

ABSTRACT
Conformational conversion of the cellular isoform of prion protein, PrPC, into the abnormally folded, amyloidogenic isoform, PrPSc, is a key pathogenic event in prion diseases, including Creutzfeldt-Jakob disease in humans and scrapie and bovine spongiform encephalopathy (BSE) in animals. We previously reported that the octapeptide repeat (OR) region could be dispensable for converting PrPC into PrPSc after infection with RML prions. We demonstrated that mice transgenically expressing mouse PrP with deletion of the OR region on the PrP knockout background, designated Tg(PrPΔOR)/Prnp0/0 mice, did not show reduced susceptibility to RML scrapie prions, with abundant accumulation of PrPScΔOR in their brains. We show here that Tg(PrPΔOR)/Prnp0/0 mice were highly resistant to BSE prions, developing the disease with markedly elongated incubation times after infection with BSE prions. The conversion of PrPΔOR into PrPScΔOR was markedly delayed in their brains. These results suggest that the OR region may have a crucial role in the conversion of PrPC into PrPSc after infection with BSE prions. However, Tg(PrPΔOR)/Prnp0/0 mice remained susceptible to RML and 22L scrapie prions, developing the disease without elongated incubation times after infection with RML and 22L prions. PrPScΔOR accumulated only slightly less in the brains of RML- or 22L-infected Tg(PrPΔOR)/Prnp0/0 mice than PrPSc in control wild-type mice. Taken together, these results indicate that the OR region of PrPC could play a differential role in the pathogenesis of BSE prions and RML or 22L scrapie prions.
IMPORTANCE Structure-function relationship studies of PrPC conformational conversion into PrPSc are worthwhile to understand the mechanism of the conversion of PrPC into PrPSc. We show here that, by inoculating Tg(PrPΔOR)/Prnp0/0 mice with the three different strains of RML, 22L, and BSE prions, the OR region could play a differential role in the conversion of PrPC into PrPSc after infection with RML or 22L scrapie prions and BSE prions. PrPΔOR was efficiently converted into PrPScΔOR after infection with RML and 22L prions. However, the conversion of PrPΔOR into PrPScΔOR was markedly delayed after infection with BSE prions. Further investigation into the role of the OR region in the conversion of PrPC into PrPSc after infection with BSE prions might be helpful for understanding the pathogenesis of BSE prions.
KEYWORDS: bovine spongiform encephalopathy, BSE, octapeptide repeat, prion, prion protein, scrapie
INTRODUCTION
Prions are causative agents of prion diseases, a group of fatal neurodegenerative disorders that include Creutzfeldt-Jakob disease and Gerstmann-Sträussler-Scheinker disease in humans and scrapie and bovine spongiform encephalopathy (BSE) in animals (1). Prions are believed to consist of the abnormally folded, relatively proteinase K (PK)-resistant isoform of prion protein, designated PrPSc, and propagate through conformational conversion of the PK-sensitive, cellular isoform of PrP, PrPC, into PrPSc (1). PrPC is a membrane glycoprotein tethered to the cell surface via a glycosylphosphatidylinositol moiety expressed most abundantly in the central nervous system, particularly by neurons (2). We and others have shown that mice devoid of PrPC (Prnp0/0) are resistant to prions, neither developing the disease nor propagating prions even after intracerebral inoculation with the prions (3–6), clearly indicating that the conversion of PrPC into PrPSc is a key pathogenic event in prion diseases.
There is the so-called octapeptide repeat (OR) region, which consists of 5 copies of an octapeptide sequence in most mammalian species and 6 copies in a dominant population of cattle, in the N-terminal domain of PrPC (7–10). Insertional mutations of one or more extra octapeptide sequences in the OR region lead to spontaneous conversion of the mutant PrP into the pathogenic PrP. This eventually causes hereditary prion diseases in humans (11). Transgenic mice expressing mouse PrP with an insertion of 9 additional OR sequences (14 OR sequences in total), designated as Tg(PG14) mice, or bovine PrP with an insertion of an additional 4 OR sequences (10 OR sequences in total), designated as bo10ORTg mice, were shown to spontaneously develop neurodegenerative disease with accumulation of the relatively PK-resistant, but noninfectious PrP in their brains (12–14). Thus, insertion of extra octapeptide sequences in the OR region could render the mutant PrP structurally unstable, thereby causing conformational changes in the mutant PrP to form pathogenic PrP.
Insertion of extra octapeptide sequences in the OR region has also been shown to increase susceptibility to BSE prions in mice. bo10ORTg and bo7ORTg mice were reported to develop prion disease earlier than control bo6ORTg mice after infection with BSE prions (14, 15). Conversely, deletion of one octapeptide sequence in the OR region was reported to reduce susceptibility to BSE prions in mice (16). These results suggest that the OR region could have an important role in the pathogenesis of BSE prions. However, we previously showed that Prnp0/0 mice transgenic for mouse PrP with deletion of the OR region alone, designated Tg(PrPΔOR)/Prnp0/0 mice, did not have reduced susceptibility to RML scrapie prions, developing the disease without elongated incubation times after infection with RML prions (17). Taken together, these results suggest that the OR region could have a differential role in the pathogenesis of BSE and RML prions.
In the present study, to verify this possibility, we intracerebrally inoculated Tg(PrPΔOR)/Prnp0/0 mice with BSE and RML prions. We also inoculated Tg mice with 22L scrapie prions. Tg(PrPΔOR)/Prnp0/0 mice inoculated with RML and 22L prions developed the disease without elongated incubation times. In contrast, incubation times were markedly elongated in Tg(PrPΔOR)/Prnp0/0 mice inoculated with BSE prions, with delayed accumulation of PrPScΔOR in their brains. These results clearly show that the OR region plays a differential role in the pathogenesis of BSE prions and RML or 22L scrapie prions.
RESULTS
Tg(PrPΔOR)/Prnp0/0 mice are highly resistant to BSE prions, but susceptible to RML and 22L prions.
To investigate the role of the OR region in prion pathogenesis, we intracerebrally inoculated Tg(PrPΔOR)/Prnp0/0 and C57BL/6 wild-type (WT) mice with RML, 22L, and BSE prions. As we previously reported (17), RML-inoculated Tg(PrPΔOR)/Prnp0/0 mice displayed foreleg paresis in addition to other disease-specific symptoms observed in control WT mice, such as emaciation, ruffled body hair, kyphosis, crossing leg, and paralysis of the hind legs. 22L-inoculated Tg(PrPΔOR)/Prnp0/0 mice also developed foreleg paresis. Other symptoms were commonly observed in 22L-infected Tg(PrPΔOR)/Prnp0/0 and WT mice. BSE-infected Tg(PrPΔOR)/Prnp0/0 and WT mice developed similar symptoms. Consistent with our previous results (17), incubation and survival times were shortened in Tg(PrPΔOR)/Prnp0/0 mice inoculated with RML prions compared to control WT mice (P < 0.0001) (Table 1). WT mice developed the disease at 159 ± 2 (average ± standard deviation [SD]) days postinoculation (dpi) and became terminal at 175 ± 3 dpi, while incubation and survival times in Tg(PrPΔOR)/Prnp0/0 mice were shortened by 29 and 35 days, respectively (Table 1). Tg(PrPΔOR)/Prnp0/0 mice express PrPΔOR in their brains about 1.7 times more than PrPC in WT mice (Fig. 1A). The higher susceptibility of Tg(PrPΔOR)/Prnp0/0 mice to RML prions could be due to higher expression of PrPΔOR in Tg(PrPΔOR)/Prnp0/0 mice than PrPC in WT mice. Tg(PrPΔOR)/Prnp0/0 mice also showed higher susceptibility to 22L prions than WT mice. Incubation and survival times were 143 ± 1 and 156 ± 2 dpi in control WT mice but shortened by 32 and 24 days in Tg(PrPΔOR)/Prnp0/0 mice inoculated with 22L prions, respectively (P < 0.0001) (Table 1). In contrast, Tg(PrPΔOR)/Prnp0/0 mice exhibited markedly reduced susceptibility to BSE prions. Incubation and survival times were elongated by 141 and 141 days, respectively, in Tg(PrPΔOR)/Prnp0/0 mice from 172 ± 6 and 180 ± 8 dpi in WT mice inoculated with BSE prions, respectively (P < 0.0001) (Table 1). These results show that Tg(PrPΔOR)/Prnp0/0 mice are highly resistant to BSE prions but remain susceptible to RML and 22L prions, indicating that the OR region could have a crucial role in determination of the susceptibility to BSE prions in mice.
