-1 ectodomains regards the gross organization of the spike resides in the position of domain IV, which appears slightly rotated when the structures are superimposed on domains I and II. The domain IV disposition is even more evident compared to those of HCMV and EBV gBs (Fig. 2).
TABLE 2.
Sequence and structural comparison of PrV and HSV-1/HCMV/EBV gB ectodomains
| Comparison | Sequence identity (%)a | nA/nTb | RMSD (Å)c |
|---|---|---|---|
| PrV vs HSV-1 | 50.1 | 524/568 | 1.04 |
| PrV vs HCMV | 25.6 | 508/535 | 3.39 |
| PrV vs EBV | 25.3 | 496/530 | 3.72 |
Percent identity was calculated using the shorter sequence as the denominator.
nA and nT, numbers of aligned and total atoms, respectively.
The RMSDs were calculated for C-α atoms in PyMOL (103).
FIG 2.

Structural comparison of gB ectodomains. The PrV gB ectodomain structure, shown in gray cartoon representation, was superimposed on those of HSV-1 (PDB accession number 2GUM; light blue), HCMV (PDB accession number 5C6T; green), and EBV (PDB accession number 3FVC; purple) gBs, using the residue range that corresponds to domains I and II of PrV, HSV-1, HCMV, and EBV gBs (residues 167 to 458, 154 to 473, 89 to 388, and 133 to 415, respectively). The alignment was done by superimposing domains I and II to highlight the disposition of the top, domain IV, which is not as obvious when the superimposition is applied to the entire molecule. The alignments were done in PyMOL (103). Domains are labeled with Roman numbers. Each superimposition is shown in two orientations, looking at the monomer from the front (top) and at domain IV from the top of the spike (bottom). The red pentagons are placed in the same loop of the PrV gBs to serve as a reference point for the movement of the superimposed domain IV, the direction of which is indicated by the arrows.
The PrV gB ectodomain requires cholesterol in the membrane for binding.
The recombinant PrV gB ectodomain used for crystallization was tested for binding to liposomes with different compositions in coflotation experiments in density gradients, as described in detail in Materials and Methods. Briefly, the protein-liposome mixture, adjusted to 36% iodixanol (OptiPrep), was loaded below the 20% iodixanol layer, which was then overlaid with buffer and subjected to ultracentrifugation. Liposomes float and are found at the top of the gradient, while the protein alone sediments and remains at the bottom of the tube. Protein bound to liposomes cofloats with liposomes and in this case partitions to the top of the gradient. All the gradient fractions were analyzed for the presence of the protein in the control experiments, demonstrating that gB was found either at the top or the bottom of the gradient, which is why only these two fractions were analyzed further.
Liposomes containing 60% 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 40% CH were prepared. This is the same composition used for testing HSV-1 gB ectodomain interactions with membranes (54, 64). The top and bottom factions were analyzed by Western blotting (WB) with a reagent that specifically binds to the Strep affinity tag. The WT PrV gB ectodomain was found in the top fraction only when liposomes were present and sedimented to the bottom of the tube in the absence of liposomes (Fig. 3A). Complexes made of the WT PrV gB ectodomain and liposomes containing DOPC and increasing molar amounts of CH (100% DOPC, 80% DOPC plus 20% CH, or 60% DOPC plus 40% CH) were prepared and tested next. gB ectodomains bound only to the latter type of liposomes (Fig. 3B), suggesting that there is a threshold CH concentration required for the association. To determine if the lack of binding at lower CH concentrations was due to the requirement for another lipid rather than insufficient CH concentration, a series of liposomes made of mixtures of DOPC, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), and sphingomyelin (SM) was prepared in the presence of 40% CH or without CH. DOPE alone or mixed with CH does not form liposomes but assembles into lipid nanotubes due to its inverted hexagonal shape (65), which is why these lipid mixtures were omitted. The results of the flotation experiments showed that WT gB was present in the top fraction, i.e., bound to the liposomes only in the presence of CH (Fig. 3C), reinforcing the conclusion that CH is essential for PrV gB ectodomain interactions with liposomes.
FIG 3.
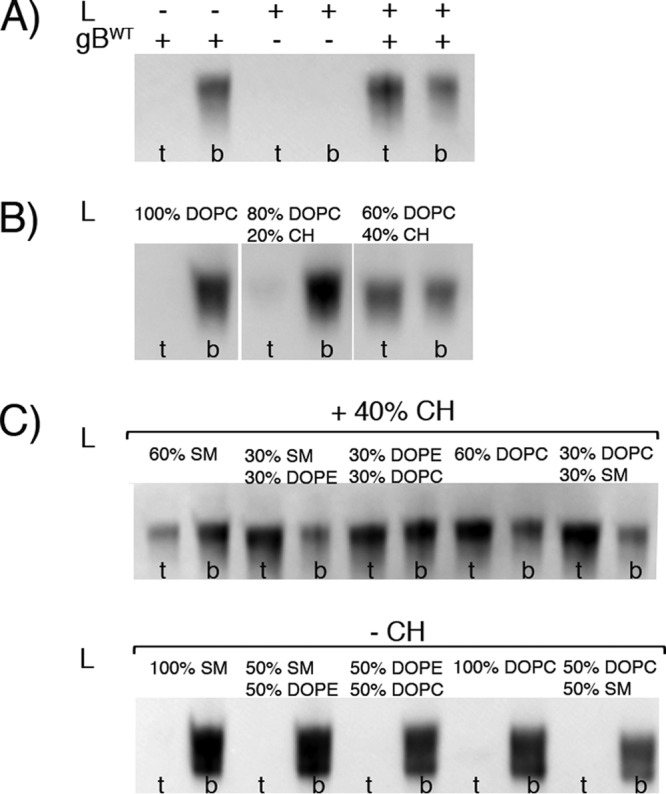
WT PrV gB ectodomain interactions with liposomes. (A) WT gB ectodomains bind to the DOPC-CH liposomes. The presence (+) of liposomes (L) and protein is indicated. Aliquots of the top (t), i.e., liposome, and bottom (b), i.e., unbound protein, fractions were analyzed by SDS-PAGE, and the Strep-tag present on gB was detected by WB using Streptactin-HRP conjugate. The WT protein is found in the top fraction only in the presence of liposomes with 60% DOPC and 40% CH. (B) Coflotation of WT gB and DOPC liposomes with increasing amounts of cholesterol. The liposome composition is indicated at the top. gB is detected in the top fraction only when 40% CH is present. (C) Coflotation of WT gB and liposomes made of combinations of lipids found in the plasma membrane. The CH concentration was fixed at 40% or 0% (top and bottom, respectively), and one or two more lipids were added, resulting in a total of 5 lipid compositions. The protein was detected bound to all of the CH-containing liposomes, while it was found in the unbound fraction in the absence of CH.
A sample of gB mixed with liposomes was visualized by negative-stain electron microscopy (EM) (Fig. 4A), revealing liposomes decorated by protein arrays, forming honeycomb-like structures in which the individual trimers appeared to be in contact with each other. Cryo-EM images of the same sample (Fig. 4B) showed individual gB trimers packing tightly and inserting into the liposomes via the spike end that contains the FLs (domain I), as was observed for the HSV-1 protein (55).
FIG 4.
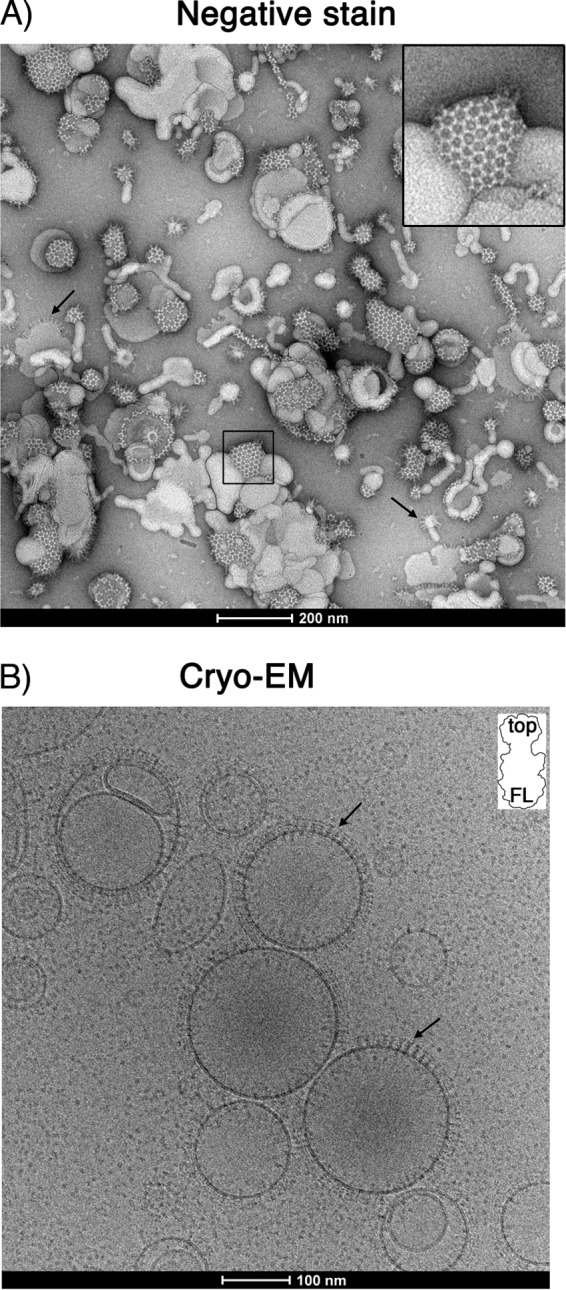
(A) Negative-stain EM image of liposomes incubated with PrV gB ectodomains. Characteristic hexagonal structures made of gB covering the liposomes (60% DOPC, 40% CH) are observed, representing the top view on the gB spikes. Individual trimers establish an extensive network of lateral interactions, giving rise to the array appearance of the protein coat. Liposomes showing side views of the gB trimers projecting away from the liposome surface are marked by arrows. An enlarged view of the boxed area is shown in the upper right corner. (B) Cryo-EM of PrV gB ectodomains bound to liposomes. The arrows indicate individual gB trimers in which the top end of the spike is better resolved, indicating that the molecule binds to the liposomes via the other end, i.e., the one carrying the FLs. (Inset) Shape of a gB trimer, with “top” representing the domain IV end of the spike and “FL” marking the base of domain I and the location of FLs.
Selection of PrV gB FL residues and design of mutagenesis studies.
