

ABSTRACT
Rubella virus (RuV) causes a systemic infection, and transplacental fetal infection causes congenital rubella syndrome. In this study, we showed that treatment of cells with sphingomyelinase inhibited RuV infection. Assays using inhibitors of serine palmitoyl transferase and ceramide transport protein demonstrated the contribution of sphingomyelin (SM) to RuV infection. Compelling evidence for direct binding of RuV to lipid membranes at neutral pH was obtained using liposome coflotation assays. The absence of either SM or cholesterol (Chol) abrogated the RuV-liposome interaction. SM and Chol (SM/Chol) were also critical for RuV binding to erythrocytes and lymphoid cells. Removal of Ca2+ from the assay buffer or mutation of RuV envelope E1 protein Ca2+-binding sites abrogated RuV binding to liposomes, erythrocytes, and lymphoid cells. However, RuV bound to various nonlymphoid adherent cell lines independently of extracellular Ca2+ or SM/Chol. Even in these adherent cell lines, both the E1 protein Ca2+-binding sites and cellular SM/Chol were essential for the early stage of RuV infection, possibly affecting envelope-membrane fusion in acidic compartments. Myelin oligodendrocyte glycoprotein (MOG) has recently been identified as a cellular receptor for RuV. However, RuV bound to MOG-negative cells in a Ca2+-independent manner. Collectively, our data demonstrate that RuV has two distinct binding mechanisms: one is Ca2+ dependent and the other is Ca2+ independent. Ca2+-dependent binding observed in lymphoid cells occurs by the direct interaction between E1 protein fusion loops and SM/Chol-enriched membranes. Clarification of the mechanism of Ca2+-independent RuV binding is an important next step in understanding the pathology of RuV infection.
IMPORTANCE Rubella has a significant impact on public health as infection during early pregnancy can result in babies being born with congenital rubella syndrome. Even though effective rubella vaccines are available, rubella outbreaks still occur in many countries. We studied the entry mechanism of rubella virus (RuV) and found that RuV binds directly to the host plasma membrane in the presence of Ca2+ at neutral pH. This Ca2+-dependent binding is specifically directed to membranes enriched in sphingomyelin and cholesterol and is critical for RuV infection. Importantly, RuV also binds to many cell lines in a Ca2+-independent manner. An unidentified RuV receptor(s) is involved in this Ca2+-independent binding. We believe that the data presented here may aid the development of the first anti-RuV drug.
KEYWORDS: cholesterol, lipid, rubella, sphingomyelin, virus entry
INTRODUCTION
Because measles and rubella have a significant impact on public health, the eradication of these diseases is the ultimate goal of the international community. At the World Health Assembly in May 2012, the 194 United Nations member states endorsed the Global Vaccine Action Plan, which set a goal to eliminate measles and rubella by 2020 in at least five of the six regions grouped by the World Health Organization (1).
Rubella is an acute infectious disease caused by rubella virus (RuV). The disease is characterized by low-grade fever, a generalized maculopapular rash, and lymphadenopathy. It is usually mild but causes multiple organ defects, known as congenital rubella syndrome (CRS), in neonates born from mothers who suffered rubella infection during the early phase of pregnancy. Cataracts, sensorineural hearing loss, and cardiovascular defects are the typical triad of CRS. A recent systematic review estimated that 105,000 cases of CRS occurred globally in 2010 (2). Even though safe and highly effective live attenuated rubella vaccines are available, it has proven difficult to achieve vaccination coverage high enough to interrupt RuV transmission in most countries. Antiviral drugs may support the interruption of RuV transmission and eradicate the spread of RuV. However, no effective drug has been developed for RuV, partly because our knowledge of the molecular mechanisms of RuV infection is incomplete. Understanding the entry mechanism is essential to understanding viral pathology and will contribute to the development of anti-RuV drugs.
RuV belongs to the genus Rubivirus in the family Togaviridae. The family consists of two genera, Rubivirus and Alphavirus. RuV is the sole member of the genus Rubivirus, whereas more than 30 viruses are classified in the genus Alphavirus (alphaviruses), including Chikungunya virus, Semliki Forest virus (SFV), and Sindbis virus (SINV). They all are enveloped viruses with positive-stranded RNA genomes. The RuV virions contain the E1 and E2 glycoproteins, which form a heterodimer (E1-E2 heterodimer) on the lipid envelope. The RuV E1 protein has a structure and functions strikingly similar to those of the E1 proteins of the alphaviruses (3–6). The E1 protein is responsible for viral binding and membrane fusion, allowing viral entry, and the E2 protein supports the folding, transport, and functions of the E1 protein. RuV enters cells via endocytosis and causes low-pH-triggered membrane fusion in early endosomes (7). Previous studies in 1989 and 1990 (8, 9) suggested that membrane lipids play a receptor role for RuV infection. However, the detailed mechanism remains to be determined. Cholesterol (Chol) is necessary and sufficient for the binding of SFV to the target membrane, whereas both sphingolipids and Chol are necessary for SFV-induced membrane fusion (10–15). The requirement for specific lipids is similar in SINV (16).
Myelin oligodendrocyte glycoprotein (MOG) has recently been identified as a cellular receptor for RuV (17). However, systemic infection with RuV (18) cannot be explained solely by the expression pattern of MOG because MOG is expressed exclusively in the central nervous system (19). In this study, we demonstrate that RuV has two distinct binding mechanisms which show different Ca2+ dependencies. Our data show that RuV binds directly to sphingomyelin (SM) and Chol (SM/Chol)-enriched membranes in a Ca2+-dependent manner and also suggest that RuV interacts with specific receptor molecules on certain cell types even in the absence of Ca2+.
RESULTS
SM and Chol of erythrocytes are important for Ca2+-dependent RuV HA.
Many viruses induce hemagglutination (HA) when they bind to erythrocytes. For example, influenza virus and measles virus (MeV) display HA activities when they interact with their receptor molecules, sialic acid and CD46, respectively, on erythrocytes. RuV also shows HA activity, but the molecule on the erythrocyte that binds to RuV remains to be identified. RuV hemagglutinates erythrocytes in a variety of animals, but the levels of HA differ greatly between the erythrocytes of different animals. A high level of RuV HA activity was observed when goose erythrocytes were used, whereas the activity was low when the erythrocytes of guinea pigs or of African green monkeys were used (Table 1). Ca2+ is required by RuV to induce HA (Table 1). The treatment of erythrocytes with trypsin completely abolishes the HA activity of MeV (Table 1) because MeV induces HA by binding to the proteinaceous receptor CD46. Surprisingly, the treatment of the erythrocytes of guinea pigs and African green monkeys with trypsin resulted in a >10-fold enhancement of the RuV HA activity (Table 1). A far smaller increase (∼2-fold) was also observed in goose erythrocytes, in which the HA activity probably reached its nearly maximal level even without trypsin (Table 1).
TABLE 1.
Assay of hemagglutination by rubella virus and measles virus
| Virus and erythrocyte type | HA titer by conditiona |
||||
|---|---|---|---|---|---|
| No treatment |
With Ca2+ and the indicated treatment |
||||
| With Ca2+ | Without Ca2+ | SMase (5 mU/ml)b | Trypsin (1.25%) | MβCD (10 mM) | |
| Rubella virus | |||||
| Goose | 64 | 2 | <2 | 128 | 4 |
| Guinea pig | <2 | ND | ND | 64 | ND |
| AGMc | 2 | ND | ND | 32 | ND |
| Measles virus | |||||
| AGM | 32 | ND | ND | <2 | ND |
Titers were determined in erythrocytes cultured in medium with or without Ca2+ and either left untreated or treated as indicated. ND, not determined.