TABLE 1.
Incubation and survival times of WT and Tg(PrPΔOR)/Prnp0/0 mice inoculated with various prions
| Prions | Recipient mice | PrP expression (fold)a | No. of diseased mice/total | Incubation time (days)b | Survival time (days)c | P valued |
|---|---|---|---|---|---|---|
| RML | WT | 1 | 18/18 | 159 ± 2 | 175 ± 3 | <0.0001 |
| Tg(PrPΔOR)/Prnp0/0 | 1.7 | 15/15 | 130 ± 7 | 140 ± 9 | ||
| 22L | WT | 1 | 13/13 | 143 ± 1 | 156 ± 2 | <0.0001 |
| Tg(PrPΔOR)/Prnp0/0 | 1.7 | 16/16 | 111 ± 9 | 132 ± 14 | ||
| BSE | WT | 1 | 11/11 | 172 ± 6 | 180 ± 8 | <0.0001 |
| Tg(PrPΔOR)/Prnp0/0 | 1.7 | 21/21 | 313 ± 4 | 321 ± 4 |
Expression levels were compared to those of PrPC in WT mice using Western blotting (Fig. 1A).
Time to onset of disease (average ± SD).
Time to terminal stage of disease (average ± SD).
P values indicate significance of comparison of incubation and survival times between WT and Tg(PrPΔOR)/Prnp0/0 mice by log rank (Mantel-Cox) test.
FIG 1.

PrPΔOR expression in the brains of Tg(PrPΔOR)/Prnp0/0 and Tg(PrPΔOR-3608)/Prnp0/0 mice. (A, left panel) Western blotting with 6D11 anti-PrP Ab of the brains of WT (n = 3), Tg(PrPΔOR)/Prnp0/0 (n = 3), and Prnp0/0 (n = 3) mice. (Right panel) Expression levels of PrPΔOR in Tg(PrPΔOR)/Prnp0/0 mice compared to PrPC in WT mice. (B, left panel) Western blotting of the brains of WT (n = 3), Prnp+/0 (n = 3), Tg(PrPΔOR-3608)/Prnp0/0 (n = 3), and Prnp0/0 (n = 3) mice with 6D11 anti-PrP Ab. (Right panel) Expression levels of PrPC in Prnp+/0 mice and PrPΔOR in Tg(PrPΔOR-3608)/Prnp0/0 mice compared to PrPC in WT mice. AU, arbitrary units.
To confirm that the reduced susceptibility to BSE prions in Tg(PrPΔOR)/Prnp0/0 mice is not a specific phenotype in the Tg line used, we produced another line of Tg(PrPΔOR)/Prnp0/0 mice, here referred to as Tg(PrPΔOR-3608)/Prnp0/0. They expressed PrPΔOR in their brains at a similar level to PrPC in Prnp+/0 mice (Fig. 1B). The expression levels of PrPΔOR in Tg(PrPΔOR-3608)/Prnp0/0 mice and PrPC in Prnp+/0 mice are at 45% ± 9% and 51% ± 5% of those of PrPC in WT mice, respectively (P = 0.29). We thus intracerebrally inoculated BSE prions into Tg(PrPΔOR-3608)/Prnp0/0 mice and control Prnp+/0 mice as controls. Longer incubation times were also observed in Tg(PrPΔOR-3608)/Prnp0/0 mice after inoculation with BSE prions, compared to control Prnp+/0 mice (P < 0.0001) (Table 2). Prnp+/0 mice developed the disease at 274 ± 6 dpi and became terminal at 290 ± 10 dpi, while Tg(PrPΔOR-3608)/Prnp0/0 mice succumbed to the disease at 335 ± 26 dpi, becoming terminal at 343 ± 27 dpi (Table 2). The lower susceptibility of Tg(PrPΔOR-3608)/Prnp0/0 mice to BSE prions than Prnp+/0 mice despite the similar expression of PrPΔOR in Tg(PrPΔOR-3608)/Prnp0/0 mice to PrPC in Prnp+/0 mice reinforces that the OR region could have an important role in determination of the susceptibility to BSE prions.
TABLE 2.
Incubation and survival times of Prnp+/0 and Tg(PrPΔOR-3608)/Prnp0/0 mice inoculated with BSE prions
| Recipient mice | PrP expression (fold)a | No. of diseased mice/total | Incubation time (days)b | Survival time (days)c | P valued |
|---|---|---|---|---|---|
| Prnp+/0 | 0.5 | 13/13 | 274 ± 6 | 290 ± 10 | <0.0001 |
| Tg(PrPΔOR-3608)/Prnp0/0 | 0.5 | 12/12 | 335 ± 26 | 343 ± 27 |
Expression levels were compared to those of PrPC in WT mice using Western blotting (Fig. 1B).
Time to onset of disease (average ± SD).
Time to terminal stage of disease (average ± SD).
The P value indicates the significance of comparison of incubation and survival times between Tg(PrPΔOR)/Prnp0/0 and Prnp+/0 mice by log rank (Mantel-Cox) test.
The OR region is not essential for the conversion of PrPC into PrPSc and brain pathologies.
To investigate the role of the OR region in the conversion of PrPC into PrPSc, we investigated the brains of terminally ill Tg(PrPΔOR)/Prnp0/0 mice infected with RML, 22L, and BSE prions for PrPScΔOR. To detect PrPScΔOR, the brain homogenates were treated with PK and then subjected to Western blotting with 6D11 anti-PrP antibody (Ab), which recognizes residues 93 to 109 of mouse PrP. Consistent with our previous results (17), PrPScΔOR was detected slightly but significantly less in the brains of RML-infected, terminally ill Tg(PrPΔOR)/Prnp0/0 mice than PrPSc in control WT mice (P = 0.006) (Fig. 2A). PrPScΔOR was also detected in the brains of 22L-infected, terminally ill Tg(PrPΔOR)/Prnp0/0 mice slightly less than PrPSc in control WT mice (P = 0.136) (Fig. 2B). However, PrPScΔOR accumulated in the brains of BSE-infected, terminally ill Tg(PrPΔOR)/Prnp0/0 mice more abundantly than PrPSc in control WT mice (P = 0.028) (Fig. 2C). Immunohistochemistry revealed indistinguishable staining for PrPSc and PrPScΔOR throughout the brain slices of terminally ill WT and Tg(PrPΔOR)/Prnp0/0 mice infected with RML, 22L, and BSE prions (Fig. 3A to D). These results indicate that while the OR region is not essential for the conversion of PrPC into PrPSc after infection with prions, it could affect the final accumulation levels of PrPSc in brains in a strain-dependent manner.
FIG 2.

Different levels of PrPScΔOR accumulated in the brains of Tg(PrPΔOR)/Prnp0/0 mice infected with RML, 22L, and BSE prions at terminal stages. Western blotting with 6D11 anti-PrP Ab of the brains of terminally ill WT (n = 3) and Tg(PrPΔOR)/Prnp0/0 mice (n = 3) infected with RML (A), 22L (B), and BSE (C) prions after treatment with (+) or without (−) PK. The right panels show levels of PrPScΔOR in Tg(PrPΔOR)/Prnp0/0 mice compared to PrPSc in WT mice. AU, arbitrary units; ns, not significant; *, P < 0.05; **, P < 0.01.
FIG 3.

Indistinguishable distribution of PrPSc and PrPScΔOR accumulated in the brains of terminally ill WT and Tg(PrPΔOR)/Prnp0/0 mice. Brain slices from uninfected (A) and RML (B)-, 22L (C)-, and BSE (D)-infected terminally ill WT (n = 3 in each mouse group) and Tg(PrPΔOR)/Prnp0/0 (n = 3 in each mouse group) mice were immunohistochemically stained for PrPSc and PrPScΔOR by 6D11 anti-PrP Ab using the HCl autoclaving method. Three sections from each mouse brain were subjected to investigation of PrPSc and PrPScΔOR distribution. Cx, cerebral cortex; Hp, hippocampus; Th, thalamus; Cb, cerebellum. Size bars, 100 μm.