The side chains of Trp and Phe, as well as smaller hydrophobic residues, such as Leu or Ile, have favorable energies for penetration into the hydrophobic membrane core, made exclusively of lipid tails (66), while amphipathic Tyr and His side chains preferentially partition in the interfacial membrane region, localized between the hydrocarbon core and the aqueous phase (66, 67). Trp is also often found in this region (68). The interfacial membrane region is amphipathic itself due to the presence of carbons from the beginnings of the apolar lipid tails and of polar phosphate and glycerol groups (69). A Phe side chain can penetrate into the membrane core and could thus be emulated by Trp, but not by Tyr, due to the presence of a polar hydroxyl group at the distal tip of the side chain. The interfacial region, on the other hand, would be expected to accommodate aromatic residues, such as Tyr, Trp, Phe, or His (the last in its unprotonated form).
To shed light on how the side chains of individual FL residues interact with membranes, several changes were introduced. Each of the aromatic or hydrophobic residues found in PrV gB FLs (Trp187, Tyr192, Tyr267, Ile270, Phe275, Tyr276, and His277) (Fig. 5) was individually mutated to Ala, a change that resulted in the replacement of the bulky hydrophobic side chain with a methyl group, or to a different aromatic residue with similar chemical structure: Trp was changed to Phe or His, Phe to Trp or Tyr, His to Trp, and Tyr to Phe (Table 3). This was done to probe if the residue was more likely to insert more deeply into the hydrocarbon core or to remain in the polar region of the membrane. Three of the mutagenized residues are unique to PrV gB (Ile270, Tyr276, and His277), Tyr267 is conserved but has not been mutated, and the changes introduced at the conserved positions were not analyzed previously in HSV-1 gB.
FIG 5.
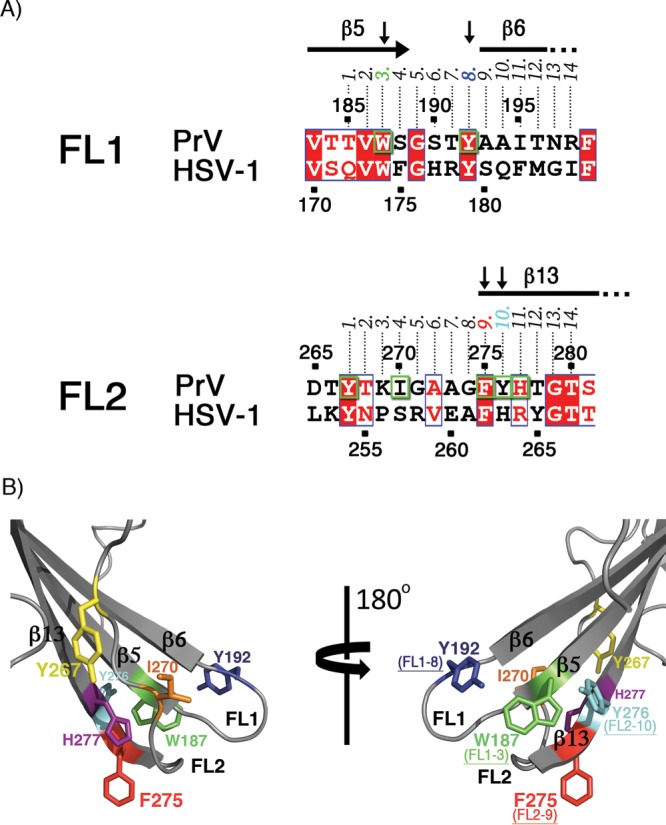
(A) Sequence alignment of PrV and HSV-1 gB fusion loop regions. Identical residues are shown as white letters on a red background and residues with similar physicochemical properties in red on a white background. Residues mutated in this study (Trp187, Tyr192, Tyr267, Ile270, Phe275, Tyr276, and His277) are boxed in green. The numbers 1 to 14 above the alignment indicate the position of each residue in FL1 or FL2. The residues found to be important for binding to liposomes are indicated by vertical arrows, and their corresponding numbers (3 and 8 in FL1 and 9 and 10 in FL2) are colored according to the scheme used in panel B. (B) Structural representation of the mutated PrV gB FL residues. The aromatic residues in FL1 and FL2 are indicated with their side chains in stick representation. These residues were individually mutated as shown in Table 3. For clarity, two views of the FLs are presented, and the remainder of the protein is omitted. The residues shown to be required for binding to liposomes are also indicated, with their corresponding positions within FL1 and FL2.
TABLE 3.
PrV gB mutants with single mutations in fusion loops in liposome binding and functional assays

Single point mutations introduced in FL1 and FL2 of PrV gB are indicated. The variants containing mutations to alanine are shaded.
2L indicates that liposomes were made of 2 lipids: 60% DOPC and 40% CH. ± indicates the presence of a faint band in the liposome fraction (Fig. 7A), indicating weak binding of the protein to liposomes.
4L indicates that liposomes were made of 4 lipids: 20% DOPC, 20% DOPE, 20% SM, and 40% CH.
The expression yield and purity of the recombinant ectodomains are indicated relative to those of the WT protein; NC, no change.
± indicates marginal complementation of PrV-ΔgB virus (Fig. 9A).
NT, not tested.
Side chains of PrV gB FL residues Trp187, Tyr192, Phe275, and Tyr276 mediate binding to liposomes and form a hydrophobic patch.
To facilitate generation of mutant proteins, recombinant PrV gB ectodomains were expressed after transient transfection of Expi293F mammalian cells, and the same type of liposomes (60% DOPC and 40% CH) as reported for the HSV-1 gB ectodomain variants were used in the liposome flotation assay (54). The variants did not differ from the WT protein in terms of expression yields, behavior during affinity and size exclusion chromatography, and cleavage by cellular furin (Fig. 6).
FIG 6.

SDS-PAGE analysis of purified PrV gB ectodomain variants. Shown is SDS-PAGE analysis of the PrV gB recombinant ectodomains under nonreducing (top) and reducing (bottom) conditions. Two micrograms of each purified protein was loaded on a 4 to 20% gradient gel. The proteins were stained with Coomassie blue. The three bands resolved under reducing conditions correspond to the uncleaved protein and the N-terminal and C-terminal fragments produced by furin cleavage. There were no observable differences in purity and cleavage patterns between the FL variants and the WT PrV gB.
Replacement of the selected residues with alanine had three consequences (Table 3 and Fig. 7A). The W187A, Y192A, F275A, and Y276A variants completely lost the ability to bind to liposomes and were present exclusively in the bottom fraction. I270A and H277A floated like the WT protein, indicating that the mutation exerted no effect on liposome binding. Y267A showed intermediate behavior, with very low binding to liposomes, at the limit of WB detection.
FIG 7.

(A) Liposome binding of PrV gB FL variants. Shown is the WB analysis of gradient fractions. t, top fraction (liposomes and bound gB); b, bottom fraction (unbound protein). The liposomes were made of 60% DOPC and 40% CH. The arrows indicate the presence of very weak signals in the top fractions observed for Y192F, Y267A, and Y276F. Streptactin-HRP conjugate was used for gB detection. (B) Solvent-accessible surface representation of the bottom tip of the PrV gB spike carrying fusion loops. (Left) The FL residues mutated in this study are shown in the same colors as in Fig. 5B. The stars indicate the positions of the conserved Gly274. The black contour indicates the edges of the continuous hydrophobic surface formed by the side chains of Trp187, Tyr192, Gly274, Phe275, and Tyr276. (Middle) Solvent-accessible surface colored according to hydrophobicity. The same PrV gB surface is shown, with the colors corresponding to the hydrophobicity of the individual side chains calculated using the Eisenberg hydrophobicity scale (104), with white corresponding to the most hydrophobic and dark yellow to the most hydrophilic/charged side chains. (Right) Electrostatic solvent-accessible surface. The same surface is shown, colored according to the electrostatic potential calculated with the ABPS tool (105), with blue and red corresponding to negative and positive potentials, respectively. All the images were generated in PyMOL (103).
Mutation to other aromatic residues also had different outcomes. Trp187 was essential for association of the ectodomain with liposomes, and changes to Phe or His yielded a protein that did not cofloat with liposomes. Y192F was only weakly detected in the liposome fraction, similar to what was observed for Y267A, while Y267F and F275W bound to liposomes comparably to WT gB. Interestingly, in contrast to F275W, F275Y did not associate with liposomes, indicating that the presence of the hydroxyl group in F275Y may impair the ability of the protein to interact with lipids. Y276F showed very weak binding, resembling that of Y192F and Y267A. The amino acid at position 277 does not seem to be important for liposome binding, since the substitutions H277A and H277W had no effect (Fig. 7A).
The four residues Trp187, Tyr192, Phe275, and Tyr276, which did not tolerate substitutions to alanine and must have a bulky aromatic side chain in order to bind to liposomes, form a continuous hydrophobic and electrostatically neutral patch at the surface of the trimeric postfusion spike, as shown in Fig. 7B. The phenyl group of Phe275 appears protrude the most, while Trp187, Tyr192, and Tyr276 form a rim above (Fig. 5B).
PrV gB FL variant binding to liposomes correlates with fusion activity.
The fusogenic potentials of a subset of PrV gB FL mutants, including all the Ala variants, were assessed in eukaryotic cells using the corresponding full-length gB constructs, generated by site-directed mutagenesis of pcDNA-gB. Correct mutagenesis was verified by sequencing, and protein expression and processing were analyzed, respectively, by indirect immunofluorescence of permeabilized gB-expressing cells (Fig. 8A) and WB of whole-cell lysates (Fig. 8B). The PrV gB FL variants revealed WT-like behavior in both subcellular localization and processing by furin, with the exception of W187A and Y192A gBs. These two variants accumulated in larger structures in the cytoplasm and showed impaired furin cleavage, as indicated by lower levels of the furin-cleaved subunit (gBb) and more abundant uncleaved gB (gBa) (Fig. 8B).
FIG 8.

Expression of full-length PrV gB variants in RK13 cells. (A) Subcellular localization. RK13 cells were transfected with expression plasmids for wild-type gB (gB Ka) or the PrV gB FL mutants and analyzed by indirect immunofluorescence. One day after transfection, the cells were fixed with 3% paraformaldehyde and permeabilized with 0.1% Triton X-100. gB was detected using a gB-specific rabbit antiserum and Alexa Fluor 488-conjugated secondary antibodies. Green fluorescence was excited at 488 nm and recorded with a laser scanning confocal microscope (SP5; Leica, Mannheim, Germany). (B) Western blot analyses. Lysates of RK13 cells transfected with expression plasmids for gB Ka or gB variants containing FL single mutations were separated by SDS-PAGE under reducing conditions. Cells transfected with the empty vector pcDNA3 served as a negative control (Mock). The blots were incubated with the gB-specific monoclonal antibody C15-b1. Signals of uncleaved gB (gBa) or a furin-cleaved gB subunit (gBb) are labeled by arrows, and the molecular masses of marker proteins are indicated. As a loading control, the blot was incubated with an anti-alpha-tubulin monoclonal antibody.