SMase, sphingomyelinase.
AGM, African green monkey.
The enhanced HA activity of RuV in trypsin-treated erythrocytes led us to hypothesize that RuV binds to a nonproteinaceous component(s) on the cell surface to induce HA activity. Sphingolipids and Chol are the major constituents of the plasma membranes of mammalian cells and are often involved in the infection and proliferation of intracellularly invasive pathogens (20–22). When goose erythrocytes were treated with sphingomyelinase ([SMase] which hydrolyzes SM to ceramide and phosphocholine), the HA activity of RuV was completely abolished (Table 1). Moreover, when Chol was removed from the erythrocytes with the Chol-adsorbing reagent methyl-β-cyclodextrin (MβCD), the HA activity of RuV decreased (Table 1). All of these experiments were performed at neutral pH. These data suggested that RuV exerts Ca2+-dependent HA activity at neutral pH by binding to the SM and Chol (SM/Chol)-enriched membranes of erythrocytes.
SM of host cells is essential for RuV infection.
We next examined whether the SM of host cells is essential for RuV infection using the wild-type (WT) RVi/Hiroshima.JPN/01.03 (HS) and TO-336 vaccine strains of RuV. When RK13 (Fig. 1A) or Vero cells (Fig. 1B) were pretreated with SMase, the infectivity of RuV declined in a dose-dependent manner. Vero cells were more sensitive to SMase treatment than RK13 cells (Fig. 1A and B). In contrast, SINV infection was not affected by SMase treatment of the cells (Fig. 1A), consistent with a previous study that showed that SM is not essential for the binding of SINV to cells (16). Infection by MeV and human metapneumovirus (HMPV) was also unaffected by the treatment of their host cells with SMase (Fig. 1B). Therefore, the inhibition of RuV infection by SMase treatment is not attributable to nonspecific effects against viral infection.
FIG 1.
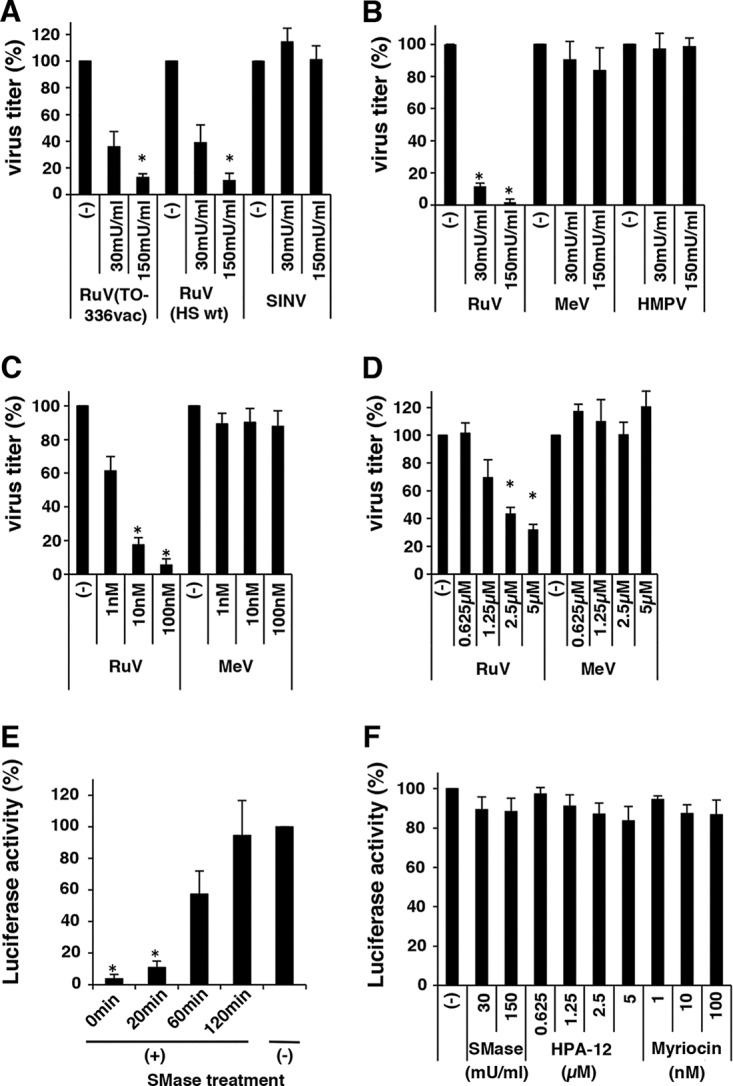
Effects of sphingomyelinase (SMase), myriocin, and HPA-12 on rubella virus (RuV) infection. (A) RK13 cells, untreated or treated with various doses of SMase for 1 h, were infected with RuV (HS wild-type or TO-336 vaccine strain) and Sindbis virus (SINV), and then standard plaque assays were performed. (B) Similar experiments were performed using Vero cells and green fluorescent protein (GFP)-expressing recombinant RuV (RuV-rHS/p150-AG1), measles virus, (MeV-IC323/Ed-H-EGFP), and human metapneumovirus (HMPV-rJPS02-76EGFP). Plaque numbers of GFP-expressing recombinant viruses were counted under a fluorescence microscope. (C) Vero cells, untreated or treated with various doses of myriocin for 48 h, were infected with RuV-rHS/p150-AG1 or MeV-IC323/Ed-H-EGFP, and standard plaque assays were performed. (D) Vero cells, untreated or treated with various doses of HPA-12 for 48 h, were infected with RuV-rHS/p150-AG1 or MeV-IC323/Ed-H-EGFP, and standard plaque assays were performed. For panels A to D, the average infectious titer of each virus in untreated cells was set to 100%. Asterisks indicate significant differences based on a t test (P < 0.01). (E) Assay using RuV virus-like particles (RuV-VLPs). Vero cells were cultured with RuV-VLPs at 4°C for 2 h and then incubated in culture medium at 37°C. Immediately thereafter or after 20, 60, or 120 min, the culture medium was supplemented with SMase (150 mU/ml). The cells were incubated at 35°C for a further 3 days, and the luciferase activity in the cells was measured. The average luciferase activity in the untreated (−) cells was set to 100%. Asterisks indicate significant differences based on a t test (P < 0.01). (F) Viability of SMase-, myriocin-, and HPA-12-treated cells was measured with a CellTiter-Glo, version 2.0, luminescent cell viability assay. The average luciferase activity in the untreated cells was set to 100%. Graphs show the means and standard deviations of three independent experiments.
Because the treatment of cells with SMase produces ceramide from SM, the results described above may have resulted from an increase in the level of ceramide rather than from a reduction in the level of SM. To test this possibility, we used a pharmacological tool, myriocin/ISP-1, a specific inhibitor of serine palmitoyl transferase, which catalyzes the first enzymatic step in the sphingolipid biosynthetic pathway and therefore reduces all types of sphingolipids in cells following treatment (23). When cells were treated with myriocin, RuV infectivity declined in a dose-dependent manner, whereas MeV infectivity was unchanged (Fig. 1C), ruling out the possibility that the inhibitory effect of SMase treatment on RuV infectivity is primarily attributable to an increase in the level of ceramide. We also used another pharmacological tool, HPA-12, a selective inhibitor of the ceramide transport protein (CERT), which mediates the transfer of ceramide from the endoplasmic reticulum to the Golgi compartment, where the de novo synthesis of SM mainly occurs (24), and therefore blocks the synthesis of new SM but not of glycosphingolipids (25). When cells were treated with HPA-12, RuV infectivity declined in a dose-dependent manner, whereas that of MeV was unchanged (Fig. 1D). These results show that SM in the host cells is essential for RuV infection.