We also pathologically investigated the brains of RML-, 22L-, and BSE-infected, terminally ill Tg(PrPΔOR)/Prnp0/0 and WT mice for vacuolation. No significant difference in the number of vacuoles was detected in each brain area between Tg(PrPΔOR)/Prnp0/0 and WT mice infected with RML, 22L, and BSE prions (Fig. 4A to D and Fig. 5A to D), suggesting that PrPScΔOR and PrPSc might be similarly pathogenic in brains.
FIG 4.
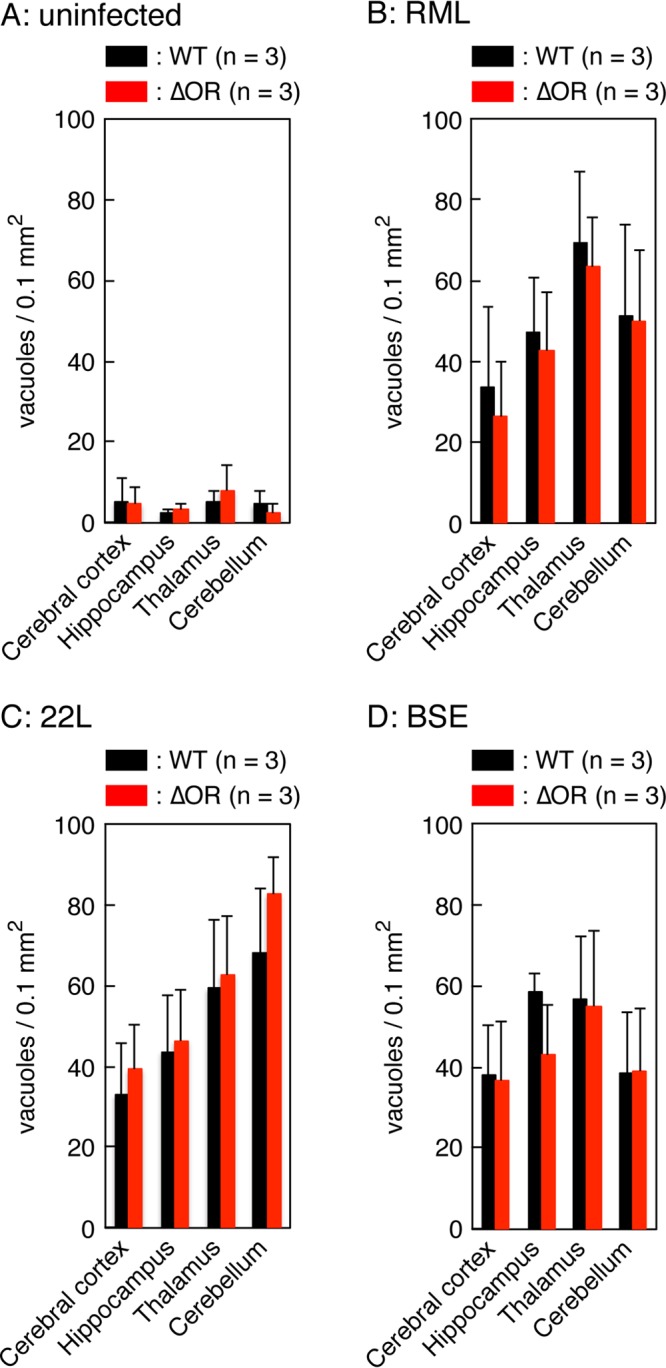
Similar vacuolation in the brains of terminally ill WT and Tg(PrPΔOR)/Prnp0/0 mice. Brain slices from uninfected (A) and RML (B)-, 22L (C)-, and BSE (D)-infected terminally ill WT (n = 3 in each mouse group) and Tg(PrPΔOR)/Prnp0/0 (n = 3 in each mouse group) mice were subjected to hematoxylin-eosin staining, and vacuoles in 0.1-mm2 areas were counted in various brain regions, including the cerebral cortex, hippocampus, thalamus, and cerebellum, respectively. Three sections from each mouse brain were subjected to counting of vacuoles.
FIG 5.

Similar pathologies in the brains of terminally ill WT and Tg(PrPΔOR)/Prnp0/0 mice infected with RML, 22L, or BSE prions. Brain slices from uninfected (A) and RML (B)-, 22L (C)-, and BSE (D)-infected terminally ill WT (n = 3 in each mouse group) and Tg(PrPΔOR)/Prnp0/0 (n = 3 in each mouse group) mice were subjected to hematoxylin-eosin staining. Three sections from each mouse brain were used for the pathological examinations. Cx, cerebral cortex; Hp, hippocampus; Th, thalamus; Cb, cerebellum. Size bars, 100 μm.
Delayed brain accumulation of PrPScΔOR in BSE-infected Tg(PrPΔOR)/Prnp0/0 mice.
To gain insights into the reduced susceptibility of Tg(PrPΔOR)/Prnp0/0 mice to BSE prions, we sacrificed WT and Tg(PrPΔOR)/Prnp0/0 mice at 190 dpi with BSE prions and compared the levels of PrPScΔOR accumulated in the brains of Tg(PrPΔOR)/Prnp0/0 mice to those of PrPSc in WT mice. WT mice were terminally ill around 190 dpi. However, Tg(PrPΔOR)/Prnp0/0 mice were still healthy by 190 dpi. PrPScΔOR was detected at very low levels in Tg(PrPΔOR)/Prnp0/0 mice compared to those of PrPSc in WT mice (P = 0.0007) (Fig. 6A and B). These results indicate that the conversion of PrPΔOR into PrPScΔOR is less efficient than that of full-length PrPC into PrPSc after infection with BSE prions, suggesting that the OR region could be important for BSE prions to convert PrPC into PrPSc. It is therefore possible that the reduced susceptibility to BSE prions in Tg(PrPΔOR)/Prnp0/0 mice could be attributable to the inefficient conversion of PrPΔOR into PrPScΔOR after infection with BSE prions.
FIG 6.
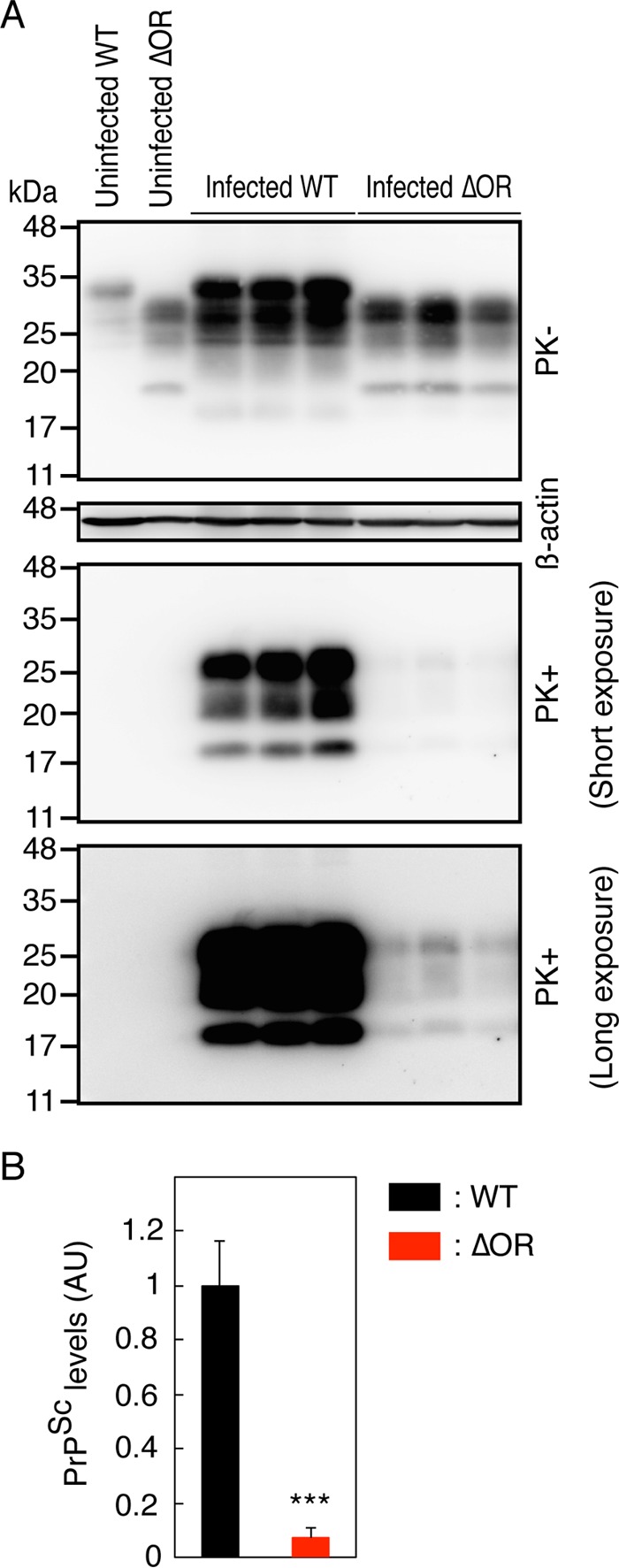
Delayed accumulation of PrPScΔOR in the brains of Tg(PrPΔOR)/Prnp0/0 mice infected with BSE prions. (A) Brain homogenates from WT (n = 3) and Tg(PrPΔOR)/Prnp0/0 mice (n = 3) sacrificed at 190 dpi with BSE prions were treated with (+) or without (−) PK and then subjected to Western blotting with the 6D11 anti-PrP Ab. (B) PrPSc and PrPScΔOR levels in the lower panels of panel A. AU, arbitrary units; ***, P < 0.001.