To test the fusogenic potential of the mutated gB proteins, RK13 cells were cotransfected with plasmids encoding WT gB (Ka) or the mutated gB and gH/gL, as described previously (70, 71). In addition, an enhanced green fluorescent protein (EGFP) expression plasmid was cotransfected to facilitate evaluation of the assays by fluorescence microscopy (72). Transfection with plasmids encoding the WT proteins served as a positive control, and the results were set as 100%, while the empty expression vector pcDNA3 was used as a negative control.
Fusion assays revealed good correlation between the ability of the protein to bind to liposomes in vitro and its activity in fusion (Table 3 and Fig. 9A). Most of the variants that did not show association with liposomes were unable to induce cell-cell fusion, and all the variants with liposome binding properties supported efficient cell-cell fusion. Surprisingly, the three variants that showed only weak binding to liposomes, Y192F, Y267A, and Y276F, mediated fusion to an extent similar to that of WT gB.
FIG 9.
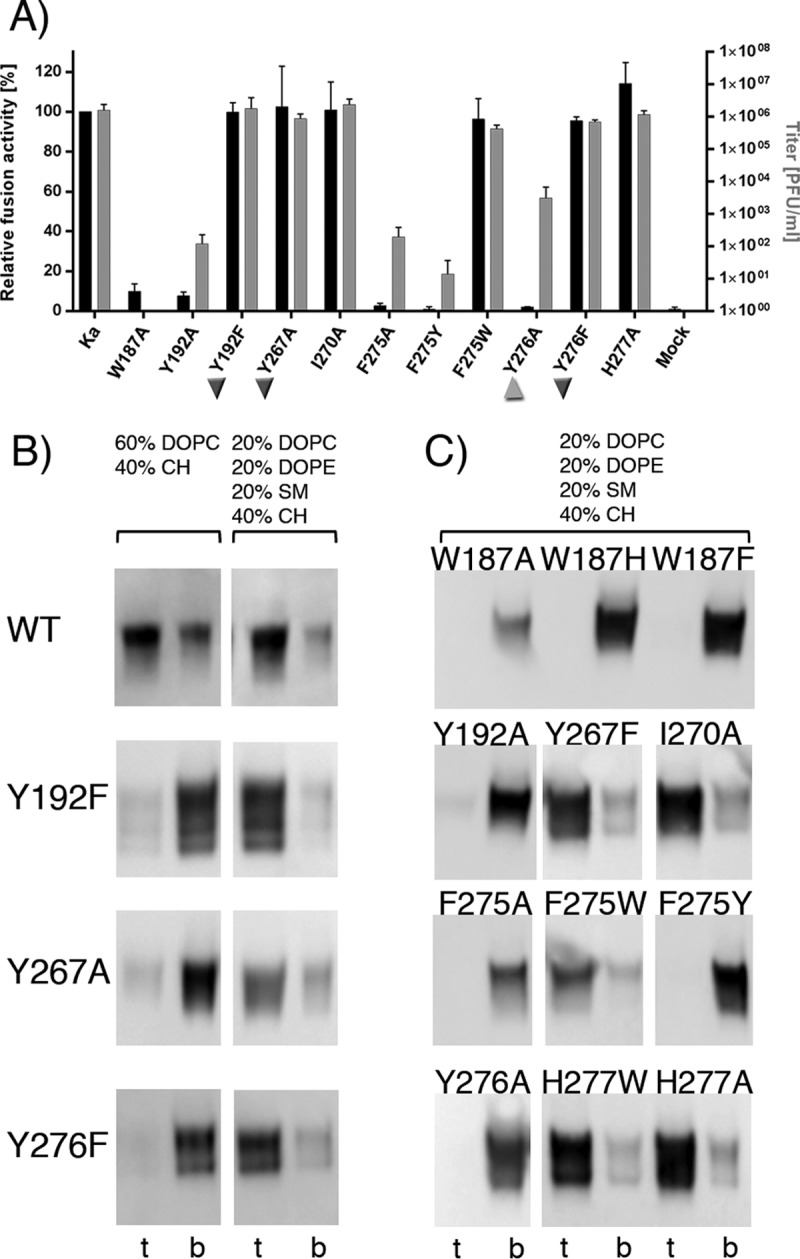
(A) Functional data obtained for the PrV gB FL variants. The results of the cell-cell fusion assay are plotted as black bars and expressed as percentages of the activity of the WT gB in combination with gH/gL, which was set to 100% (left y axis). Viral titers obtained in the trans-complementation assay are shown as gray bars (right y axis). Ka, PrV strain Kaplan, the WT protein, was used as a positive control. The inverted triangles mark the three variants that showed poor binding to liposomes while being functional in fusion and complementation assays. The triangle labels Y276A, which had a fusion-null phenotype in the cell-cell assay but was partially functional in the virus complementation assay. The error bars indicate standard deviations from three independent experiments. (B) Effect of liposome composition on binding of the WT and the Y192F, Y267A, and Y276F PrV gB variants. Two types of liposomes were used, 2L (60% DOPC and 40% CH) (left) and 4L (20% DOPC, 20% DOPE, 20% SM, and 40% CH) (right), and the top and bottom fractions were analyzed by WB, showing larger amounts of all the variants bound to the more complex 4L liposomes. (C) Flotation of PrV gB variants whose binding to liposomes is not affected by lipid composition. These variants showed the same pattern of binding to 4L liposomes as to 2L liposomes (Fig. 7A). Aliquots from the top and bottom fractions were analyzed by WB.
To test whether this discrepancy was due to the liposome composition used in the in vitro experiments (60% DOPC and 40% CH, referred to as “2L,” for two lipids present), the coflotation experiments were repeated with liposomes whose composition more closely resembled the composition of the plasma membrane (20% DOPC, 20% DOPE, 20% SM, and 40% cholesterol, referred to as “4L,” for four lipids present) (73–75). The three variants Y192F, Y267A, and Y276 indeed floated with 4L liposomes, like the WT protein (Fig. 9B), demonstrating that the previously observed weak association was due to the inadequate liposome composition and not the mutated protein. The association of the other FL variants did not change as a function of liposome composition (Fig. 9C). These data demonstrate for the first time significant differences in the capabilities of gB mutants to bind to liposomes as a function of lipid composition, underlining the importance of testing different liposome compositions and cross-checking biochemical data with functional data.
Since W187A and Y192A variants were inactive and showed impaired furin cleavage, we wanted to establish whether furin processing was important for the function of PrV gB in the particular functional assays used in this study. A gB variant with a 5-residue (RRARR) deletion in the furin site (gB-Δfurin) was created. WB analysis of gB-Δfurin expressed in RK13 cells confirmed that the protein was not cleaved, as indicated by a prominent band for uncleaved gB (gBa) and absence of bands corresponding to the furin-cleaved subunits (Fig. 10). gB-Δfurin was functional in cell-cell fusion assays and was able to complement gB-negative PrV to levels comparable to those of WT-gB (Fig. 10C). These data demonstrate that furin cleavage of PrV gB is not necessary for its function in cell-cell fusion and complementation. The observed fusion deficiency of the W187A and Y192A variants thus cannot be attributed to the reduced furin cleavage alone but could still be caused by impaired trafficking and low surface expression. Unlike HSV-1 gB, which is highly abundant at the cell surface, WT PrV gB exhibits rather low surface expression. Only around 4% of WT PrV gB is targeted to the plasma membrane, as determined by fluorescence-activated cell sorting (FACS) analysis (data not shown), making robust quantification complicated.
FIG 10.

Functional characterization of gB-Δfurin. (A) Western blot analysis. Lysates of RK13 cells transfected with expression plasmids for wild-type gB (gB Ka) or the gB furin deletion mutant (gB-Δfurin) were separated by SDS-PAGE under reducing conditions. Cells transfected with the empty vector pcDNA3 served as a negative control (Mock). The blots were incubated with the gB-specific monoclonal antibody C15-b1. Signals of uncleaved gB (gBa) or a furin-cleaved gB subunit (gBb) are labeled by arrows, and the molecular masses of marker proteins are indicated. Signals of α-tubulin served as a loading control. (B) Subcellular localization. RK13 cells were transfected with expression plasmids for gB Ka or gB-Δfurin mutants and analyzed by indirect immunofluorescence. gB was detected using a gB-specific rabbit antiserum and Alexa Fluor 488-conjugated secondary antibodies. Green fluorescence was excited at 488 nm and recorded with a laser scanning confocal microscope (SP5; Leica, Mannheim, Germany). (C) Functional data. Results from the cell-cell fusion assay are plotted as black bars and expressed as the percentage of the signal measured for the WT protein, set to 100% (left y axis). The viral titers obtained in the trans-complementation assay are shown as gray bars (right y axis). PrV strain Kaplan (Ka) was used as a control. pcDNA-3-transfected cells were used as a negative control (Mock). The error bars indicate standard deviations from three independent experiments.
Next, the functions of the mutated proteins during virus entry were tested in a trans-complementation assay. RK13 cells were transfected with the different gB expression plasmids and infected 1 day later with a PrV mutant lacking the gB gene (PrV-ΔgB) (76). Cells and supernatant were harvested 24 h postinfection (p.i.), and progeny virus titers were determined on RK13-gB cells (Fig. 9A). Cells transfected with expression plasmids for gBs that efficiently mediated cell-cell fusion also complemented the defect of PrV-ΔgB during entry comparably to WT gB. In contrast, no infectious progeny was derived from cells transfected with the empty vector. Infectious progeny was also not produced after infection of cells transfected with the expression plasmid for W187A, indicating that this gB mutant is not functional. Surprisingly, Y192A, F275A, and F275Y gB mutants, which were unable to mediate cell-cell fusion, supported production of a low titer of infectious virions (102 PFU/ml), while expression of Y276A resulted in titers of 104 PFU/ml, indicating that the presence of other viral proteins might have partially compensated for the impairment in membrane binding and fusion.
DISCUSSION
Comparison with available gB structures from other herpesviruses.