To clarify the stage at which SM is involved in the RuV infection process, we used a virus-like particle of RuV (RuV-VLP), which allowed us to monitor the early steps of RuV infection. The RuV-VLP undergoes only a single round of infection and expresses luciferase (Luc) when it enters a cell. Thus, the infection can be assessed as luciferase activity. When cells were treated with SMase immediately after infection or 20 min after infection, infection was completely blocked, producing only background levels of luciferase activity (Fig. 1E). Infection was reduced by ∼40% when SMase treatment was started 60 min after infection (Fig. 1E). The treatment was no longer effective at 120 min after infection (Fig. 1E). In contrast, cell viability was reduced by only ∼10% by SMase, HPA-12, and myriocin treatment at the concentrations used for the experiments described above (Fig. 1F). These results suggest that SM is crucial for the early events of RuV infection. When cells were transiently transfected with a replicon RuV-Luc genome synthesized in vitro (Fig. 2A), the replication of the replicon was not affected by SMase treatment (Fig. 2B). Furthermore, when Vero cells stably expressing an RuV replicon (Vero-HS-Rep-C-P2R cells) were used, gene expression was not affected by SMase and was slightly (∼20%) decreased by HPA-12 or myriocin (Fig. 2C). In part, inhibition by these drugs was probably attributable to the small (∼10%) adverse effects on cell viability (Fig. 1F). Interferon (IFN) was used as a control and strongly inhibited gene expression in the Vero-HS-Rep-C-P2R cells (Fig. 2C). Collectively, these results indicate that the SM in the host cells is required for the early events (including entry), but not the late events (including genome replication), in the infection and proliferation of RuV.
FIG 2.
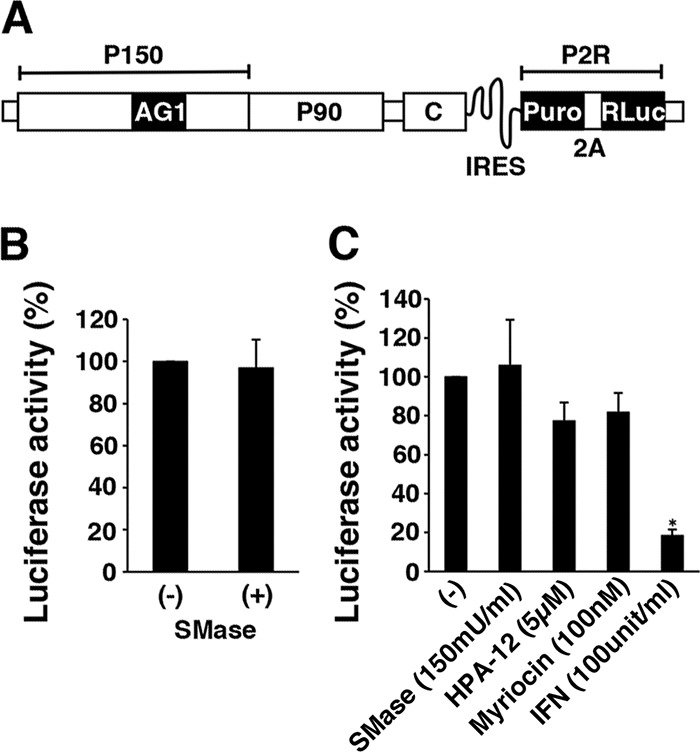
Effects of SMase, myriocin, HPA-12, and IFN on RuV replicon systems. (A) Structure of the subgenomic replicon RuV-Luc genome (HS-Rep-C-P2R). P2R, a reporter fusion protein composed of puromycin N-acetyl-transferase (Puro), the foot-and-mouth disease virus 2A self-cleavage domain, and Renilla luciferase (RLuc); IRES, internal ribosome entry site sequence of encephalomyocarditis virus; P150 and P90, RuV nonstructural proteins; C, RuV capsid protein; AG1, monomeric Aami-Green1. (B) The replicon RuV-Luc genome was synthesized in vitro, and Vero cells were transfected with the in vitro-synthesized replicon RuV-Luc genome. At 4 h posttransfection, the cells were left untreated or treated with SMase (150 mU/ml) and incubated for 72 h at 35°C. The luciferase activity in the cells was then measured. (C) RuV-RNA replicon cells (Vero-HS-Rep-C-P2R cells) were left untreated or treated with SMase (150 mU/ml), myriocin (100 nM), HPA-12 (5 μM), or IFN (100 units/ml) and incubated for 2 days at 35°C. The Renilla luciferase activity in the cells was then measured. For panels B and C, the average luciferase activity in untreated cells (−) was set to 100%. The asterisk indicates a significant difference based on a t test (P < 0.01). Graphs show the means and standard deviations of three independent experiments.
Both SM and Chol are essential for Ca2+-dependent RuV binding to cells.
As shown in Table 1, the HA activity of RuV was highly dependent on extracellular Ca2+. We next examined whether the binding of RuV to various cell lines (African monkey kidney-derived Vero, rabbit kidney-derived RK13, human embryonic kidney-derived 293T, rhesus monkey kidney derived LLC-MK2, human cervical carcinoma-derived HeLa, human immortalized T lymphocyte Jurkat, and human immortalized B lymphocyte Raji) is also dependent on Ca2+. RuV binding to Jurkat and Raji cells was highly dependent on Ca2+, whereas its binding to other cell lines was only partly dependent on Ca2+ (Fig. 3A and B). RuV binding to Jurkat cells in the presence of Ca2+ declined to a baseline level when the cells were treated with SMase or MβCD (Fig. 3C to F). In contrast, RuV binding to Vero cells in the presence of Ca2+ declined only slightly following these treatments (Fig. 3C to F). Notably, in Vero cells, SMase treatment and MβCD treatment reduced the level of RuV binding less significantly than the removal of Ca2+ (Fig. 3) although RuV infection of Vero cells was strongly inhibited by SMase (Fig. 1B). Therefore, Ca2+-dependent binding to lymphoid cells is highly dependent on SM/Chol-enriched membranes, as observed for erythrocytes, whereas the binding to nonlymphoid adherent cells appeared to be more complicated. Both Ca2+-dependent and -independent binding occurred in adherent cells, and the Ca2+-dependent binding was only partly dependent on SM/Chol, unlike the case in lymphoid cells. These results suggest that RuV particles bind to cells by at least two distinct mechanisms and that these mechanisms are Ca2+ dependent and independent.
FIG 3.

Effects of Ca2+, SMase, and MβCD on RuV binding to mammalian cells. (A and B) Vero, RK13, 293T, LLC-MK2, HeLa, Jurkat, and Raji cells were incubated with RuV antigens at 4°C in DMEM containing 2 mM CaCl2 or in Ca2+-free DMEM for 1 h. The bound RuV antigens were detected by flow cytometry using a MAb specific for the RuV E1 protein and a PE-conjugated secondary antibody. (A) Representative histograms of three independent experiments. Black and red lines indicate cells incubated with RuV in DMEM containing 2 mM CaCl2 and in Ca2+-free DMEM, respectively. Shaded areas indicate cells incubated without RuV antigens. (B) The geometric mean fluorescence intensity (MFI) and standard deviation of triplicate samples. (C and D) Vero and Jurkat cells cultured in medium supplemented with 2 mM CaCl2 were treated with 150 mU/ml SMase for 1 h or remained untreated. The cells were then incubated with RuV antigens at 4°C for 1 h. The bound RuV antigens were detected by flow cytometry. Representative histograms of three independent experiments are shown (C). Black lines indicate cells with or without SMase pretreatment and then incubated with RuV. Shaded areas indicate cells incubated without RuV antigens. The geometric MFIs and standard deviations of triplicate samples were determined (D). (E and F) Vero and Jurkat cells cultured in medium supplemented with 2 mM CaCl2 were treated with MβCD (10 mM and 5 mM, respectively) for 15 min or remained untreated. The cells were then incubated with RuV antigens at 4°C for 1 h. The bound RuV antigens were detected by flow cytometry. Representative histograms of three independent experiments are shown (E). Black lines indicate cells with or without MβCD pretreatment and then incubated with RuV. Shaded areas indicate cells incubated without RuV antigens. The geometric MFIs and standard deviations of triplicate samples were determined (F).