The pre-OR region in PrPScΔOR is PK resistant.
We previously showed that the pre-OR region consisting of residues 23 to 50 of PrPScΔOR produced in the brains of RML-infected Tg(PrPΔOR)/Prnp0/0 mice forms a PK-resistant conformation (17). To investigate whether or not 22L and BSE prions could also convert the pre-OR region into a PK-resistant structure upon the conversion of PrPΔOR into PrPScΔOR, we treated 22L- and BSE-infected as well as RML-infected Tg(PrPΔOR)/Prnp0/0 brain homogenates with PK. They were then subjected to Western blotting with IBL-N anti-PrP Abs, which were raised against a pre-OR synthetic peptide comprising residues 24 to 37 (18). The Abs exhibited no PK-resistant signals in the RML-, 22L-, and BSE-infected WT brain homogenates (Fig. 7A to C). This is consistent with the pre-OR region of full-length PrPSc being PK sensitive. However, PK-resistant signals were observed in the RML-, 22L-, and BSE-infected Tg(PrPΔOR)/Prnp0/0 brain homogenates (Fig. 7A to C), indicating that the pre-OR region of PrPScΔOR is converted into a PK-resistant structure after infection with RML, 22L, and BSE prions.
FIG 7.
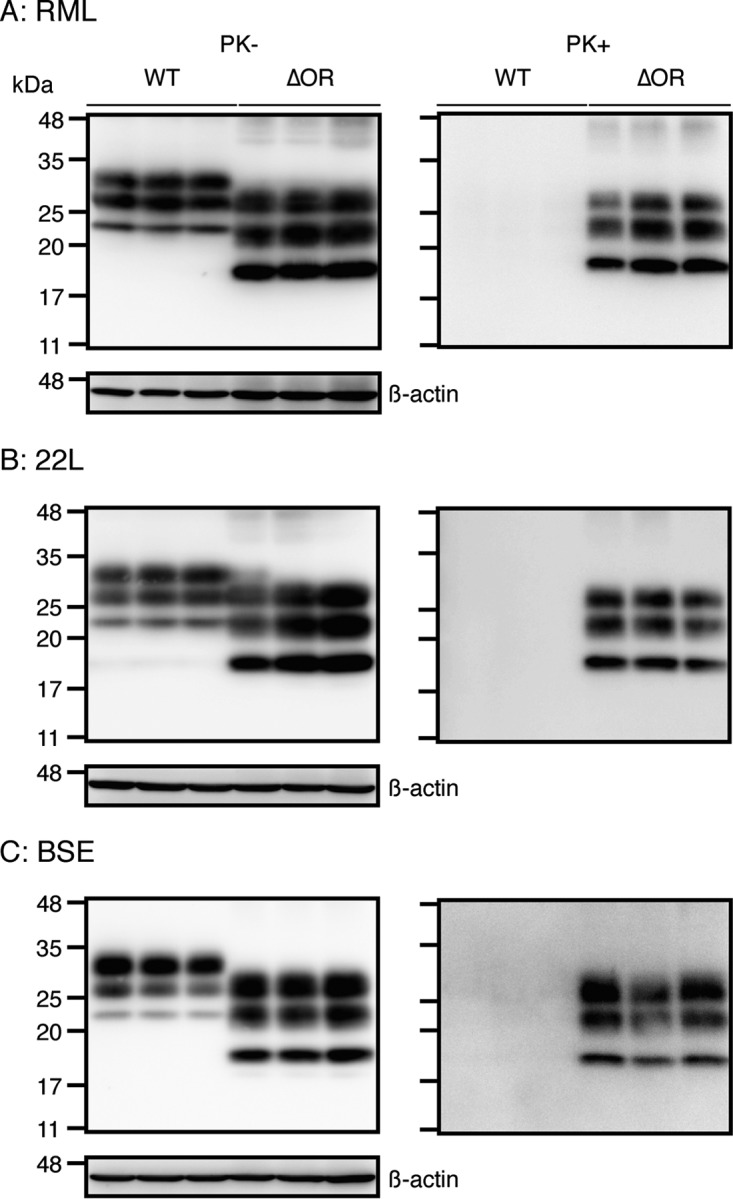
The pre-OR region of PrPScΔOR is PK resistant, but not in WT PrPSc. Brain homogenates from terminally ill WT (n = 3) and Tg(PrPΔOR)/Prnp0/0 mice (n = 3) infected with RML (A), 22L (B), and BSE (C) prions were treated with (+) or without (−) PK and then subjected to Western blotting with IBL-N anti-PrP Abs.
Tg(PrPΔOR)/Prnp0/0 mice are also highly resistant to secondarily inoculated PrPScΔOR-BSE prions.
Differences in the primary sequence between PrPC in recipient animals and PrPSc in an inoculum often create the so-called “prion transmission barrier” leading to elongation of incubation times in recipient animals (19). If a prion transmission barrier is responsible for longer incubation times in primary inoculated mice, secondary inoculation into mice with the same genotype causes shorter incubation times. To investigate whether or not PrPΔOR might create a prion transmission barrier against full-length PrPSc and thereby render Tg(PrPΔOR)/Prnp0/0 mice less susceptible to BSE prions, we secondarily inoculated Tg(PrPΔOR)/Prnp0/0 and WT mice by the intracerebral route with brain homogenates from BSE-infected, terminally ill Tg(PrPΔOR)/Prnp0/0 mice containing PrPScΔOR. We also secondarily inoculated Tg(PrPΔOR)/Prnp0/0 and WT mice with brain homogenates from RML- and 22L-infected, terminally ill Tg(PrPΔOR)/Prnp0/0 mice. Similar clinical symptoms were observed in Tg(PrPΔOR)/Prnp0/0 mice as well as WT mice after primary and secondary inoculation with RML-, 22L-, or BSE-infected Tg(PrPΔOR)/Prnp0/0 brain homogenate. Incubation times were slightly but not significantly shorter in Tg(PrPΔOR)/Prnp0/0 mice secondarily inoculated with RML-infected Tg(PrPΔOR)/Prnp0/0 brain homogenates compared to control WT mice (P > 0.3) (Table 3). 22L-infected Tg(PrPΔOR)/Prnp0/0 brain homogenates caused significantly shorter incubation times in Tg(PrPΔOR)/Prnp0/0 mice than in WT mice (P < 0.001) (Table 3). However, incubation times were still longer in Tg(PrPΔOR)/Prnp0/0 mice than in WT mice even after secondary inoculation with the BSE-infected Tg(PrPΔOR)/Prnp0/0 brain homogenate (Table 3). WT mice developed the disease at 165 ± 8 dpi, whereas Tg(PrPΔOR)/Prnp0/0 mice succumbed to the disease with longer incubation times of 307 ± 9 dpi (P < 0.0001) (Table 3). These results indicate that the different primary sequence between PrPΔOR and WT PrPSc does not create a transmission barrier for BSE prions.
TABLE 3.