The structural alignment of PrV gB with the homologous HSV-1, EBV, and HCMV proteins demonstrated that the postfusion conformation is well preserved, with the main difference residing in the positioning of domain IV relative to the rest of the protein (Fig. 2). The low-resolution structure obtained for the full-length HSV-1 gB in a conformation distinct from the postfusion conformation localizes domain IV to the interior of the spike (30), implying that a large movement occurs during the conformational change. Insertion of a foot-and-mouth disease virus (FMDV) epitope in PrV gB domain IV, right after the residue Arg685, resulted in high titers of neutralizing antibodies against FMDV (77), suggesting that at least some of the antigenic determinants on PrV gB domain IV are presented at the surface when the protein is expressed in its prefusion form. It is also possible, however, that the epitope is available because a fraction of gB at the virion surface may have adopted the postfusion conformation.
Cholesterol dependence.
HSV-1 gB binding to membranes was previously concluded to be cholesterol dependent based on experiments using binary liposomes made of DOPC and CH (54). As we show in this study, the ability of PrV gB to bind to liposomes is influenced not only by its FL residues, but also by the lipid membrane composition. The WT PrV gB ectodomains, as well as Y192F, Y267A, and Y276F variants, showed increased binding to 4L compared to 2L liposomes (Fig. 9B), whose composition does not emulate a real biological membrane well. We show here that liposomes, either 2L or 4L, need to contain at least 40% CH for gB to bind (Fig. 3C), which is in agreement with 30 to 40% CH being present in the plasma membrane and secretory vesicles (75) and with PrV entering cells via fusion with the plasma membrane. In the case of herpesviruses that enter cells by endocytosis, the requirement for a high CH concentration might also indicate that the fusion would occur within the internal compartments enriched in CH. A possible role for CH could be to fill the voids in the leaflet introduced by displacement of the bulkier head groups of the other lipids upon insertion of the FLs.
Cholesterol has been shown previously to induce lipid curvature and to promote formation of lipid stalks in fusion intermediates (25). While CH depletion was reported not to affect PrV attachment to cells, the virus was observed to be stalled at the plasma membrane, and entry was significantly reduced (58). It was speculated that the particles might have been blocked due to an inability to resolve the hemifusion intermediate in the absence of cholesterol, which is contrary to our observation that CH is required for the initial insertion of FLs into the membrane, i.e., for an early event that must take place prior to reaching the hemifusion state.
HSV-1 gB full-length protein expressed on cells was shown to associate specifically with CH- and SM-enriched lipid rafts (60), in which the two lipids are organized in the so-called ordered lipid domains (Lo) (78). The borders between these Lo and the more fluid, disordered membrane domains (Ld) were identified as the preferred sites for insertion of the HIV Env class I fusion protein FP (79), presumably because they impose the smallest energy penalty for insertion due to the line tension caused by the discontinuity between the domains. Desplanques et al. had previously observed a significant fraction of PrV virions juxtaposed with the lipid raft marker GM1 (58), which could, in the light of the Env insertion mode, indicate that PrV may use Lo-Ld boundaries as sites of attachment, as well. Although the FP of HIV Env is a short hydrophobic sequence that has a free N terminus, in contrast to the bipartite internal FLs of gB, it is tempting to speculate that despite the different natures of the inserted segments, similar energetic constraints might apply to membrane insertion of these divergent proteins. Further experimental studies on herpesvirus gB are needed to test these hypotheses and to establish if CH is required for binding of herpesviruses to membranes, for membrane fusion, or for both. It is worth noting that our EM data show an even distribution of the PrV gB ectodomains bound to liposomes, opposing the idea of gB having preferred membrane insertion sites. This could be due to the properties of the liposomes used for EM, which were made of 60% DOPC and 40% CH, a binary mixture that would be expected to form the Lo phase only (80).
Furin processing.
Proteolytic cleavage by cellular furin had been previously reported to be dispensable for gB function in herpesviruses encoding cleavable gBs, as its absence did not have an effect on viral replication or penetration kinetics, although smaller syncytia were detected (81–85). A similar observation was made when gB was expressed in LoVo cells, which are naturally deficient in furin, suggesting that PrV gB cleavage may play a role in cell-cell fusion (83). Our data demonstrate that furin cleavage of PrV gB was not required for its activity in cell-cell fusion and the virus transcomplementation assays used in this study (Fig. 10). The discrepancy between our results regarding the gB-Δfurin variant function in cell-cell fusion and those reported by Okazaki (83) could be due to the different cell types used for fusion.
PrV gB FL variants in liposome binding and functional assays.
The liposome binding experiments performed with the 15 recombinant gB ectodomain variants studied here allowed the selection of a subset of variants to be followed up by studies in the context of the full-length protein, both in cell-cell fusion and after incorporation into virus particles. The gB ectodomains in interaction with liposomes provided snapshots of the protein already in the postfusion form (Fig. 7A and 9B and C). The functional assays, on the other hand, illuminated the functions of full-length protein mutants expressed at the cell surface or incorporated into virus particles in their prefusion state (Fig. 9A). This may explain certain discrepancies, for instance, with variants that were expressed well as recombinant ectodomains but that failed to be folded and transported to the cell surface in cells. The selected variants of full-length gB were assessed for the ability to mediate fusion in two functional assays: a virus-free cell-cell fusion assay and trans-complementation of gB-negative PrV (18, 19, 21, 71).
To simplify comparisons with gBs from other herpesviruses, we numbered the FL residues from 1 to 14 (Fig. 5A). Our mutagenesis data are consistent with a model in which the residues Trp187 (FL1-3), Tyr192 (FL1-8), and Tyr276 (FL2-10) form an aromatic surface compatible with insertion into the polar region of the membrane, establishing an interfacial rim structure that would provide multiple interactions with the lipid head groups, while Phe275 (FL2-9) would reach deeper into the hydrocarbon core (Fig. 11A). gB sequence conservation in alphaherpesviruses shows that residues with similar membrane-partitioning preferences are found at these four positions (see Fig. S1 in the supplemental material). Hydrophobic residues compatible with insertion into the membrane core (Phe, Val, Leu, and Trp) are present at FL2-9, while amphipathic side chains that would favor the interfacial region (Tyr, His, and Trp) are found at positions FL1-3, FL1-8, and FL2-10. This suggests that a common mode of gB insertion into membranes may have evolved within the alphaherpesvirus subfamily, although the residues interacting with lipids may not be identical. Charged residues, such as Arg and Glu, are present in the FLs of HSV-1 but not of PrV gB (Fig. 5A), highlighting intrinsic differences that exist despite potentially shared principles for insertion of aromatic and hydrophobic residues.
FIG 11.

(A) Model of PrV gB ectodomain interactions with membranes. The model displays the base of the PrV gB trimer, illustrating putative locations of the FL residues investigated in this study, shown with their side chains as sticks and colored as in Fig. 5B. The arrows and numbers are used to indicate the deeper insertion of Phe275 (FL2-9) into the apolar region containing the lipid tails (1) and the interfacial rim made of Tyr192, Trp187, Tyr276 (2) (FL1-8, FL1-3, and FL2-10) that would be located within the more polar part of the membrane. A single molecule of DOPC and its putative localization in the membrane are shown in stick representation on the right. Carbon, oxygen, phosphorus, and nitrogen atoms are colored white, red, orange, and blue, respectively. (B) Model of HCMV and EBV gB interactions with membranes. HCMV (PDB accession number 5CXF), and EBV (PDB accession number 3FVC) gB structures were superimposed on the PrV structure using the Dali pairwise alignment algorithm (106). The last two structures contain WT residues (YIY155–157 and WLY240–242 in HCMV gB; WY112–113 and WLIW193–196 in EBV gB) that were modeled back onto the FL residues that had been mutated in the crystallized constructs (GHR155–157 and ATH240–242 in HCMV and HR112–113 and RVEA193–196 in EBV). The residues at positions FL1-8, FL1-3, FL2-10, and FL2-9 are indicated and colored as in Fig. 5B, with their side chains shown as sticks. Trp residues found at FL2-8 in HCMV and EBV gB are colored salmon, and Tyr side chains at positions FL1-10 and FL1-7 in HCMV and EBV gBs are colored light blue. The gray shading highlights the putative interfacial rim in each structure. The side chain of Trp193 inserted between residues FL2-5 and FL2-6 in EBV gB is labeled FL2-5*6 and colored magenta. Other hydrophobic residues are not shown as sticks for clarity. (C) Sequence alignment of the FL regions. Residues with similar physicochemical properties that are 75% or more conserved are colored red. Secondary-structure elements corresponding to the PrV gB ectodomain structure are displayed at the top, and residues identified as membrane contact sites in PrV gB (FL1-3, FL1-8, FL2-9, and FL2-10) are indicated with black arrows. The numbering at the top corresponds to that of the PrV gB protein. The alignment was generated in Clustal Omega (107) and displayed using ESPript (108).
The FL2-9 position is always occupied by a hydrophobic side chain, such as Phe275 in PrV gB. Mutation of the corresponding residue in HSV-1 gB (Phe262) to Asp or Leu resulted, respectively, in a poorly expressed protein and a protein that could mediate fusion at 70% of the level measured for the WT protein (51, 54). The tolerance for Leu (but not for Asp) in HSV-1 gB and for Trp (but not for Tyr) in PrV gB indicates that FL2-9 has the potential to insert into the membrane core, i.e., deeper than the interfacial region.
Positions FL1-3 (Trp187), FL1-5 (Gly189), and FL1-8 (Tyr192) are occupied by the best-conserved residues in FL1 of alphaherpesviruses (Fig. 12; see Fig. S1 in the supplemental material). FL1-5 Gly has already been shown to be important for the function of HSV-1 gB (54) and was not tested here. Mutation of HSV-1 gB FL1-3 (Trp) to Tyr resulted in a protein that was 50% active in fusion (51). When the same residue was changed to Phe in PrV gB, the resulting ectodomain did not cofloat with liposomes. It is possible that the FL1-3 position requires Trp because its indole ring may be involved in interactions with a specific lipid and/or because its central location between FL1-8 and FL2-10 might be important for the structural integrity and docking of the trio of rim residues into the membrane interface. HSV-1 gB variants containing Ser and Arg at positions FL1-3 and FL1-8, respectively, were shown to be defective in fusion and in liposome binding (54). These variants were subsequently crystallized, demonstrating that the mutations did not introduce any structural changes in FL1 and that the loss of binding to liposomes was solely due to the elimination of the hydrophobic side chains (86). While we anticipate that the same is true for the PrV gB ectodomains with mutations in FL1-3 and FL1-8, it is important to note that when expressed in cells as full-length proteins, W187A and Y192A accumulated in intracellular compartments and showed impaired processing (Fig. 8). We speculate that the furin sites in the misfolded gB variants may not be accessible for cleavage, giving rise to the observed decrease in the amount of cleaved protein. The possibility therefore remains that these two FL1 variants were not active in functional assays due to the altered cellular localization, even though production of ectodomains with the same mutations in insect cells led to efficient protein secretion into the medium, suggesting that these residues are at least not important for correct folding of the postfusion ectodomain.