RuV binds to SM/Chol-enriched lipid membranes in a Ca2+-dependent manner.
In a series of experiments using cell membranes (i.e., erythrocytes and live cells) as the acceptor membranes for RuV (described above), we did not formally exclude the possibility that the host SM and Chol alter the structure or behavior of a host membrane protein(s) that acts as the real RuV receptor, thereby indirectly affecting RuV binding. To obtain compelling evidence that RuV directly binds to SM/Chol-enriched lipid membranes, we analyzed the binding of RuV particles to artificial phospholipid vesicles (liposomes) composed only of pure lipids. For this, we devised a coflotation assay with liposomes of different lipid compositions (Fig. 4). For example, 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) was used as the matrix phospholipid because phosphatidylcholine is the most abundant type of phospholipid in the plasma membrane. SM, Chol, or both were added to the DOPC matrix in a ratio partly mimicking the ratio in the membranes of mammalian erythrocyte ghosts (26). The experiments were performed at neutral pH, which is optimal for the first step in RuV binding. The results clearly showed that RuV specifically binds to SM/Chol-enriched liposomes. Both the envelope E1 protein and the internal C protein representing RuV particles were distributed among fractions 5 to 7 in the samples using liposomes formed with the DOPC matrix alone or containing either SM or Chol (Fig. 4B). In these samples (Fig. 4B, rows b to d), the peak fraction containing the RuV particles was fraction 6, which was near the interface between 30% and 40% Nycodenz before ultracentrifugation (Fig. 4A). A similar E1/C protein distribution pattern was also observed in the absence of liposomes (Fig. 4B, row f). In contrast, when SM/Chol-enriched liposomes were used, the RuV particles were redistributed to less dense fractions (fractions 3 and 4) (Fig. 4B, row a). The RuV-containing peak fraction was 4 (Fig. 4B), which was near the interface between 0% and 30% Nycodenz before ultracentrifugation (Fig. 4A). The absence of either SM or Chol (Fig. 4B, rows b and c) or both (Fig. 4B, row d) in the liposomes abrogated the redistribution of RuV to the less dense fractions. Importantly, in the absence of Ca2+, this redistribution to a less dense fraction was not observed even when SM/Chol-enriched liposomes were used (Fig. 4B, row e). The distribution of liposomes per se was not affected by Ca2+ (Fig. 4C). The lower fractions (fractions 10 to 12) contained only the C protein, suggesting that they represented aggregates of the C protein (Fig. 4B). From these results, we conclude that RuV has the ability to bind directly to SM/Chol-enriched lipid membranes in a Ca2+-dependent manner at neutral pH.
FIG 4.
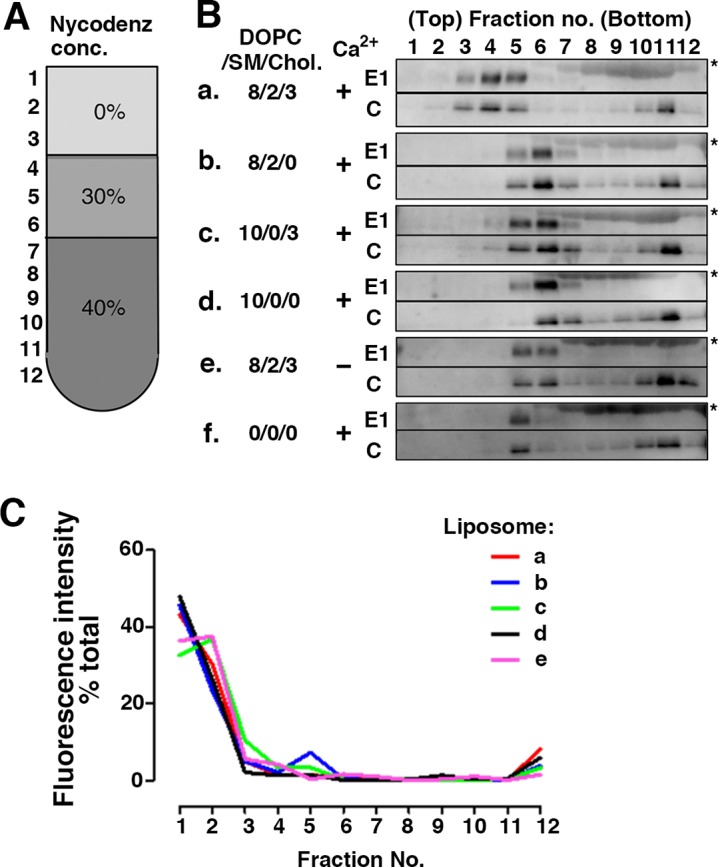
SM and Chol requirement for RuV binding to liposomes. (A) Nycodenz-containing preloaded layers and obtained fractions in flotation assays. conc, concentration. (B) RuV antigens (UV-inactivated RuV virions) were incubated with (a to e) or without (f) various compositions of liposomes and then subjected to liposome flotation assays on a Nycodenz gradient. Viral E1 and C proteins in each fraction of the gradient were detected with Western blotting. The values indicated are the amounts (in nanomoles) of DOPC, SM, and Chol added per microliter of RuV antigen. All of the liposomes also contained 0.05 nmol of Rhod PE per μl of RuV antigen. In the experiment shown in row e, calcium ions were omitted throughout the experiment. Asterisks indicate bovine serum albumin, which was an additive to RuV antigens. The experiments were repeated twice, and very similar results were obtained. Representative immunoblots are shown. (C) Distribution of liposomes monitored using the fluorescence intensity of Rhod PE. Values are expressed as the percentage of the total fluorescence intensity of Rhod PE retrieved from each fraction of the gradient. Lipid compositions of the liposome in rows a to e are the same as those indicated for panel B. The experiments were repeated twice, and very similar results were obtained. Representative data of repeated experiments are shown.
Both SM and Chol in the target membrane are crucial for the functional interaction by RuV envelope proteins to produce low-pH-triggered membrane fusion.