Tg(PrPΔOR)/Prnp0/0 mice are still susceptible to RML and 22L prions, but highly resistant to BSE prions, even after secondary inoculation with RML-, 22L-, and BSE-infected Tg(PrPΔOR)/Prnp0/0 brain homogenates
| Tg(PrPΔOR)/Prnp0/0 brain homogenate inoculum | Recipient mice | No. of diseased mice/total | Incubation time (days)a | Survival time (days)b | P valuec |
|---|---|---|---|---|---|
| RML | WT | 10/10 | 144 ± 3 | 176 ± 7 | >0.3 |
| Tg(PrPΔOR)/Prnp0/0 | 10/10 | 145 ± 7 | 164 ± 15 | ||
| 22L | WT | 10/10 | 148 ± 6 | 155 ± 4 | <0.0001 |
| Tg(PrPΔOR)/Prnp0/0 | 10/10 | 104 ± 7 | 109 ± 3 | ||
| BSE | WT | 6/6 | 165 ± 8 | 179 ± 9 | <0.001 |
| Tg(PrPΔOR)/Prnp0/0 | 6/6 | 307 ± 9 | 331 ± 20 |
Time to onset of disease (average ± SD).
Time to terminal stage of disease (average ± SD).
P values indicate significance of comparison of incubation and survival times between Tg(PrPΔOR)/Prnp0/0 and WT mice by log rank (Mantel-Cox) test.
WT mice infected with PrPSc- and PrPScΔOR prions accumulate PrPSc with the same biochemical properties.
To assess the pathogenic effects of PrPScΔOR prions on the conversion of PrPC into PrPSc, we investigated accumulation levels, the PK-resistant core size, and glycosylation patterns of PrPSc molecules in the brains of WT mice inoculated with RML-, 22L-, or BSE-infected Tg(PrPΔOR)/Prnp0/0 and WT brain homogenates containing PrPScΔOR and PrPSc, respectively. Western blotting with 6D11 anti-PrP Ab revealed that similar amounts of PrPSc accumulated in terminally ill WT mice inoculated with PrPSc and PrPScΔOR of RML, 22L, or BSE prions (Fig. 8A). Similar migration patterns were also observed for PrPSc in WT mice inoculated with RML-, 22L-, or BSE-PrPSc and -PrPScΔOR (Fig. 8A). The deglycosylated, PK-resistant fragment of PrPSc in these brains also showed the same migration distance (Fig. 8B). These results suggest that the PK cleavage site is the same for PrPSc produced after inoculation with RML-, 22L-, or BSE-PrPSc and -PrPScΔOR. We also investigated glycosylation patterns of PrPSc in these brains. The ratio of di-, mono-, and unglycosylated forms of PrPSc in WT mice inoculated with RML-, 22L-, or BSE-PrPScΔOR were similar to those of PrPSc in WT mice inoculated with RML-, 22L-, or BSE-PrPSc (Fig. 8C). Taken together, these results indicate that the biochemical properties of PrPSc produced in WT mice after inoculation with PrPSc- and PrPScΔOR prions are similar, suggesting that PrPScΔOR prions might have the same pathogenic properties as PrPSc prions.
FIG 8.

Biochemical characterization of PrPSc produced in WT mice after inoculation with full-length PrPSc and PrPScΔOR prions. (A) Western blotting with 6D11 anti-PrP Ab of the brains of terminally ill WT mice inoculated with RML-, 22L-, or BSE-infected WT and Tg(PrPΔOR)/Prnp0/0 brain homogenates. (B) Western blotting with 6D11 anti-PrP Ab of the brains of terminally ill WT mice inoculated with RML-, 22L-, or BSE-infected WT and Tg(PrPΔOR)/Prnp0/0 brain homogenates after treatment with PNGase F. (C) Percentage of the diglycosylated, monoglycosylated, and unglycosylated forms of PrPSc in the brains of terminally ill WT mice inoculated with RML-, 22L-, or BSE-infected WT and Tg(PrPΔOR)/Prnp0/0 brain homogenates.
DISCUSSION
In the present study, we studied the role of the OR region for PrPC to support prion infection by inoculating RML, 22L, and BSE prions into Tg(PrPΔOR)/Prnp0/0 mice, which express PrP with a deletion of the OR region alone on the Prnp0/0 background. Compared to control WT mice, Tg(PrPΔOR)/Prnp0/0 mice had accelerated disease, exhibiting shorter incubation times after infection with RML and 22L prions. Shorter incubation times in RML-inoculated Tg(PrPΔOR)/Prnp0/0 mice are consistent with our previous results (17). The length of incubation times inversely correlates to the expression levels of PrPC in animals infected with prions (20). Tg(PrPΔOR)/Prnp0/0 mice express PrPΔOR in their brains more than PrPC in WT mice. Therefore, the higher susceptibility of Tg(PrPΔOR)/Prnp0/0 mice to RML and 22L prions could be due to the higher expression of PrPΔOR in their brains, suggesting that the OR region of PrPC might be dispensable for RML and 22L infection in mice. In contrast, in spite of the higher expression of PrPΔOR, Tg(PrPΔOR)/Prnp0/0 mice were highly resistant to BSE prions, exhibiting markedly elongated incubation times after infection with BSE prions, indicating that the OR region could play a crucial role for PrPC to support BSE infection in mice.
The so-called prion transmission barrier often occurs when PrPSc in an inoculum and PrPC in recipient animals differ in primary sequence, interfering with prion infection and eventually causing elongated incubation times in the recipient animals (21, 22). However, Tg(PrPΔOR)/Prnp0/0 mice remained highly resistant to BSE prions, developing the disease with longer incubation times than WT mice, even after secondary inoculation with brain homogenates from BSE-infected, terminally ill Tg(PrPΔOR)/Prnp0/0 mice, which contain PrPScΔOR. The longer incubation times of Tg(PrPΔOR)/Prnp0/0 mice secondarily inoculated with PrPScΔOR-associated BSE prions indicate that the reduced susceptibility of Tg(PrPΔOR)/Prnp0/0 mice to full-length PrPSc-associated BSE prions primarily inoculated is not due to the different primary sequences between PrPSc in the inoculum and PrPΔOR in the recipient mice. This reinforces the crucial role of the OR region in BSE infection.
Similar strain-dependent different susceptibility has been reported in mice transgenically expressing PrP with specific mutations or deletions in the sequence. Prnp0/0 mice transgenic for mouse PrP with a serine residue at codon 170, designated Tg(PrP-170S)/Prnp0/0 mice, became highly resistant to RML and 79A prions, but were still susceptible to 22L and ME7 prions (23). Tg(OvPrP-V136)/Prnp0/0 mice expressing ovine PrP with a valine residue at codon 136 on the Prnp0/0 background were still susceptible to SSBP1 prions, but became resistant to CH1641 prions (24). We previously reported that Tg(MHM2Δ23-88)/Prnp0/0 mice, which express mouse-hamster chimeric PrP with the deletion of residues 23 to 88, became highly resistant to RML, but were still susceptible to 22L prions (25). Elucidation of the mechanism for the strain-dependent susceptibility would be important for understanding of the pathogenesis of prion diseases.
The conversion efficiency of PrPC into PrPSc is a key factor determining prion susceptibility. Indeed, the pathogenic PrPs were undetectable or much less accumulated in the brains of Tg(PrP-170S)/Prnp0/0, Tg(OvPrP-V136)/Prnp0/0, or Tg(MHM2Δ23-88)/Prnp0/0 mice inoculated with resistant prion strains, but not with susceptible prion strains (23–25). We also showed that PrPScΔOR was accumulated much less in the brains of Tg(PrPΔOR)/Prnp0/0 mice than PrPSc in control WT mice at 190 dpi with BSE prions, indicating that, compared to the conversion of PrPC into PrPSc, the conversion of PrPΔOR into PrPScΔOR is much more inefficient after infection with BSE prions. However, RML- or 22L-inoculated Tg(PrPΔOR)/Prnp0/0 mice accumulated PrPScΔOR in their brains only slightly less than or similarly to PrPSc in control WT mice, respectively, suggesting that RML and 22L prions could efficiently convert PrPΔOR into PrPScΔOR. The different levels of efficiency of conversion of PrPΔOR into PrPScΔOR after infection with BSE prions and RML or 22L prions indicate that the OR region might be differently involved in the conversion of PrPC into PrPSc after infection with BSE prions and RML or 22L prions. It is thus conceivable that the different role of the OR region in the conversion of PrPC into PrPSc could underlie the different susceptibilities of Tg(PrPΔOR)/Prnp0/0 mice to BSE prions and RML or 22L prions.