FIG 12.

Conservation of gB in alphaherpesviruses plotted on the PrV gB ectodomain structure. (A) Solvent-accessible surface of PrV gB in multiple orientations. Residues showing identity lower than 75% are colored white, while the residues that are strictly conserved in more than 75% of the analyzed sequences are colored in an increasing purple gradient. The alignment shown in Fig. S1 in the supplemental material was used. (B) PrV gB FLs. Fusion loop residues investigated in this study are shown with the side chains in stick representation. Two orientations are presented for clarity.
The FL2-10 residue is predicted to insert into the interfacial region, and while PrV gB Y276F was functional, Y276A did not associate with liposomes and failed to mediate cell-cell fusion, although it unexpectedly partially rescued gB-null virus in the complementation assay. This result emphasizes distinct functional requirements for cell-cell fusion and viral entry processes and that the two do not follow identical mechanisms. This is highlighted by the requirement for gD in PrV entry but not for cell-cell spread (18, 87). The presence of other viral proteins during infection may compensate for the reduced membrane binding activity, and/or the protein may be present at higher density in the virion envelope than at the cell surface, favoring the fusion function.
FL2-1 is the most conserved FL2 residue in alphaherpesviruses, being Tyr in 75% of the sequences (Fig. 12; see Fig. S1 in the supplemental material). This residue was not mutagenized in HSV-1 gB but was analyzed here for PrV gB, revealing, to our surprise, that changes to Ala or Phe had no effect. This suggests that Tyr267 is not directly involved in interactions with liposomes and is not essential for the protein function in fusion. Interestingly, Y267F bound indiscriminately to 2L and 4L liposomes, while Y267A was sensitive to the lipid composition and associated better with the 4L liposomes. Membrane composition has been shown to also influence binding of fusion proteins of flaviviruses (88, 89), bunyaviruses (90), influenza virus (91), and HIV (92). In the last case, the FP of gp41 was shown to adopt an α-helical or β-strand structure, depending on the CH concentration, indicating alternative modes of insertion into membranes of the same protein.
Position FL2-11 is occupied by Arg in most alphaherpesviruses and by His in PrV gB. The substitution of this residue for Ala or the larger Trp in PrV gB resulted in a functional protein that associated with liposomes, indicating that the aromatic side chain is not important for interactions with lipids. In contrast, Arg present at the same position in HSV-1 gB and in 26 other analyzed sequences (see Fig. S1 in the supplemental material) was implicated in binding to liposomes, possibly by interacting with the negatively charged phosphate head groups of membrane lipids (54). The His and Arg side chains present at position FL2-11 obviously play different roles in binding to lipids.
Model of PrV gB interactions with lipids.
We show here that PrV gB residues Trp187, Tyr192, Gly274, Phe275, and Tyr276 form a continuous, electrostatically neutral hydrophobic surface (Fig. 7B) reminiscent of the hydrophobic patch reported for HSV-1 gB (54). EBV and HCMV gB ectodomains used for crystallization had polar and charged residues inserted in place of the hydrophobic WT FL residues, but similar hydrophobic patches at the base of the trimers were observed when the WT residues were modeled back on the structure (data not shown). This strongly suggests functional conservation of the areas involved in membrane interactions. Closer inspection of the HCMV and EBV gB FL residues found at the positions identified as the membrane contact sites in PrV gB revealed that two of the three rim positions, FL1-3 and FL2-10, are occupied by Tyr, similar to gBs of alphaherpesviruses, while Thr and Ala are found at FL1-8 in HCMV and EBV gBs, respectively (Fig. 11B and C). The Tyr residues at FL1-3 and FL2-10 are highly conserved in beta- and gammaherpesviruses (data not shown), while FL1-8 is not. In addition, the side chain of Tyr, found at the neighboring FL1-10 in HCMV and at FL1-7 in HCMV and EBV gBs, could reach the plane in which FL1-3 and FL2-10 side chains insert, contributing to formation of the interfacial rim. FL2-9, which according to our model inserts more deeply, is Leu in HCMV gB and Thr in EBV gB, while Trp is found at FL2-8 in both proteins. This Trp is strictly conserved within betaherpesviruses and partially within gammaherpesviruses, while the latter have in addition a well-conserved Trp inserted between FL2-5 and FL2-6 (designated FL2-5*6 in Fig. 11B). There are also other hydrophobic residues in the FLs on HCMV and EBV gBs whose side chains could insert into the lipid bilayer (FL1-4, FL1-5, and FL1-6 in HCMV and FL1-6, FL2-6, and FL2-7 in EBV gB) (not shown in Fig. 11B for clarity). Thus, although they have more hydrophobic residues at the tips of the FLs, our model suggests that beta- and gammaherpesvirus gBs interact with the membranes similarly to alphaherpesvirus gB, with exposed hydrophobic residues in FL2 penetrating into the hydrocarbon core (FL2-9 and/or FL2-8 and/or FL2-5*6), while the rim formed by residues from both FLs (FL1-3, FL1-7, and FL2-10) would secure protein insertion into the interface. The exact molecular mechanisms through which this is achieved may vary, pointing to differences between the herpesvirus subfamilies that might be related to the different host cells and the specific compositions of the target membranes.
MATERIALS AND METHODS
Expression and purification of the PrV gB ectodomain in insect cells for crystallization studies.
The synthetic gene encoding gB of suid herpesvirus 1 strain Kaplan (GenBank accession number AEM64049.1) was codon optimized for protein expression in Drosophila S2 cells and was purchased from GenScript. The gene segment encoding the ectodomain (PrV gB residues 59 to 756) was cloned into the expression vector pT350 (61) so that the ectodomain was flanked by the Drosophila Bip secretion signal, driving efficient protein secretion (93) at the N terminus, and the enterokinase-cleavable double Strep-tag II (sequence SRFESDDDDKAGWSHPQFEKGGGSGGGSGGGSWSHPQFEK) at the C terminus. Protein expression was induced by the addition of 0.5 mM CuSO4, and the protein was harvested from the culture supernatant 7 days postinduction (the detailed protocol is available in reference 62). Standard protocols were applied to purify the protein by affinity (Streptactin resin; IBA Technologies) and then by size exclusion chromatography using 10 mM Tris, 50 mM NaCl, pH 8, buffer and a Superdex S200 column. An extinction coefficient of 1.2 ml/mg for protein absorbance at 280 nm was used for calculation of the protein concentration.
Crystallization and structure determination of the PrV gB ectodomain.
Purified PrV gB ectodomains were deglycosylated with Endo D from Streptococcus pneumoniae (94) by overnight incubation of gB and Endo D in a 10:1 (wt/wt) ratio in 50 mM sodium-phosphate buffer, pH 7.5, at 25°C. The reaction mixture was then loaded onto a Superdex S200 column to separate deglycosylated gB from Endo D, using 10 mM Tris, 50 mM NaCl, pH 8, buffer. Fractions corresponding to gB were pooled, and the protein was concentrated to 9.2 mg/ml in a 50-kDa-cutoff Vivaspin concentrator.
Crystals were grown by vapor diffusion in hanging drops in 0.1 M Tris, pH 8.5, 7% PEG 4000, 0.6 M LiCl and then flash frozen in liquid nitrogen using 20% glycerol as the cryoprotectant. Data were collected at the European Synchrotron Radiation Facility (ESRF) synchrotron source ID29 beamline and processed using XDS (95). Molecular replacement was done with Phaser (96) using HSV-1 gB structure (Protein Data Bank [PDB] accession number 2GUM) as a search model. The structure was refined using BUSTER (97).
Electron microscopy.
Samples for negative-stain EM were prepared by mixing 0.5 μM protein and 1 mM liposomes as described in more detail under “Liposome Flotation Assay” below. Liposomes were made of 60% DOPC and 40% CH. Staining was done with 2% uranyl acetate, and images were collected on a Tecnai G2 Spirit Biotwin microscope 5 (FEI) operating at an accelerating voltage of 120 kV. Samples for cryo-EM analysis were made by incubating 4 μM protein and 2 mM liposomes. Cryofixation was done using Lacey grids (Leica EMGP, Austria). Data were recorded on a Tecnai F20 operating at 200 kV, equipped with a Falcon II direct detector (FEI) under low-dose conditions.
Generation of expression constructs and production of PrV gB recombinant ectodomains in mammalian cells.
The gene encoding PrV gB ectodomain residues 59 to 756, followed by a double Strep-tag II, was cloned into the pcDNA4 (Invitrogen) expression vector using Gibson Assembly master mix (New England BioLabs). QuikChange mutagenesis was applied to introduce single point mutations in the FLs, following the standard protocols (Agilent Technologies; QuikChange II site-directed mutagenesis kit). The presence of desired mutations was confirmed by DNA sequencing. The expression constructs were transfected into Expi293F mammalian cells (Thermo Fisher Scientific) using Expifectamine transfection agent as described in the manufacturer's manual (Thermo Fisher Scientific). Cell supernatants were collected 5 days posttransfection, and the protein was purified by affinity chromatography on Streptactin resin (IBA Technologies), followed by SEC.
Liposome flotation assay.
Lipids, DOPC, DOPE, SM (from bovine brain), and CH (from ovine wool) were purchased from Avanti Polar Lipids. Liposomes were prepared by the freeze-thaw and extrusion method (98), using 200-nm membranes for extrusion. 2L liposomes were made of 60 mol% DOPC and 40 mol% CH, and 4L liposomes were 20 mol% DOPC, 20 mol% DOPE, 20 mol% SM, and 40 mol% CH. Recombinant 0.5 μM gB ectodomains were mixed with 1 mM liposomes in phosphate-buffered saline (PBS) in a 100-μl volume, incubated overnight (12 to 15 h) at 4°C and then at 37°C for 15 min (the overnight incubation can be omitted and was done for convenience). OptiPrep medium (Axis-Shield PoC AS, Oslo, Norway) was used for density gradient preparation. Protein-liposome samples were adjusted to a volume of 300 μl by addition of 30 μl of PBS and 170 μl of 60% OptiPrep (final OptiPrep concetration, 36%) and deposited with a syringe on the bottom of the ultracentrifuge tube already containing 4.5 ml of 20% OptiPrep solution in PBS. Two hundred microliters of PBS was added to the top, and the samples were centrifuged for 1 h at 40,000 × g and 4°C in an SWTi 55 swinging-bucket rotor in a Beckman Coulter ultracentrifuge. The 5-ml gradients were fractionated into two 400-μl fractions (the first one representing the top fraction), three 1.2-ml fractions, and one 600-μl (bottom) fraction. Thirty-microliter aliquots of the top and bottom fractions were analyzed on SDS-PAGE 4 to 20% gradient gels (GenScript), and Western blotting to detect the Strep-tag II on gB was done with a Streptactin-horseradish peroxidase (HRP) conjugate (IBA Technologies) following the manufacturer's instructions.