In Vero cells, cellular SM was required for RuV infection but not for binding (at neutral pH) (Fig. 1 and 3). The requirement for SM during RuV infection was specifically during the early stage of infection (Fig. 1E). After binding, RuV enters the cells by endocytosis and releases its genome into the host cytosol by low-pH-triggered fusion of the viral envelope with the host membranes in early endosomes. Thus, we examined the possibility that SM/Chol-enriched membranes are involved in RuV-mediated membrane fusion under acidic conditions. To assess the requirement for SM and Chol in RuV-mediated membrane fusion at acidic pH, a quantitative cell-cell fusion assay using a dual-split-protein (DSP) system (27, 28) was performed. The DSP assay uses a pair of chimeric reporter proteins (DSP1–7 and DSP8–11) composed of split Renilla luciferase and split green fluorescent protein (GFP). DSP1–7 and DSP8–11 exhibit both Renilla luciferase and GFP activities when they associate with each other. Thus, by using cell lines that constitutively express DSP1–7 and DSP8–11 (293CD4/DSP1–7 and 293FT/DSP8–11 cells, respectively), cell-cell fusion can be monitored by detecting Renilla luciferase and GFP activities (29). When the RuV E1 and E2 glycoproteins were expressed in the system, cell-to-cell fusion was efficiently triggered by low pH, and treatment of the cells with SMase completely abrogated the low-pH-triggered cell-to-cell fusion (Fig. 5A). The depletion of Chol by MβCD also inhibited cell-to-cell fusion (Fig. 5B). This was confirmed by another cell-to-cell fusion assay in which syncytium formation was visualized with Aequorea coerulescens green fluorescent protein (AcGFP) expressed simultaneously with the RuV E1 and E2 (E1/E2) glycoproteins (Fig. 4C and D). These data indicate that both SM and Chol are crucial for the low-pH-triggered membrane fusion mediated by the RuV E1/E2 glycoproteins.
FIG 5.

Effects of SMase and MβCD on cell-to-cell fusion by RuV E2 and E1 proteins. (A and B) 293CD4/DSP1–7 and 293FT/DSP8–11 cells constitutively expressing DSP1–7 and DSP8–11, respectively, were mixed and cultured together and transfected with the E1/E2 expression plasmid or the empty vector together with pcDNA3.1-FLuc. At 32 h posttransfection, the cells were left untreated or treated with SMase (150 mU/ml) (A) or MβCD (10 mM) (B) and incubated in low-pH (pH 6.0) culture medium for 15 min and then in a standard culture medium for 8 h at 37°C. The Renilla luciferase activity and the firefly luciferase activity in the cells were then measured with a Dual-Luciferase Reporter Assay System (Promega). The expression of Renilla luciferase was normalized to the expression of firefly luciferase. The average luciferase activity in the RuV E1/E2-expressing untreated cells, in which cell-to-cell fusion was triggered by low pH, was set to 100%. (C and D) 293T cells were transfected with the E1 and E2 (E1/E2) expression plasmid or an empty vector together with an AcGFP-expressing plasmid. At 32 h posttransfection, the cells were left untreated or treated with SMase (150 mU/ml) and incubated in a low-pH (pH 6.0) culture medium for 15 min and then in a standard culture medium for 8 h at 37°C. The cells were observed with a fluorescence microscope. (C) Representative images with nuclear DNA counterstained with DAPI are shown. Bar, 50 μm. (D) Lower-magnification images. Bar, 200 μm.
The Ca2+-dependent RuV interaction with SM/Chol-enriched membranes is associated with the fusion loops on E1 protein.
The E1 protein of RuV uniquely contains two fusion loops in which Ca2+ is incorporated and contributes to the formation of a wide hydrophobic surface area on the E1 protein (30). A mutant E1 protein, with alanine substitutions at positions 88 and 136 (N88A and D136A, respectively) fails to incorporate Ca2+ (30). RuV-VLPs with or without the N88A and D136A mutations were generated. The mutations did not affect the production of RuV-VLPs (Fig. 6A). RuV-VLPs with the N88A and D136A mutations showed little HA activity (Table 2). The RuV-VLPs with the N88A and D136A mutations also showed little Ca2+-dependent binding activity to Jurkat cells (Fig. 6B and C), in which RuV uses only an SM/Chol-dependent and Ca2+-dependent binding mechanism. The binding of the mutant RuV-VLPs to LLC-MK2 cells in the absence of Ca2+ was comparable to that of the WT RuV-VLPs (Fig. 6B and C), indicating that these mutations did not affect Ca2+-independent binding. These mutations also did not affect extracellular Ca2+-dependent binding of RuV-VLPs to LLC-MK2 cells (Fig. 6B and C). We interpreted these results to mean that an N88 and D136 residue-independent binding of RuV occurs in the absence of Ca2+ in LLC-MK2 cells and that this binding is enhanced by extracellular Ca2+. These observations may indicate the existence of an as yet unidentified host molecule(s) other than SM/Chol, as discussed below. We next determined whether the N88 and D136 residues of E1 protein are required for membrane fusion at low pH (pH 6.0) in a cell-to-cell fusion assay (Fig. 6D to G). The levels of E1 protein expressed on the cell surface were not affected by the N88A/D136A double mutation (Fig. 6D and E), but no fusion activity was observed for the E1 N88A/D136A mutant (Fig. 6F and G). Cell-to-cell fusion at pH 6 was abrogated by treatment of the cells with SMase and MβCD (Fig. 5). Collectively, these data suggested that the Ca2+-coordinating E1 protein fusion loops and SM/Cho-enriched membranes were required for membrane fusion of RuV in acidic compartments, even in cells to which RuV binding occurs independently of E1 Ca2+-binding residues (N88 and D136) or SM/Chol.
FIG 6.
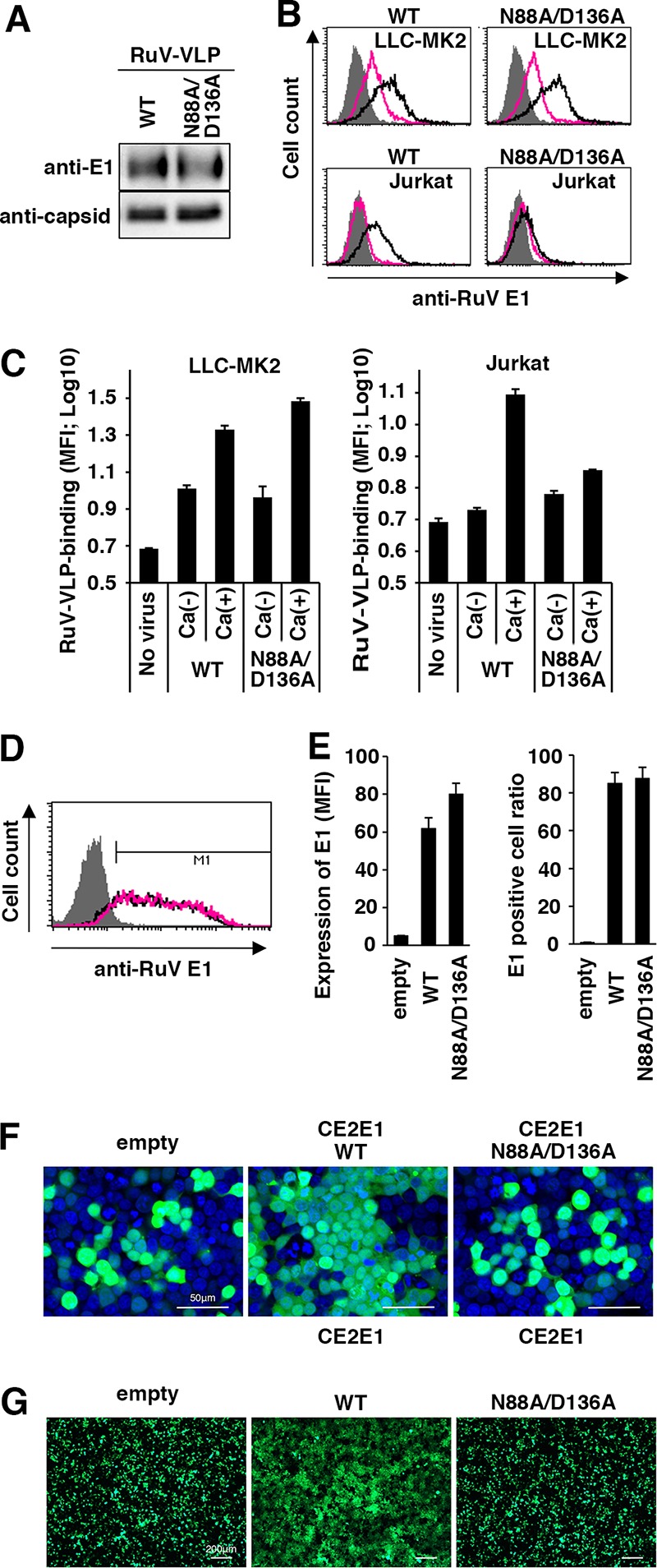
Effects of the N88A and D136A mutations in the E1 protein. (A) Detection of RuV-VLPs possessing the wild-type (WT) or mutant E1 protein (N88A/D136A) with Western blotting using a MAb specific for RuV E1 or capsid protein. (B and C) Cell-binding assays using RuV-VLPs. LLC-MK2 or Jurkat cells were incubated with RuV-VLPs possessing the WT or mutant (N88A/D136A) E1 protein shown in the blot in panel A at 4°C in DMEM containing 2 mM CaCl2 or in Ca2+-free DMEM. The bound RuV antigens were detected by flow cytometry. (B) Representative histograms of three independent experiments. Black and red lines indicate cells incubated with RuV-VLPs in DMEM containing 2 mM CaCl2 and in Ca2+-free DMEM, respectively. Shaded areas indicate cells incubated without RuV-VLPs. (C) The geometric MFIs and standard deviations of triplicate samples. (D to G) 293T cells were transfected with the C/E2/E1 protein-expressing plasmid or an empty vector together with an AcGFP-expressing plasmid. The expression plasmid encoded either WT or mutant (N88A/D136A) E1 protein. Cell surface expression of the WT or mutant (N88A/D136A) E1 protein was detected by flow cytometry using a MAb specific for RuV E1. (D) Representative histograms of two independent experiments are shown (D). Black and red lines indicate cells expressing WT E1 protein and mutant (N88A/D136A) E1 protein, respectively. The geometric MFI and positive cell ratio for the E1 protein with the standard deviation of triplicate samples were determined (E). At 32 h posttransfection, the cells were incubated with low-pH (pH 6.0) culture medium for 15 min. The cells were observed with a fluorescence microscope. (F) Representative images with nuclear DNA counterstained with DAPI are shown (F). Bar, 50 μm. Lower-magnification images are shown in panel G. Bar, 200 μm.