The conformational selection model has been proposed as a mechanism to explain the strain-dependent conversion of PrPC into PrPSc. PrPSc molecules from different strains are believed to adopt different conformations (21, 22). Indeed, two different prion strains of transmissible mink encephalopathy, HY and DY, have been shown to produce PrPSc with strain-specific, different PK cleavage sites (26). DY-PrPSc has a shorter PK-resistant fragment than HY-PrPSc (27). The different PK cleavage sites of DY-PrPSc and HY-PrPSc indicate different protein conformations of both molecules. The PK-resistant fragment of BSE-PrPSc is shorter than that of RML- and 22L-PrPSc, indicating that BSE-PrPSc forms a different conformation from RML- and 22L-PrPSc. The conformational selection model postulates that inoculated PrPSc could select host PrPC as a substrate for conversion on the basis of its conformational compatibility with the host PrPC. Conformational incompatibility between inoculated PrPSc and host PrPC leads to unsuccessful or insufficient conversion of the host PrPC into PrPSc and vice versa: thereby, inoculation of PrPSc converts host PrPC into PrPSc in a strain-dependent manner (21, 22). PrPΔOR might adopt a different conformation from WT PrPC, and the adopted conformation of PrPΔOR might still be compatible with RML- and 22L-PrPSc, but not with BSE-PrPSc, with PrPΔOR therefore being insufficiently converted into PrPScΔOR. Structural studies have shown that the N-terminal domain of PrP, including the OR region, transiently interacts with the C-terminal globular domain, suggesting that the transient interaction might confer structural stability within the C-terminal globular domain (28, 29). Lack of the OR region might render the C-terminal globular domain of PrPΔOR structurally unstable, thereby reducing the conformational compatibility of PrPΔOR with BSE-PrPSc, but not with RML- and 22L-PrPSc. Alternatively, since the OR region binds Cu2+ ions via histidine residues (30), lack of Cu2+ ions might cause conformational incompatibility of PrPΔOR with BSE-PrPSc.
Other mechanisms might also be possible. Upon the conversion of PrPC into PrPSc, the OR region undergoes conformational changes to form a trypsin-resistant structure (31). It is thus possible that the conformational changes of the OR region might be important for BSE prions to convert PrPC into PrPSc; therefore, lack of the OR region reduces the conversion of PrPC into PrPSc after infection with BSE prions. The N-terminal domain of PrPC, including the OR region, is highly flexible and displays a marked conformational heterogeneity (29, 32, 33). Therefore, it is also possible that lack of the OR region might reduce the N-terminal conformational heterogeneity in PrPΔOR, rendering PrPΔOR resistant to BSE prions, but not to RML and 22L prions. The conversion of PrPC into PrPSc has been suggested to take place on the cell surface and/or along the endocytic pathway to lysosomes (34, 35). The OR region has been shown to be important for internalization of PrPC (36). Defective internalization of PrPΔOR might disturb conversion into PrPScΔOR, specifically after infection with BSE prions. Further studies are needed to elucidate the mechanism of the strain-specific conversion of PrPC into PrPSc. Elucidation of the exact role of the OR region in the conversion of PrPC into PrPSc after infection with BSE prions might be helpful for understanding strain-specific conversion of PrPC into PrPSc.
At terminal stages, PrPScΔOR was higher in the brains of BSE-infected Tg(PrPΔOR)/Prnp0/0 mice than PrPSc in control WT mice. However, PrPScΔOR accumulated at levels slightly less than or similar to those in the brains of RML- or 22L-infected, terminally ill Tg(PrPΔOR)/Prnp0/0 mice, respectively, compared to PrPSc in control WT mice. This is consistent with our previous results that PrPScΔOR was slightly lower in the brains of Tg(PrPΔOR)/Prnp0/0 mice than WT mice after infection with RML prions (17). Tg(PrPΔOR)/Prnp0/0 mice developed the disease earlier than WT mice after infection with RML and 22L prions, whereas they succumbed to the disease much later than WT mice after infection with BSE prions. Therefore, the different incubation times might affect the final levels of PrPScΔOR in the brains of Tg(PrPΔOR)/Prnp0/0 mice infected with RML, 22L, and BSE prions.
The pre-OR region was converted into a PK-resistant structure upon the conversion of PrPΔOR into PrPScΔOR, but not upon the conversion of full-length PrPC into PrPSc. We showed that PrPSc- and PrPScΔOR-associated BSE prions were highly pathogenic in WT mice but poorly so in Tg(PrPΔOR)/Prnp0/0 mice. We also showed that PrPScΔOR could have similar pathogenic properties to full-length PrPSc. Similar amounts of PrPSc, with the same PK-resistant core size and the same glycosylation patterns, were detected between the brains of terminally ill WT mice inoculated with PrPSc- and PrPScΔOR-associated prions. These results suggest that the PK-resistant pre-OR region might not affect the pathogenic properties of prions.
We showed that the OR region could be differentially involved in the conversion of PrPC into PrPSc after infection with BSE prions and RML or 22L prions, suggesting that PrPC might be converted into PrPSc through an OR region-dependent or -independent mechanism in a strain-dependent way. The major PK cleavage site in PrPSc is usually located either within the C-terminal part of the OR region or in the region C-terminal to the OR region (37). BSE-PrPSc has a PK cleavage site outside the OR region (37), therefore producing a shorter C-terminal fragment after PK treatment. In contrast, RML- and 22L-PrPScs have a longer PK-resistant fragment, indicating that the PK cleavage site of RML- and 22L-PrPScs is within the OR region. It is thus interesting to speculate that PrPSc carrying a PK cleavage site outside the OR region, like BSE-PrPSc, might convert PrPC into PrPSc through the OR region-dependent mechanism. In contrast, PrPSc with a PK cleavage site within the OR region might convert PrPC into PrPSc in the OR region-independent way. Investigation of other prions for the relationship between the role of the OR region in the conversion of PrPC into PrPSc and the location of the PK cleavage site in the corresponding PrPScs might be worthwhile for further understanding the mechanism for conversion of PrPC into PrPSc.
MATERIALS AND METHODS
Ethics statements.
The Ethics Committees of Animal Care and Experimentation of the University of Occupational and Environmental Health and Tokushima University approved this study (approval no. AE08-013 and T28-100). Animals were cared for in accordance with The Guiding Principle for Animal Care and Experimentation of the University of Occupational and Environmental Health and Tokushima University and with Japanese Law for Animal Welfare and Care.
Antibodies.
The antibodies used in this study are as follows: 6D11 mouse anti-PrP Ab (SIG-399810; BioLegend, San Diego, CA), IBL-N rabbit anti-PrP Ab (18635; Immuno-Biological Laboratories, Gunma, Japan), mouse anti-β-actin Ab (A5441; Sigma-Aldrich, St. Louis, MO), anti-mouse IgG horseradish peroxidase (HRP)-linked Ab (NA931; GE Healthcare, Little Chalfont, England), and anti-rabbit IgG HRP-linked Ab (NA934; GE Healthcare).
Animals.
Tg(PrPΔOR)/Prnp0/0 mice with the C57BL/6 background were produced elsewhere (18). In brief, a transgene construct encoding PrPΔOR was injected into the zygotes of C57BL/6 mice to generate Tg(PrPΔOR) mice as described elsewhere (38, 39). The resulting Tg(PrPΔOR) mice were successively mated with Zrch I Prnp0/0 mice, which had been backcrossed with C57BL/6 mice at least 9 times, to produce the line of Tg(PrPΔOR)/Prnp0/0 mice. A new line of Tg(PrPΔOR)/Prnp0/0 mice, designated Tg(PrPΔOR-3608)/Prnp0/0 mice, were similarly produced in this study. Prnp+/0 mice were produced by mating of Zrch I Prnp0/0 mice with C57BL/6 mice. C57BL/6 mice were purchased from Charles River Laboratories Japan (Kanagawa, Japan). CD-1 mice were purchased from Japan SLC, Inc. (Shizuoka, Japan).
Prion inoculation.