Viruses and cells.
Rabbit kidney (RK13) and RK13-gB cells were grown in Dulbecco's modified Eagle's minimum essential medium (MEM) supplemented with 10% fetal calf serum (FCS) at 37°C and 5% CO2. A PrV mutant lacking gB (PrV-ΔgB) (76), which was derived from PrV strain Kaplan (PrV-Ka), was propagated in RK13-gB cells.
Expression plasmids for cell-cell fusion assays.
Generation of expression plasmids for PrV-Ka gB, gH, and gL has been described previously (70). The expression plasmid pcDNA-gBKa containing the gB open reading frame (ORF UL27) was used for site-directed mutagenesis (QuikChange II XL kit; Agilent) with the complementary pair of oligonucleotide primers. Correct mutagenesis was verified by sequencing.
In vitro cell-cell fusion assays.
Fusion activities of the different gB mutants were analyzed after transient transfection of RK13 cells as described recently (72). Briefly, approximately 1.8 × 105 RK13 cells per well were seeded onto 24-well cell culture plates. On the following day, the cells were transfected with 200 ng each of expression plasmids for EGFP (pEGFP-N1; Clontech) and for PrV glycoprotein gBKa or mutant gB, gLKa, and gHKa (21, 70, 99) in 100 μl Opti-MEM using 1 μl Lipofectamine 2000 (Thermo Fisher Scientific). An empty vector (pcDNA3) served as a negative control. The mixture was incubated for 20 min at room temperature and added to the cells. After 3 h, the cells were washed with PBS and incubated in MEM supplemented with 2% FCS for another 24 h at 37°C. Thereafter, the cells were fixed with 3% paraformaldehyde (PFA). Syncytium formation was analyzed using an Eclipse Ti-S fluorescence microscope and NIS Elements imaging software (Nikon). Total fusion activity was determined by multiplication of the area of cells with three or more nuclei by the number of syncytia within 10 fields of view (5.5 mm2 each). The experiment was repeated three times, and average percent values of positive-control transfections, as well as standard deviations, were calculated.
trans-complementation assay.
The functions of the different gB mutants in virus entry were determined by trans-complementation of PrV-ΔgB (76). Approximately 1.8 × 105 RK13 cells per well were seeded onto 24-well cell culture plates. On the following day, the cells were transfected with 200 ng of the corresponding gB expression plasmid as described above. One day posttransfection, the cells were infected with phenotypically gB-complemented PrV-ΔgB at a multiplicity of infection (MOI) of 3 and consecutively incubated on ice for 1 h and at 37°C for 1 h. Subsequently, the inoculum was removed, nonpenetrated virus was inactivated by low-pH treatment (100), and 1 ml fresh medium was added. After 24 h at 37°C, the cells were harvested together with the supernatants and lysed by freeze-thawing (−80°C and 37°C). Progeny virus titers were determined on PrV gB-expressing cells (RK13-gB) (76). The mean values of three independent experiments with the corresponding standard deviations are shown.
Indirect immunofluorescence tests.
To test for subcellular localization of the mutated proteins, RK13 cells were transfected with the gB expression plasmids as described above. After 24 h, the cells were fixed with 3% PFA for 20 min and permeabilized in PBS containing 0,1% Triton X-100 for 10 min at room temperature. Subsequently, the cells were incubated with a rabbit antiserum specific for PrV gB (101), which was diluted 1:1,000 in PBS. After 1 h at room temperature, bound antibody was detected with Alexa 488-conjugated goat anti-rabbit antibodies (1:1,000 in PBS; Invitrogen). After each step, the cells were washed repeatedly with PBS. Green fluorescence was excited at 488 nm and recorded with a laser scanning confocal microscope (SP5; Leica, Mannheim, Germany).
Western blot analyses.
RK13 cells were harvested 24 h after transfection with the different gB expression plasmids, as described above. The cells were lysed, and protein samples were separated by SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was incubated with a monoclonal gB antibody (C15-b1 at 1:500 dilution) (101, 102). Binding of peroxidase-conjugated secondary antibody (Jackson ImmunoResearch) was detected with Clarity Western ECL substrate (Bio-Rad) and recorded with a VersaDoc 4000 MP imager (Bio-Rad).
Accession number(s).
The PrV gB coordinates have been deposited in the RCSB Protein Data Bank (PDB) under accession code 6ESC.
Supplementary Material
ACKNOWLEDGMENTS
Work done by M.B., D.B., M.-C.V., P.G.-C., and F.A.R. was supported by Institut Pasteur and CNRS recurrent funding and the Pasteur-Weizmann/Servier International Prize (F.A.R., 2015). Work done by M.V., B.G.K., and T.C.M. was supported by DFG, grant Me 854/11-2.
We acknowledge assistance of the core facilities for crystallization and microscopy at Institut Pasteur and also thank M. Nilges and the Equipex CACSICE for providing the Falcon II direct detector. We thank the European Synchrotron Radiation Facility for help during data collection.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01203-17.
REFERENCES
- 1.Pellet PE, Roizman B. 2014. Herpesviridae, p 1802–1822. In Knipe DM, Howley PM (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, New York, NY. [Google Scholar]
- 2.Mettenleiter TC. 1996. Immunobiology of pseudorabies (Aujeszky's disease). Vet Immunol Immunopathol 54:221–229. doi: 10.1016/S0165-2427(96)05695-4. [DOI] [PubMed] [Google Scholar]
- 3.Pomeranz LE, Reynolds AE, Hengartner CJ. 2005. Molecular biology of pseudorabies virus: impact on neurovirology and veterinary medicine. Microbiol Mol Biol Rev 69:462–500. doi: 10.1128/MMBR.69.3.462-500.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. 2012. Herpes virus fusion and entry: a story with many characters. Viruses 4:800–832. doi: 10.3390/v4050800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nicola AV. 2016. Herpesvirus entry into host cells mediated by endosomal low pH. Traffic 17:965–975. doi: 10.1111/tra.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li A, Lu G, Qi J, Wu L, Tian K, Luo T, Shi Y, Yan J, Gao GF. 2017. Structural basis of nectin-1 recognition by pseudorabies virus glycoprotein D. PLoS Pathog 13:e1006314. doi: 10.1371/journal.ppat.1006314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spear PG. 2004. Herpes simplex virus: receptors and ligands for cell entry. Cell Microbiol 6:401–410. doi: 10.1111/j.1462-5822.2004.00389.x. [DOI] [PubMed] [Google Scholar]
- 8.Vanarsdall AL, Chase MC, Johnson DC. 2011. Human cytomegalovirus glycoprotein gO complexes with gH/gL, promoting interference with viral entry into human fibroblasts but not entry into epithelial cells. J Virol 85:11638–11645. doi: 10.1128/JVI.05659-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mullen MM, Haan KM, Longnecker R, Jardetzky TS. 2002. Structure of the Epstein-Barr virus gp42 protein bound to the MHC class II receptor HLA-DR1. Mol Cell 9:375–385. doi: 10.1016/S1097-2765(02)00465-3. [DOI] [PubMed] [Google Scholar]
- 10.Krummenacher C, Carfi A, Eisenberg RJ, Cohen GH. 2013. Entry of herpesviruses into cells: the enigma variations. Adv Exp Med Biol 790:178–195. doi: 10.1007/978-1-4614-7651-1_10. [DOI] [PubMed] [Google Scholar]
- 11.Sathiyamoorthy K, Chen J, Longnecker R, Jardetzky TS. 2017. The COMPLEXity in herpesvirus entry. Curr Opin Virol 24:97–104. doi: 10.1016/j.coviro.2017.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agelidis AM, Shukla D. 2015. Cell entry mechanisms of HSV: what we have learned in recent years. Future Virol 10:1145–1154. doi: 10.2217/fvl.15.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Giovine P, Settembre EC, Bhargava AK, Luftig MA, Lou H, Cohen GH, Eisenberg RJ, Krummenacher C, Carfi A. 2011. Structure of herpes simplex virus glycoprotein D bound to the human receptor nectin-1. PLoS Pathog 7:e1002277. doi: 10.1371/journal.ppat.1002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lazear E, Whitbeck JC, Zuo Y, Carfi A, Cohen GH, Eisenberg RJ, Krummenacher C. 2014. Induction of conformational changes at the N-terminus of herpes simplex virus glycoprotein D upon binding to HVEM and nectin-1. Virology 448:185–195. doi: 10.1016/j.virol.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gianni T, Amasio M, Campadelli-Fiume G. 2009. Herpes simplex virus gD forms distinct complexes with fusion executors gB and gH/gL in part through the C-terminal profusion domain. J Biol Chem 284:17370–17382. doi: 10.1074/jbc.M109.005728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Atanasiu D, Saw WT, Cohen GH, Eisenberg RJ. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J Virol 84:12292–12299. doi: 10.1128/JVI.01700-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmidt J, Klupp BG, Karger A, Mettenleiter TC. 1997. Adaptability in herpesviruses: glycoprotein D-independent infectivity of pseudorabies virus. J Virol 71:17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rauh I, Mettenleiter TC. 1991. Pseudorabies virus glycoproteins gII and gp50 are essential for virus penetration. J Virol 65:5348–5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peeters B, de Wind N, Broer R, Gielkens A, Moormann R. 1992. Glycoprotein H of pseudorabies virus is essential for entry and cell-to-cell spread of the virus. J Virol 66:3888–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klupp BG, Fuchs W, Weiland E, Mettenleiter TC. 1997. Pseudorabies virus glycoprotein L is necessary for virus infectivity but dispensable for virion localization of glycoprotein H. J Virol 71:7687–7695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schröter C, Vallbracht M, Altenschmidt J, Kargoll S, Fuchs W, Klupp BG, Mettenleiter TC. 2015. Mutations in pseudorabies virus glycoproteins gB, gD, and gH functionally compensate for the absence of gL. J Virol 90:2264–2272. doi: 10.1128/JVI.02739-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roche S, Bressanelli S, Rey FA, Gaudin Y.. 2006. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G Science 313:187–191. [DOI] [PubMed] [Google Scholar]
- 23.Kadlec J, Loureiro S, Abrescia NG, Stuart DI, Jones IM. 2008. The postfusion structure of baculovirus gp64 supports a unified view of viral fusion machines. Nat Struct Mol Biol 15:1024–1030. doi: 10.1038/nsmb.1484. [DOI] [PubMed] [Google Scholar]