TABLE 2.
Assay of hemagglutination by VLPs of rubella virus
| VLP type | HA titer in medium with Ca2+a |
|---|---|
| E1-wild type | 32 |
| E1-N88A/D136A | <2 |
Titers were determined in goose erythrocytes.
DISCUSSION
In this study, to test the binding activity of RuV to various adherent and nonadherent cell lines, erythrocytes, and liposomes, we performed the experiments at neutral pH because the initial binding of RuV to cells must occur at neutral pH. Consequently, we found that SM/Chol- and extracellular Ca2+-dependent RuV binding at neutral pH predominantly occurred in lymphoid cells, erythrocytes, and liposomes. Moreover, we found that RuV bound to nonlymphoid adherent cell types via a different mechanism(s), which was SM/Chol- and E1 Ca2+-binding-residue independent. Of note, RuV infection of nonlymphoid adherent cell types also required SM/Chol at the early stage of infection. This is probably attributable to the stage of virus envelope-membrane fusion in the endosomes of host cells because both SM/Chol- and E1 Ca2+-binding residues were found to be essential for E1/E2-dependent membrane fusion at low pH (Fig. 5 and 6). The E1 protein undergoes conformational changes for fusion at acidic pH (31). Thus, the prefusion form of the E1 protein may interact with the SM/Chol-enriched plasma membrane at neutral pH. These observations were consistent with the data of a previous study (30) showing that the interaction between RuV and the liposome is not strictly dependent on low-pH conditions although a low pH promotes the RuV-liposome interaction (30).
This lipid interaction at neutral pH may be unique to RuV. The RuV E1 protein has a structure strikingly similar to that of the alphavirus E1 proteins (3–6), whereas the liposome interactions with alphaviruses SINV and SFV are low-pH dependent (10, 16, 32). The E1 protein of RuV uniquely contains two fusion loops (33). By accommodating Ca2+, the two fusion loops generate a wide hydrophobic surface area, which may interact with the target membrane (30, 33). The Ca2+-binding site in E1 is located between the two fusion loops, contributing to the formation of the surface structure (30, 33). The importance of the wide hydrophobic surface area of the E1 protein for the binding of RuV to SM/Chol-enriched membranes was confirmed by analysis of the N88A and D136A mutations, which abolish the binding of Ca2+ to the E1 protein. Thus, the unique feature of RuV, namely, its capability for binding to cells at neutral pH, may be attributable to the wide hydrophobic surface area formed by the Ca2+-requiring fusion loops.
As described above, RuV has two distinct binding mechanisms, a Ca2+-dependent and a Ca2+-independent mechanism. Ca2+-dependent binding was observed in all of the tested cell lines, erythrocytes, and liposomes, whereas Ca2+-independent binding was observed in nonlymphoid adherent cells (Vero, RK13, 293T, LLC-MK2, and HeLa cells) but not in lymphoid cells (Jurkat and Raji cells), erythrocytes, or liposomes. Interestingly, nonlymphoid adherent cells showed RuV binding in the absence of extracellular Ca2+. Even in these cell types, extracellular Ca2+ enhanced the level of RuV binding, but this enhancement by Ca2+ was not abrogated by the E1 N88A/D136A mutations, which ablate Ca2+ coordination into the E1 loop (30, 33). Recently, we demonstrated that human nonimmune cell lines are susceptible to RuV entry, while immune cell lines are much less susceptible (34). Indeed, the immune cell-derived Jurkat and Raji cells, which do not demonstrate Ca2+-independent RuV binding, cannot be infected with RuV (34). Therefore, it seems that Ca2+-dependent binding to SM/Chol-enriched domains at the plasma membrane is not sufficient for RuV infection, whereas RuV infection of cells that showed Ca2+-independent binding was still inhibited by treatments that reduced SM (Fig. 1). Mutation of the E1 protein Ca2+-binding sites abrogated E1 membrane fusion activity. These data suggest that the Ca2+-dependent and -independent binding mechanisms work concurrently during RuV entry. It remains unclear why the SM/Chol- and E1 fusion loop-dependent binding of RuV was not obviously detected in the adherent cells although SM/Chol-enriched domains presumably exist at the plasma membrane in all mammalian cell types. It is likely that RuV uses a specific molecule(s) for binding via the Ca2+-independent mechanism. Ca2+-independent binding was observed in many cell lines, including Vero, RK13, 293T, LLC-MK2, and HeLa cells, but these cell lines do not necessarily express MOG, a known receptor for RuV (17). Therefore, MOG may be responsible for the Ca2+-independent binding of RuV to some, but not all, cell lines. With LLC-MK2 cells, the binding of mutant RuV-VLPs abrogating Ca2+ incorporation into the fusion loops of the E1 protein was comparable to that of wild-type RuV-VLPs even in the presence of Ca2+. We interpreted these results to suggest that RuV binding to a molecule(s) that was used in the Ca2+-independent mechanism was enhanced in the presence of Ca2+. If this interpretation is correct, Jurkat and Raji cells would potentially be suitable cell types for the identification of RuV receptors in the Ca2+-independent pathway by a gain-of-function screening approach after transfection of genome-wide cDNAs derived from RuV-susceptible cells. As for alphaviruses, lipid components are also important for the interaction with the target membrane and the induction of membrane fusion (16, 35, 36), but several proteins and heparan sulfate have been shown to function as receptors or attachment factors (37, 38). These data suggest that the use of membrane lipids together with specific receptors is a common feature of the togaviruses (RuV and alphaviruses).