BSE prions originate from the classical type of BSE and have been maintained in CD-1 WT mice by successive intracerebral inoculations (37). RML and 22L prions are passaged in C57BL/6 WT mice. Brains were removed from terminally ill mice infected with RML, 22L, or BSE prions. A single brain was homogenized (10% [wt/vol]) in phosphate-buffered saline (PBS [11482-15; Nakalai Tesque, Osaka, Japan]) using a Multibeads shocker (Yasui Kikai, Osaka, Japan) and then diluted 1% with PBS. Two brain homogenates from RML-, 22L-, or BSE-infected mice were mixed in equal amounts to prepare a brain homogenate inoculum, and the resulting inoculum was intracerebrally inoculated into 5- to 6-week-old C57BL/6 WT, Prnp+/0, or Tg(PrPΔOR)/Prnp0/0 mice with a 20-μl aliquot. Mice were diagnosed as sick when they developed more than five of the following features: emaciation, decreased locomotion, ruffled body hair, ataxic gait, kyphosis, priapism, upright tail, crossing leg, hind leg paresis, and foreleg paresis. Mice were also diagnosed as terminal when they became akinetic.
Protease K and PNGase F treatment.
Brain homogenates (10% [wt/vol]) were prepared in lysis buffer (50 mM Tris-HCl [pH 7.4], containing 0.5% Triton X-100, 0.5% sodium deoxycholate, 150 mM NaCl) using a Multibeads shocker (Yasui Kikai). The protein concentration was determined by a bicinchoninic acid (BCA) protein assay kit (23225; Pierce, Rockford, IL) using bovine serum albumin (23209; Pierce) as a standard, and the homogenates were adjusted to 5 mg of protein/ml with the lysis buffer. For sample preparation for analysis of PrPSc, aliquots of 100 μl of the lysates were digested with 10 μg proteinase K (PK [165-21043; Wako Pure Chemical Industries, Osaka, Japan]) at 37°C for 30 min. Peptide N-glycosidase F (PNGase F [P0704L; New England BioLabs, Beverly, MA]) was used according to the manufacturer's protocol. In brief, total proteins were denatured in glycoprotein denaturing buffer (B1704S; New England BioLabs, Beverly, MA) by being heated at 100°C for 10 min and incubated with PNGase F (New England BioLabs) in a reaction buffer containing GlycoBuffer 2 (B3704S; New England BioLabs) and 1% NP-40 (B2704S; New England BioLabs) at 37°C for 1 h. The samples were finally mixed with sodium dodecyl sulfate (SDS) sample buffer (62.5 mM Tris-HCl [pH 6.8], containing 5% SDS, 4% β-mercaptoethanol, 5% glycerol, 0.04% bromophenol blue, 3 mM EDTA) and heated at 95°C for 10 min before being subjected to Western blotting.
Western blotting.
Proteins were resolved by SDS-polyacrylamide gel electrophoresis and electrically transferred to an Immobilon-P polyvinylidene difluoride (PVDF) membrane (IPVH00010; Millipore, Billerica, MA). After blocking with 1% nonfat dry milk in TBST (10 mM Tris-HCl [pH 7.4] containing 0.05% Tween 20, and 150 mM NaCl) at room temperature (RT) for 1 h, the membranes were washed 3 times with TBST at RT for 5 min and incubated with the first Ab at 4°C overnight in TBST containing 0.5% nonfat dry milk. The membranes were then washed 3 times with TBST at RT for 5 min and incubated with horseradish peroxidase-conjugated secondary Ab at RT for 2 h in TBST containing 0.5% nonfat dry milk. After washing 3 times with TBST at RT for 5 min, immunoreactive proteins were visualized using Immobilon Western chemiluminescent HRP substrate (WBKLS0500; Millipore) and detected by the LAS-4000 mini-chemiluminescence imaging system (Fuji Film, Tokyo, Japan). Signal intensities were determined by Image Gauge software (Fuji Film).
Hematoxylin-eosin staining.
Paraffin-embedded samples were sectioned at 5 μm. The sectioned samples were deparaffinized, rehydrated, and stained with Mayer's hematoxylin solution (131-09665; Wako Pure Chemical Industries) and 1% eosin Y solution (051-06515; Wako Pure Chemical Industries). After washing, the samples were mounted with Softmount (192-16301; Wako Pure Chemical Industries).
Immunohistochemistry.
Paraffin-embedded samples were sectioned at 5 μm. After being deparaffinized and rehydrated, the samples were autoclaved in 1 mM HCl at 121°C for 5 min and subsequently washed with PBS. The samples were digested with 50 μg/ml PK in PBS at 37°C for 30 min, treated with 3 M guanidine thiocyanate at RT for 10 min, and then washed with PBS. After blocking with 5% FBS in PBS at RT for 1 h, the samples were incubated with 6D11 anti-PrP Ab at RT for 2 h and washed with PBS. The samples were then treated with ImmPRESS reagent anti-mouse IgG (MP-7402; Vector Laboratories, Burlingame, CA) at RT for 1 h. After washing with PBS, the samples were incubated with ImmPACT DAB peroxidase substrate (SK-4105; Vector Laboratories) for 180 s for staining.
Statistical analysis.
Survival and incubation times were analyzed using the log rank (Mantel-Cox) test. Other data were analyzed using the Student t test.
ACKNOWLEDGMENTS
We thank Stanley B. Prusiner for providing Zrch I Prnp0/0 mice.
This work was supported in part by JSPS KAKENHI 26293212, MEXT KAKENHI 15H01560 and 17H05701, and the Practical Research Project for Rare/Intractable Diseases of the Japan Agency for Medical Research and Development (AMED) to S.S. and by JSPS KAKENHI 25870479 to H.H.
REFERENCES
- 1.Prusiner SB. 1998. Prions. Proc Natl Acad Sci U S A 95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stahl N, Borchelt DR, Hsiao K, Prusiner SB. 1987. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 51:229–240. doi: 10.1016/0092-8674(87)90150-4. [DOI] [PubMed] [Google Scholar]
- 3.Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. 1993. Mice devoid of PrP are resistant to scrapie. Cell 73:1339–1347. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- 4.Prusiner SB, Groth D, Serban A, Koehler R, Foster D, Torchia M, Burton D, Yang SL, DeArmond SJ. 1993. Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies. Proc Natl Acad Sci U S A 90:10608–10612. doi: 10.1073/pnas.90.22.10608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manson JC, Clarke AR, McBride PA, McConnell I, Hope J. 1994. PrP gene dosage determines the timing but not the final intensity or distribution of lesions in scrapie pathology. Neurodegeneration 3:331–340. [PubMed] [Google Scholar]
- 6.Sakaguchi S, Katamine S, Shigematsu K, Nakatani A, Moriuchi R, Nishida N, Kurokawa K, Nakaoke R, Sato H, Jishage K, Kuno J, Noda T, Miyamoto T. 1995. Accumulation of proteinase K-resistant prion protein (PrP) is restricted by the expression level of normal PrP in mice inoculated with a mouse-adapted strain of the Creutzfeldt-Jakob disease agent. J Virol 69:7586–7592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldmann W, Hunter N, Martin T, Dawson M, Hope J. 1991. Different forms of the bovine PrP gene have five or six copies of a short, G-C-rich element within the protein-coding exon. J Gen Virol 72:201–204. doi: 10.1099/0022-1317-72-1-201. [DOI] [PubMed] [Google Scholar]
- 8.Neibergs HL, Ryan AM, Womack JE, Spooner RL, Williams JL. 1994. Polymorphism analysis of the prion gene in BSE-affected and unaffected cattle. Anim Genet 25:313–317. doi: 10.1111/j.1365-2052.1994.tb00364.x. [DOI] [PubMed] [Google Scholar]
- 9.Ferguson NM, Donnelly CA, Woolhouse ME, Anderson RM. 1997. A genetic interpretation of heightened risk of BSE in offspring of affected dams. Proc Biol Sci 264:1445–1455. doi: 10.1098/rspb.1997.0201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schlapfer I, Saitbekova N, Gaillard C, Dolf G. 1999. A new allelic variant in the bovine prion protein gene (PRNP) coding region. Anim Genet 30:386–387. doi: 10.1046/j.1365-2052.1999.00526-5.x. [DOI] [PubMed] [Google Scholar]
- 11.Prusiner SB. 1993. Genetic and infectious prion diseases. Arch Neurol 50:1129–1153. doi: 10.1001/archneur.1993.00540110011002. [DOI] [PubMed] [Google Scholar]