- 24.Harrison SC. 2015. Viral membrane fusion. Virology 479-480:498–507. doi: 10.1016/j.virol.2015.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chernomordik LV, Kozlov MM. 2008. Mechanics of membrane fusion. Nat Struct Mol Biol 15:675–683. doi: 10.1038/nsmb.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220. doi: 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- 27.Backovic M, Longnecker R, Jardetzky TS. 2009. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc Natl Acad Sci U S A 106:2880–2885. doi: 10.1073/pnas.0810530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chandramouli S, Ciferri C, Nikitin PA, Calo S, Gerrein R, Balabanis K, Monroe J, Hebner C, Lilja AE, Settembre EC, Carfi A. 2015. Structure of HCMV glycoprotein B in the postfusion conformation bound to a neutralizing human antibody. Nat Commun 6:8176. doi: 10.1038/ncomms9176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burke HG, Heldwein EE. 2015. Crystal structure of the human cytomegalovirus glycoprotein B. PLoS Pathog 11:e1005227. doi: 10.1371/journal.ppat.1005227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zeev-Ben-Mordehai T, Vasishtan D, Hernandez Duran A, Vollmer B, White P, Prasad Pandurangan A, Siebert CA, Topf M, Grunewald K. 2016. Two distinct trimeric conformations of natively membrane-anchored full-length herpes simplex virus 1 glycoprotein B. Proc Natl Acad Sci U S A 113:4176–4181. doi: 10.1073/pnas.1523234113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roche S, Rey FA, Gaudin Y, Bressanelli S. 2007. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 315:843–848. doi: 10.1126/science.1135710. [DOI] [PubMed] [Google Scholar]
- 32.Gallagher JR, Atanasiu D, Saw WT, Paradisgarten MJ, Whitbeck JC, Eisenberg RJ, Cohen GH. 2014. Functional fluorescent protein insertions in herpes simplex virus gB report on gB conformation before and after execution of membrane fusion. PLoS Pathog 10:e1004373. doi: 10.1371/journal.ppat.1004373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chowdary TK, Cairns TM, Atanasiu D, Cohen GH, Eisenberg RJ, Heldwein EE. 2010. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat Struct Mol Biol 17:882–888. doi: 10.1038/nsmb.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsuura H, Kirschner AN, Longnecker R, Jardetzky TS. 2010. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc Natl Acad Sci U S A 107:22641–22646. doi: 10.1073/pnas.1011806108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xing Y, Oliver SL, Nguyen T, Ciferri C, Nandi A, Hickman J, Giovani C, Yang E, Palladino G, Grose C, Uematsu Y, Lilja AE, Arvin AM, Carfi A. 2015. A site of varicella-zoster virus vulnerability identified by structural studies of neutralizing antibodies bound to the glycoprotein complex gHgL. Proc Natl Acad Sci U S A 112:6056–6061. doi: 10.1073/pnas.1501176112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Backovic M, DuBois RM, Cockburn JJ, Sharff AJ, Vaney MC, Granzow H, Klupp BG, Bricogne G, Mettenleiter TC, Rey FA. 2010. Structure of a core fragment of glycoprotein H from pseudorabies virus in complex with antibody. Proc Natl Acad Sci U S A 107:22635–22640. doi: 10.1073/pnas.1011507107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heldwein EE. 2016. gH/gL supercomplexes at early stages of herpesvirus entry. Curr Opin Virol 18:1–8. doi: 10.1016/j.coviro.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.White JM, Delos SE, Brecher M, Schornberg K. 2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol 43:189–219. doi: 10.1080/10409230802058320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ito H, Watanabe S, Sanchez A, Whitt MA, Kawaoka Y. 1999. Mutational analysis of the putative fusion domain of Ebola virus glycoprotein. J Virol 73:8907–8912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Apellaniz B, Huarte N, Largo E, Nieva JL. 2014. The three lives of viral fusion peptides. Chem Phys Lipids 181:40–55. doi: 10.1016/j.chemphyslip.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bressanelli S, Stiasny K, Allison SL, Stura EA, Duquerroy S, Lescar J, Heinz FX, Rey FA. 2004. Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. EMBO J 23:728–738. doi: 10.1038/sj.emboj.7600064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Voss JE, Vaney MC, Duquerroy S, Vonrhein C, Girard-Blanc C, Crublet E, Thompson A, Bricogne G, Rey FA. 2010. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 468:709–712. doi: 10.1038/nature09555. [DOI] [PubMed] [Google Scholar]
- 43.DuBois RM, Vaney MC, Tortorici MA, Kurdi RA, Barba-Spaeth G, Krey T, Rey FA. 2013. Functional and evolutionary insight from the crystal structure of rubella virus protein E1. Nature 493:552–556. doi: 10.1038/nature11741. [DOI] [PubMed] [Google Scholar]
- 44.Dessau M, Modis Y. 2013. Crystal structure of glycoprotein C from Rift Valley fever virus. Proc Natl Acad Sci U S A 110:1696–1701. doi: 10.1073/pnas.1217780110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guardado-Calvo P, Bignon EA, Stettner E, Jeffers SA, Perez-Vargas J, Pehau-Arnaudet G, Tortorici MA, Jestin JL, England P, Tischler ND, Rey FA. 2016. Mechanistic insight into bunyavirus-induced membrane fusion from structure-function analyses of the hantavirus envelope glycoprotein Gc. PLoS Pathog 12:e1005813. doi: 10.1371/journal.ppat.1005813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun X, Belouzard S, Whittaker GR. 2008. Molecular architecture of the bipartite fusion loops of vesicular stomatitis virus glycoprotein G, a class III viral fusion protein. J Biol Chem 283:6418–6427. doi: 10.1074/jbc.M708955200. [DOI] [PubMed] [Google Scholar]
- 47.Baquero E, Albertini AA, Gaudin Y. 2015. Recent mechanistic and structural insights on class III viral fusion glycoproteins. Curr Opin Struct Biol 33:52–60. doi: 10.1016/j.sbi.2015.07.011. [DOI] [PubMed] [Google Scholar]
- 48.Backovic M, Leser GP, Lamb RA, Longnecker R, Jardetzky TS. 2007. Characterization of EBV gB indicates properties of both class I and class II viral fusion proteins. Virology 368:102–113. doi: 10.1016/j.virol.2007.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sharma S, Wisner TW, Johnson DC, Heldwein EE. 2013. HCMV gB shares structural and functional properties with gB proteins from other herpesviruses. Virology 435:239–249. doi: 10.1016/j.virol.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Backovic M, Jardetzky TS, Longnecker R. 2007. Hydrophobic residues that form putative fusion loops of Epstein-Barr virus glycoprotein B are critical for fusion activity. J Virol 81:9596–9600. doi: 10.1128/JVI.00758-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hannah BP, Heldwein EE, Bender FC, Cohen GH, Eisenberg RJ. 2007. Mutational evidence of internal fusion loops in herpes simplex virus glycoprotein B. J Virol 81:4858–4865. doi: 10.1128/JVI.02755-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin E, Spear PG. 2007. Random linker-insertion mutagenesis to identify functional domains of herpes simplex virus type 1 glycoprotein B. Proc Natl Acad Sci U S A 104:13140–13145. doi: 10.1073/pnas.0705926104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Atanasiu D, Saw WT, Gallagher JR, Hannah BP, Matsuda Z, Whitbeck JC, Cohen GH, Eisenberg RJ. 2013. Dual split protein-based fusion assay reveals that mutations to herpes simplex virus (HSV) glycoprotein gB alter the kinetics of cell-cell fusion induced by HSV entry glycoproteins. J Virol 87:11332–11345. doi: 10.1128/JVI.01700-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hannah BP, Cairns TM, Bender FC, Whitbeck JC, Lou H, Eisenberg RJ, Cohen GH. 2009. Herpes simplex virus glycoprotein B associates with target membranes via its fusion loops. J Virol 83:6825–6836. doi: 10.1128/JVI.00301-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maurer UE, Zeev-Ben-Mordehai T, Pandurangan AP, Cairns TM, Hannah BP, Whitbeck JC, Eisenberg RJ, Cohen GH, Topf M, Huiskonen JT, Grunewald K. 2013. The structure of herpesvirus fusion glycoprotein B-bilayer complex reveals the protein-membrane and lateral protein-protein interaction. Structure 21:1396–1405. doi: 10.1016/j.str.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang ST, Kreutzberger AJ, Lee J, Kiessling V, Tamm LK. 2016. The role of cholesterol in membrane fusion. Chem Phys Lipids 199:136–143. doi: 10.1016/j.chemphyslip.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ren X, Yin J, Li G, Herrler G. 2011. Cholesterol dependence of pseudorabies herpesvirus entry. Curr Microbiol 62:261–266. doi: 10.1007/s00284-010-9700-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Desplanques AS, Nauwynck HJ, Vercauteren D, Geens T, Favoreel HW. 2008. Plasma membrane cholesterol is required for efficient pseudorabies virus entry. Virology 376:339–345. doi: 10.1016/j.virol.2008.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wudiri GA, Pritchard SM, Li H, Liu J, Aguilar HC, Gilk SD, Nicola AV. 2014. Molecular requirement for sterols in herpes simplex virus entry and infectivity. J Virol 88:13918–13922. doi: 10.1128/JVI.01615-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bender FC, Whitbeck JC, Ponce de Leon M, Lou H, Eisenberg RJ, Cohen GH. 2003. Specific association of glycoprotein B with lipid rafts during herpes simplex virus entry. J Virol 77:9542–9552. doi: 10.1128/JVI.77.17.9542-9552.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krey T, d'Alayer J, Kikuti CM, Saulnier A, Damier-Piolle L, Petitpas I, Johansson DX, Tawar RG, Baron B, Robert B, England P, Persson MA, Martin A, Rey FA. 2010. The disulfide bonds in glycoprotein E2 of hepatitis C virus reveal the tertiary organization of the molecule. PLoS Pathog 6:e1000762. doi: 10.1371/journal.ppat.1000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Backovic M, Krey T. 2016. Stable Drosophila cell lines: an alternative approach to exogenous protein expression. Methods Mol Biol 1350:349–358. doi: 10.1007/978-1-4939-3043-2_17. [DOI] [PubMed] [Google Scholar]
- 63.Culp JS, Johansen H, Hellmig B, Beck J, Matthews TJ, Delers A, Rosenberg M. 1991. Regulated expression allows high level production and secretion of HIV-1 gp120 envelope glycoprotein in Drosophila Schneider cells. Biotechnology 9:173–177. [DOI] [PubMed] [Google Scholar]