In conclusion, RuV has two distinct binding mechanisms, one that is Ca2+ dependent and one that is Ca2+ independent. Ca2+-dependent binding is mediated by the direct interaction between RuV E1 protein and SM/Chol-enriched membranes. Clarification of the mechanism of Ca2+-independent RuV binding is an important next step in understanding the pathology of RuV infection.
MATERIALS AND METHODS
Materials.
1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC; 850375C), sphingomyelin (SM) (from egg; 86006L), and l-α-phosphatidylethanolamine-N-(lissamine rhodamine B sulfonyl) (Rhod PE; 81146C) were purchased from Avanti Polar Lipids, Inc., Alabaster, AL. Cholesterol (C8667) and 5-(N-2,3-dihydroxypropylacetamido)-2,4,6-triiodo-N,N′-bis(2,3-dihydroxypropyl) isophthalamide (Nycodendz; D2158) were from Sigma-Aldrich Corp., St. Louis, MO. Protease inhibitor cocktail (259-55) was purchased from Nacalai Tesque, Inc., Kyoto, Japan. The CERT inhibitor HPA-12 (25) was purchased from Tokyo Chemical Industry Co., Ltd., Tokyo, Japan. SMase (from Bacillus cereus; S7651) and myriocin were purchased from Sigma-Aldrich Corp. The stock solutions of HPA-12 and myriocin were prepared in dimethyl sulfoxide (Hybri-Max; Sigma-Aldrich Corp.) at a concentration of 10 mM.
Viruses.
The RVi/Hiroshima.JPN/01.03 (Hiroshima) WT and TO-336 vaccine strains of RuV have been reported previously (39, 40). The M2215 strain of SINV was provided by C. K. Lim and has been reported previously (41). The recombinant RuV RVi/Hiroshima.JPN/01.03 (HS) strain expressing the green fluorescent AG1 protein (rHS/p150-AG1) has been reported previously (39). The recombinant measles virus (MeV) and human metapneumovirus (HMPV) expressing enhanced green fluorescent protein (EGFP) (MeV-IC323/Ed-H-EGFP and HMPV-rJPS02-76EGFP, respectively) have been reported previously (42, 43). The stock viruses were propagated in RK13 cells (Hiroshima WT and TO-336 vaccine strains), BHK cells (SINV) (41), Vero cells (rHS/p150-AG1) (39), Vero/hSLAM cells (MeV) (44), or Vero/TMPRSS2 cells (HMPV) (43).
Plasmid construction.
The expression plasmid encoding both the E2 and E1 proteins of the RuV HS strain (pcDNA3.1-E2E1) has been reported previously (39). The expression plasmid encoding the E2, E1, and C proteins of the RuV HS strain (pcDNA3.1-CE2E1) has been reported previously (39). Plasmid pcDNA3.1-CE2E1 expressing the E1 protein with the N88A and D136A mutations (pcDNA3.1-CE2E1-N88A/D136A) was generated by site-directed mutagenesis. The plasmid encoding residues 1 to 300 of the C protein (C1–300) of the RuV HS strain has been reported previously (34). The plasmid encoding the cDNA for a subgenomic replicon of the RuV HS strain, pHS-Rep-C-P2R, was constructed by replacing the E1 and E2 regions of the structural polyprotein gene with the open reading frame for the P2R reporter and sequences encoding a fusion protein of puromycin N-acetyl-transferase, the foot-and-mouth disease virus 2A self-cleavage domain, and Renilla luciferase, in that order (39). The internal ribosome entry site sequence of encephalomyocarditis virus was inserted between the coding regions of the RuV C protein and the P2R reporter so that both proteins were translated from the same mRNA. The pcDNA3.1-FLuc plasmid encoding firefly luciferase was generated by inserting the coding sequence of the firefly luciferase gene into the pcDNA3.1 vector.
In vitro synthesis of the subgenomic replicon RNA and the RNA encoding the full-length C protein.
As reported previously (39), the subgenomic replicon RNA was synthesized from pHS-Rep-C-P2R by in vitro RNA transcription with a mMESSAGE mMACHINE SP6 transcription kit (Life Technologies, Carlsbad, CA, USA). The RNA encoding the full-length C protein was synthesized from the plasmid encoding the C protein of the RuV HS strain (C1–300) by in vitro RNA transcription with a mMESSAGE mMACHINE T7 transcription kit (Life Technologies). The quality of the synthesized RNAs was confirmed by electrophoresis, and the amounts of RNAs were calculated spectrophotometrically.
Cells.
Vero cells (American Type Culture Collection [ATCC], Manassas, VA, USA) and BHK cells (from a preexisting cell collection) were maintained in Dulbecco's modified Eagle's medium (DMEM) (Sigma-Aldrich Corp.) containing 5% fetal bovine serum (FBS). 293T (ATCC), LLC-MK2 (ATCC), and HeLa (ATCC) cells were maintained in DMEM containing 10% FBS. RK13 cells (a gift from the Kitazato Institute) were maintained in Eagle's minimum essential medium (MEM) (Nissui Pharmaceutical, Tokyo, Japan) containing 8% bovine serum (BS). Jurkat cells (ATCC) and Raji cells (from a preexisting collection) were maintained in RPMI 1640 medium containing 10% FBS. The RuV-RNA replicon BHK cells (BHK-HS-Rep-C-P2R cells) were generated by transfecting BHK cells with the in vitro-synthesized subgenomic replicon RNA and subsequent puromycin selection (2.5 μg/ml) of stable clones carrying the RNA (34). The RuV-RNA replicon Vero cells (Vero-HS-Rep-C-P2R cells) were generated by methods similar to those described for BHK-HS-Rep-C-P2R cells using Vero cells. BHK-HS-Rep-C-P2R cells and Vero-HS-Rep-C-P2R cells were maintained in DMEM containing 10% FBS and 2.5 μg/ml puromycin. The 293CD4/DSP1–7 and 293FT/DSP8–11 cells were kindly provided by Z. Matsuda and maintained in DMEM containing 10% FBS and 1.5 μg/ml puromycin (28).
RuV-VLPs.
Virus-like particles of RuV (RuV-VLPs) were produced and concentrated as described previously, with some modifications (39). Briefly, BHK-HS-Rep-C-P2R cells were transfected with pcDNA3.1-CE2E1 or pcDNA3.1-CE2E1-N88A/D136A to produce RuV-VLPs (WT or mutant N88A/D136A, respectively). At 24 h posttransfection, the culture medium was replaced with fresh medium without FBS, and the cells were harvested after incubation for another 24 h. The culture medium was centrifuged at 10,000 × g for 10 min at 4°C. The supernatants were layered onto 10% (wt/vol) sucrose cushions and centrifuged at 113,000 × g for 2 h at 4°C to pellet the RuV-VLPs. The RuV-VLPs were resuspended in phosphate-buffered saline (PBS) overnight for use in HA and cell-binding assays. The amounts of RuV-VLPs were analyzed by Western blotting using a monoclonal antibody (MAb) specific for the RuV capsid or E1 protein.