- 12.Chiesa R, Piccardo P, Ghetti B, Harris DA. 1998. Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron 21:1339–1351. doi: 10.1016/S0896-6273(00)80653-4. [DOI] [PubMed] [Google Scholar]
- 13.Chiesa R, Piccardo P, Quaglio E, Drisaldi B, Si-Hoe SL, Takao M, Ghetti B, Harris DA. 2003. Molecular distinction between pathogenic and infectious properties of the prion protein. J Virol 77:7611–7622. doi: 10.1128/JVI.77.13.7611-7622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castilla J, Gutierrez-Adan A, Brun A, Pintado B, Salguero FJ, Parra B, Segundo FD, Ramirez MA, Rabano A, Cano MJ, Torres JM. 2005. Transgenic mice expressing bovine PrP with a four extra repeat octapeptide insert mutation show a spontaneous, non-transmissible, neurodegenerative disease and an expedited course of BSE infection. FEBS Lett 579:6237–6246. doi: 10.1016/j.febslet.2005.09.099. [DOI] [PubMed] [Google Scholar]
- 15.Castilla J, Gutierrez-Adan A, Brun A, Pintado B, Parra B, Ramirez MA, Salguero FJ, Diaz San Segundo F, Rabano A, Cano MJ, Torres JM. 2004. Different behavior toward bovine spongiform encephalopathy infection of bovine prion protein transgenic mice with one extra repeat octapeptide insert mutation. J Neurosci 24:2156–2164. doi: 10.1523/JNEUROSCI.3811-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brun A, Gutierrez-Adan A, Castilla J, Pintado B, Diaz-San Segundo F, Cano MJ, Alamillo E, Espinosa JC, Torres JM. 2007. Reduced susceptibility to bovine spongiform encephalopathy prions in transgenic mice expressing a bovine PrP with five octapeptide repeats. J Gen Virol 88:1842–1849. doi: 10.1099/vir.0.82568-0. [DOI] [PubMed] [Google Scholar]
- 17.Yamaguchi Y, Miyata H, Uchiyama K, Ootsuyama A, Inubushi S, Mori T, Muramatsu N, Katamine S, Sakaguchi S. 2012. Biological and biochemical characterization of mice expressing prion protein devoid of the octapeptide repeat region after infection with prions. PLoS One 7:e43540. doi: 10.1371/journal.pone.0043540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoshikawa D, Yamaguchi N, Ishibashi D, Yamanaka H, Okimura N, Yamaguchi Y, Mori T, Miyata H, Shigematsu K, Katamine S, Sakaguchi S. 2008. Dominant-negative effects of the N-terminal half of prion protein on neurotoxicity of prion protein-like protein/doppel in mice. J Biol Chem 283:24202–24211. doi: 10.1074/jbc.M804212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scott M, Foster D, Mirenda C, Serban D, Coufal F, Walchli M, Torchia M, Groth D, Carlson G, DeArmond SJ, Westaway D, Prusiner SB. 1989. Transgenic mice expressing hamster prion protein produce species-specific scrapie infectivity and amyloid plaques. Cell 59:847–857. doi: 10.1016/0092-8674(89)90608-9. [DOI] [PubMed] [Google Scholar]
- 20.Fischer M, Rulicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. 1996. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J 15:1255–1264. [PMC free article] [PubMed] [Google Scholar]
- 21.Collinge J, Clarke AR. 2007. A general model of prion strains and their pathogenicity. Science 318:930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 22.Wadsworth JD, Asante EA, Collinge J. 2010. Review: contribution of transgenic models to understanding human prion disease. Neuropathol Appl Neurobiol 36:576–597. doi: 10.1111/j.1365-2990.2010.01129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Striebel JF, Race B, Meade-White KD, LaCasse R, Chesebro B. 2011. Strain specific resistance to murine scrapie associated with a naturally occurring human prion protein polymorphism at residue 171. PLoS Pathog 7:e1002275. doi: 10.1371/journal.ppat.1002275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saijo E, Kang HE, Bian J, Bowling KG, Browning S, Kim S, Hunter N, Telling GC. 2013. Epigenetic dominance of prion conformers. PLoS Pathog 9:e1003692. doi: 10.1371/journal.ppat.1003692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uchiyama K, Miyata H, Yano M, Yamaguchi Y, Imamura M, Muramatsu N, Das NR, Chida J, Hara H, Sakaguchi S. 2014. Mouse-hamster chimeric prion protein (PrP) devoid of N-terminal residues 23–88 restores susceptibility to 22L prions, but not to RML prions in PrP-knockout mice. PLoS One 9:e109737. doi: 10.1371/journal.pone.0109737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bessen RA, Marsh RF. 1992. Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J Virol 66:2096–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bessen RA, Marsh RF. 1994. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol 68:7859–7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.James TL, Liu H, Ulyanov NB, Farr-Jones S, Zhang H, Donne DG, Kaneko K, Groth D, Mehlhorn I, Prusiner SB, Cohen FE. 1997. Solution structure of a 142-residue recombinant prion protein corresponding to the infectious fragment of the scrapie isoform. Proc Natl Acad Sci U S A 94:10086–10091. doi: 10.1073/pnas.94.19.10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Donne DG, Viles JH, Groth D, Mehlhorn I, James TL, Cohen FE, Prusiner SB, Wright PE, Dyson HJ. 1997. Structure of the recombinant full-length hamster prion protein PrP(29–231): the N terminus is highly flexible. Proc Natl Acad Sci U S A 94:13452–13457. doi: 10.1073/pnas.94.25.13452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R, Fraser PE, Kruck T, von Bohlen A, Schulz-Schaeffer W, Giese A, Westaway D, Kretzschmar H. 1997. The cellular prion protein binds copper in vivo. Nature 390:684–687. doi: 10.1038/37733. [DOI] [PubMed] [Google Scholar]
- 31.Yam AY, Gao CM, Wang X, Wu P, Peretz D. 2010. The octarepeat region of the prion protein is conformationally altered in PrP(Sc). PLoS One 5:e9316. doi: 10.1371/journal.pone.0009316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riek R, Hornemann S, Wider G, Glockshuber R, Wuthrich K. 1997. NMR characterization of the full-length recombinant murine prion protein, mPrP(23–231). FEBS Lett 413:282–288. doi: 10.1016/S0014-5793(97)00920-4. [DOI] [PubMed] [Google Scholar]
- 33.Peretz D, Williamson RA, Matsunaga Y, Serban H, Pinilla C, Bastidas RB, Rozenshteyn R, James TL, Houghten RA, Cohen FE, Prusiner SB, Burton DR. 1997. A conformational transition at the N terminus of the prion protein features in formation of the scrapie isoform. J Mol Biol 273:614–622. doi: 10.1006/jmbi.1997.1328. [DOI] [PubMed] [Google Scholar]
- 34.Borchelt DR, Taraboulos A, Prusiner SB. 1992. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J Biol Chem 267:16188–16199. [PubMed] [Google Scholar]
- 35.Goold R, Rabbanian S, Sutton L, Andre R, Arora P, Moonga J, Clarke AR, Schiavo G, Jat P, Collinge J, Tabrizi SJ. 2011. Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat Commun 2:281. doi: 10.1038/ncomms1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taylor DR, Watt NT, Perera WS, Hooper NM. 2005. Assigning functions to distinct regions of the N-terminus of the prion protein that are involved in its copper-stimulated, clathrin-dependent endocytosis. J Cell Sci 118:5141–5153. doi: 10.1242/jcs.02627. [DOI] [PubMed] [Google Scholar]
- 37.Hayashi HK, Yokoyama T, Takata M, Iwamaru Y, Imamura M, Ushiki YK, Shinagawa M. 2005. The N-terminal cleavage site of PrPSc from BSE differs from that of PrPSc from scrapie. Biochem Biophys Res Commun 328:1024–1027. doi: 10.1016/j.bbrc.2005.01.065. [DOI] [PubMed] [Google Scholar]
- 38.Brinster RL, Chen HY, Trumbauer ME, Yagle MK, Palmiter RD. 1985. Factors affecting the efficiency of introducing foreign DNA into mice by microinjecting eggs. Proc Natl Acad Sci U S A 82:4438–4442. doi: 10.1073/pnas.82.13.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilmut I, Hooper ML, Simons JP. 1991. Genetic manipulation of mammals and its application in reproductive biology. J Reprod Fertil 92:245–279. doi: 10.1530/jrf.0.0920245. [DOI] [PubMed] [Google Scholar]