- 64.Gianni T, Fato R, Bergamini C, Lenaz G, Campadelli-Fiume G. 2006. Hydrophobic alpha-helices 1 and 2 of herpes simplex virus gH interact with lipids, and their mimetic peptides enhance virus infection and fusion. J Virol 80:8190–8198. doi: 10.1128/JVI.00504-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sugihara K, Chami M, Derenyi I, Voros J, Zambelli T. 2012. Directed self-assembly of lipid nanotubes from inverted hexagonal structures. ACS Nano 6:6626–6632. doi: 10.1021/nn300557s. [DOI] [PubMed] [Google Scholar]
- 66.Wimley WC, White SH. 1996. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol 3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 67.MacCallum JL, Bennett WF, Tieleman DP. 2008. Distribution of amino acids in a lipid bilayer from computer simulations. Biophys J 94:3393–3404. doi: 10.1529/biophysj.107.112805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yau WM, Wimley WC, Gawrisch K, White SH. 1998. The preference of tryptophan for membrane interfaces. Biochemistry 37:14713–14718. doi: 10.1021/bi980809c. [DOI] [PubMed] [Google Scholar]
- 69.White SH, Wimley WC. 1999. Membrane protein folding and stability: physical principles. Annu Rev Biophys Biomol Struct 28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 70.Klupp BG, Nixdorf R, Mettenleiter TC. 2000. Pseudorabies virus glycoprotein M inhibits membrane fusion. J Virol 74:6760–6768. doi: 10.1128/JVI.74.15.6760-6768.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Turner A, Bruun B, Minson T, Browne H. 1998. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J Virol 72:873–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vallbracht M, Schröter C, Klupp BG, Mettenleiter TC. 2017. Transient transfection-based fusion assay for viral proteins. Bio-protocol 7(5):e2162. doi: 10.21769/BioProtoc.2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dodge JT, Phillips GB. 1967. Composition of phospholipids and of phospholipid fatty acids and aldehydes in human red cells. J Lipid Res 8:667–675. [PubMed] [Google Scholar]
- 74.Virtanen JA, Cheng KH, Somerharju P. 1998. Phospholipid composition of the mammalian red cell membrane can be rationalized by a superlattice model. Proc Natl Acad Sci U S A 95:4964–4969. doi: 10.1073/pnas.95.9.4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kalvodova L, Sampaio JL, Cordo S, Ejsing CS, Shevchenko A, Simons K. 2009. The lipidomes of vesicular stomatitis virus, Semliki Forest virus, and the host plasma membrane analyzed by quantitative shotgun mass spectrometry. J Virol 83:7996–8003. doi: 10.1128/JVI.00635-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nixdorf R, Klupp BG, Karger A, Mettenleiter TC. 2000. Effects of truncation of the carboxy terminus of pseudorabies virus glycoprotein B on infectivity. J Virol 74:7137–7145. doi: 10.1128/JVI.74.15.7137-7145.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dory D, Remond M, Beven V, Cariolet R, Backovic M, Zientara S, Jestin A. 2009. Pseudorabies virus glycoprotein B can be used to carry foot and mouth disease antigens in DNA vaccination of pigs. Antiviral Res 81:217–225. doi: 10.1016/j.antiviral.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 78.Rog T, Vattulainen I. 2014. Cholesterol, sphingolipids, and glycolipids: what do we know about their role in raft-like membranes? Chem Phys Lipids 184:82–104. doi: 10.1016/j.chemphyslip.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 79.Yang ST, Kiessling V, Tamm LK. 2016. Line tension at lipid phase boundaries as driving force for HIV fusion peptide-mediated fusion. Nat Commun 7:11401. doi: 10.1038/ncomms11401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Feigenson GW. 2006. Phase behavior of lipid mixtures. Nat Chem Biol 2:560–563. doi: 10.1038/nchembio1106-560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Oliver SL, Sommer M, Zerboni L, Rajamani J, Grose C, Arvin AM. 2009. Mutagenesis of varicella-zoster virus glycoprotein B: putative fusion loop residues are essential for viral replication, and the furin cleavage motif contributes to pathogenesis in skin tissue in vivo. J Virol 83:7495–7506. doi: 10.1128/JVI.00400-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sorem J, Longnecker R. 2009. Cleavage of Epstein-Barr virus glycoprotein B is required for full function in cell-cell fusion with both epithelial and B cells. J Gen Virol 90:591–595. doi: 10.1099/vir.0.007237-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Okazaki K. 2007. Proteolytic cleavage of glycoprotein B is dispensable for in vitro replication, but required for syncytium formation of pseudorabies virus. J Gen Virol 88:1859–1865. doi: 10.1099/vir.0.82610-0. [DOI] [PubMed] [Google Scholar]
- 84.Strive T, Borst E, Messerle M, Radsak K. 2002. Proteolytic processing of human cytomegalovirus glycoprotein B is dispensable for viral growth in culture. J Virol 76:1252–1264. doi: 10.1128/JVI.76.3.1252-1264.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kopp A, Blewett E, Misra V, Mettenleiter TC. 1994. Proteolytic cleavage of bovine herpesvirus 1 (BHV-1) glycoprotein gB is not necessary for its function in BHV-1 or pseudorabies virus. J Virol 68:1667–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stampfer SD, Lou H, Cohen GH, Eisenberg RJ, Heldwein EE. 2010. Structural basis of local, pH-dependent conformational changes in glycoprotein B from herpes simplex virus type 1. J Virol 84:12924–12933. doi: 10.1128/JVI.01750-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Peeters B, de Wind N, Hooisma M, Wagenaar F, Gielkens A, Moormann R. 1992. Pseudorabies virus envelope glycoproteins gp50 and gII are essential for virus penetration, but only gII is involved in membrane fusion. J Virol 66:894–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stiasny K, Koessl C, Heinz FX. 2003. Involvement of lipids in different steps of the flavivirus fusion mechanism. J Virol 77:7856–7862. doi: 10.1128/JVI.77.14.7856-7862.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zaitseva E, Yang ST, Melikov K, Pourmal S, Chernomordik LV. 2010. Dengue virus ensures its fusion in late endosomes using compartment-specific lipids. PLoS Pathog 6:e1001131. doi: 10.1371/journal.ppat.1001131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guardado-Calvo P, Rey FA. 2017. The envelope proteins of the Bunyavirales. Adv Virus Res 98:83–118. doi: 10.1016/bs.aivir.2017.02.002. [DOI] [PubMed] [Google Scholar]
- 91.Domanska MK, Wrona D, Kasson PM. 2013. Multiphasic effects of cholesterol on influenza fusion kinetics reflect multiple mechanistic roles. Biophys J 105:1383–1387. doi: 10.1016/j.bpj.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lai AL, Moorthy AE, Li Y, Tamm LK. 2012. Fusion activity of HIV gp41 fusion domain is related to its secondary structure and depth of membrane insertion in a cholesterol-dependent fashion. J Mol Biol 418:3–15. doi: 10.1016/j.jmb.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kirkpatrick RB, Ganguly S, Angelichio M, Griego S, Shatzman A, Silverman C, Rosenberg M. 1995. Heavy chain dimers as well as complete antibodies are efficiently formed and secreted from Drosophila via a BiP-mediated pathway. J Biol Chem 270:19800–19805. doi: 10.1074/jbc.270.34.19800. [DOI] [PubMed] [Google Scholar]
- 94.Fan SQ, Huang W, Wang LX. 2012. Remarkable transglycosylation activity of glycosynthase mutants of endo-D, an endo-beta-N-acetylglucosaminidase from Streptococcus pneumoniae. J Biol Chem 287:11272–11281. doi: 10.1074/jbc.M112.340497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kabsch W. 2010. XDS. Acta Crystallogr D Biol Crystallogr 66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bricogne G, Blanc E, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Smart OS, Vonrhein C, Womack TO. 2009. BUSTER, v2.8.0. Global Phasing Ltd., Cambridge, United Kingdom. [Google Scholar]
- 98.Castile JD, Taylor KM. 1999. Factors affecting the size distribution of liposomes produced by freeze-thaw extrusion. Int J Pharm 188:87–95. doi: 10.1016/S0378-5173(99)00207-0. [DOI] [PubMed] [Google Scholar]
- 99.Böhm SW, Eckroth E, Backovic M, Klupp BG, Rey FA, Mettenleiter TC, Fuchs W. 2015. Structure-based functional analyses of domains II and III of pseudorabies virus glycoprotein H. J Virol 89:1364–1376. doi: 10.1128/JVI.02765-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mettenleiter TC. 1989. Glycoprotein gIII deletion mutants of pseudorabies virus are impaired in virus entry. Virology 171:623–625. doi: 10.1016/0042-6822(89)90635-1. [DOI] [PubMed] [Google Scholar]
- 101.Kopp M, Granzow H, Fuchs W, Klupp BG, Mundt E, Karger A, Mettenleiter TC. 2003. The pseudorabies virus UL11 protein is a virion component involved in secondary envelopment in the cytoplasm. J Virol 77:5339–5351. doi: 10.1128/JVI.77.9.5339-5351.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pavlova SP, Veits J, Keil GM, Mettenleiter TC, Fuchs W. 2009. Protection of chickens against H5N1 highly pathogenic avian influenza virus infection by live vaccination with infectious laryngotracheitis virus recombinants expressing H5 hemagglutinin and N1 neuraminidase. Vaccine 27:773–785. doi: 10.1016/j.vaccine.2008.11.033. [DOI] [PubMed] [Google Scholar]
- 103.DeLano WL. 2002. The PyMOL Molecular Graphics System, DeLano Scientific, San Carlos, CA, USA.
- 104.Eisenberg D, Schwarz E, Komaromy M, Wall R. 1984. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J Mol Biol 179:125–142. doi: 10.1016/0022-2836(84)90309-7. [DOI] [PubMed] [Google Scholar]
- 105.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. 2001. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A 98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hasegawa H, Holm L. 2009. Advances and pitfalls of protein structural alignment. Curr Opin Struct Biol 19:341–348. doi: 10.1016/j.sbi.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 107.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gouet P, Courcelle E, Stuart DI, Metoz F. 1999. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 109.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.