Antibodies.
Mouse MAbs specific for the RuV capsid and E1 proteins were purchased from Abcam, Cambridge, United Kingdom, and U.S. Biologicals, Salem, MA, respectively. Goat anti-RuV polyclonal antibody (BP1061) was purchased from Acris Antibodies GmbH, Herford, Germany. A phycoerythrin (PE)-conjugated anti-mouse antibody and a horseradish peroxidase (HRP)-conjugated anti-goat antibody were purchased from BioLegend and Sigma-Aldrich Corp., respectively.
Analysis of the effect of SMase treatment on viral infectivity.
Monolayers of RK13 cells in six-well plates were left untreated or treated with 30 or 150 mU/ml SMase in MEM for 1 h. After the cells were washed with PBS, they were incubated with 30 to 100 PFU of RuV (HS WT strain or TO-336 vaccine strain) or SINV for 1 h at room temperature and washed with MEM. Then, the cells infected with RuV were cultured with 3 ml of MEM containing 2% BS and 0.5% agarose at 35°C for 7 days. The cells infected with SINV were cultured with 3 ml of MEM containing 2% BS and 0.8% agarose at 35°C for 1 day. After the 7-day incubation period for RuV-infected cells, 2 ml of MEM containing 0.01% neutral red and 0.5% agarose were overlaid onto the 2% BS and 0.5% agarose-containing MEM. The numbers of plaques were counted at 2 or 3 days after the overlay procedure. After the 1-day incubation period for SINV-infected cells, plaques were visualized by staining with 1 ml of PBS containing 0.1% crystal violet and 4% formalin for 4 h. After the cells were washed with distilled H2O (dH2O), the numbers of plaques were counted. Similar experiments were performed using Vero cells and AG1- or EGFP-expressing recombinant viruses (RuV-rHS/p150-AG1, MeV-IC323/Ed-H-EGFP, and HMPV-rJPS02-76EGFP). For RuV-rHS/p150-AG1 infection, monolayers of Vero cells in 12-well plates were left untreated or treated with 2.5, 5, 10, 20, 30, or 150 mU/ml SMase for 1 h. For MeV-IC323/Ed-H-EGFP and HMPV-rJPS02-76EGFP infection, monolayers of Vero cells in 12-well plates were left untreated or treated with 30 or 150 mU/ml SMase for 1 h. After the cells were washed with PBS, they were incubated with 30 to 100 fluorescent focus-forming units (FFU) of RuV-rHS/p150-AG1, MeV-IC323/Ed-H-EGFP, or HMPV-rJPS02-76EGFP for 1 h at room temperature and washed with MEM. Then, the cells were cultured with 1 ml of MEM containing 2% BS and 0.4% agarose at 35°C for 6 days. The numbers of foci expressing AG1 or EGFP were counted under a fluorescence microscope.
Analysis of the effects of HPA-12 and myriocin treatment on viral infectivity.
Subconfluent monolayers (80 to 90% confluence) of Vero cells in 12-well plates were left untreated or treated with 1, 10, or 100 nM myriocin in DMEM for 48 h. Vero cells were also left untreated or treated with 0.625, 1.25, 2.5, or 5.0 μM HPA-12 in DMEM for 48 h. After cells were washed with PBS, they were incubated with 30 to 100 FFU of AG1- or EGFP-expressing recombinant virus (RuV-rHS/p150-AG1, MeV-IC323/Ed-H-EGFP, or HMPV-rJPS02-76EGFP) for 1 h at room temperature and washed with MEM. Then, the cells were cultured with 1 ml of MEM containing 2% BS and 0.4% agarose at 35°C for 6 days. The numbers of foci expressing AG1 or EGFP were counted under a fluorescence microscope.
Analysis of the effects of SMase treatment on VLP infection.
Monolayers of Vero cells in 24-well plates were cultured with RuV-VLPs at 4°C for 2 h. After cells were washed with MEM at 4°C, they were incubated in DMEM at 37°C. Immediately thereafter or after 20, 60, or 120 min, the culture medium (DMEM) was supplemented with 150 mU/ml SMase. The cells were incubated at 35°C for a further 3 days, and the luciferase activity in the cells was measured using a Renilla luciferase assay system (Promega, Madison, WI, USA).
Analysis of the effects of SMase, HPA-12, and myriocin treatment on RuV gene expression.
Vero cells in 24-well plates were transfected with an in vitro-synthesized subgenomic replicon RNA (1 μg) and C1–300 RNA (0.5 μg) using DMRIE-C transfection reagent (Thermo Fisher Scientific) and cultured for 4 h. After cells were washed with DMEM, they were further cultured in the presence (150 mU/ml) or absence of SMase at 35°C for 3 days, and the luciferase activity in the cells was measured with a Renilla luciferase assay system (Promega). Monolayers of RuV-RNA replicon cells (Vero-HS-Rep-C-P2R cells) in 12-well plates were incubated at 35°C for 2 days in the presence of SMase (150 mU/ml), HPA-12 (5 μM), or myriocin (100 nM). The replicon cells were also treated with 100 U/ml universal type I IFN (PBL Biomedical Laboratories, Piscataway, NJ, USA) as a control. After incubation, the luciferase activity in the cells was measured with a Renilla luciferase assay system.
HA assay.
Erythrocytes from geese, guinea pigs (Nippon Bio-Test Laboratories, Inc., Saitama, Japan), and African green monkeys were washed three times with dextrose-gelatin-veronal solution (Denka Seiken Co. Ltd., Tokyo, Japan) and then suspended in dilution buffer (0.2 mg/ml CaCl2, 0.2 mg/ml MgCl2, 1 mg/ml bovine serum albumin, 0.05% gelatin in PBS, pH 6.8). Erythrocytes from African green monkeys, geese, and guinea pigs were prepared at concentrations of 0.25%, 0.5%, and 0.5%, respectively. UV-inactivated RuV particles (RuV antigens), purchased from Denka Seiken, were reconstituted with distilled water and serially diluted 2-fold in dilution buffer in 96-well round-bottom plates. The same volume of erythrocyte solution was then mixed with the RuV antigen solution in the 96-well round-bottom plates. After a 1 h-incubation at 4°C, the HA titers were determined. To assess the effects of SMase, trypsin, and MβCD, the erythrocytes were treated with SMase (5 mU/ml), trypsin (1.25%), or MβCD (10 mM) for 90 min at 37°C, washed with MEM, and suspended in a dilution buffer for the HA assay.
RuV-binding assay.
Adherent cell lines (Vero, RK13, 293T, LLC-MK2, and HeLa cells) were dissociated with PBS-based, enzyme-free, cell dissociation buffer (Thermo Fisher Scientific). To analyze the requirement for Ca2+ in RuV binding, Vero, RK13, 293T, LLC-MK2, HeLa, Jurkat, and Raji cells were incubated with RuV antigens at 4°C for 1 h in DMEM containing CaCl2 (2 mM) or in Ca2+-free DMEM (Thermo Fisher Scientific). The cells were then washed twice with DMEM with or without Ca2+. The control cells remained untreated. To analyze the effects of SMase, Vero and Jurkat cells were treated with 150 mU/ml SMase for 1 h at 37°C. The control cells remained untreated. The cells were then washed twice with DMEM or RPMI 1640 medium and incubated with RuV antigens at 4°C for 1 h. The cells were washed twice again with DMEM or RPMI 1640 medium. To analyze the effects of MβCD, Vero and Jurkat cells were treated with 10 mM and 5 mM MβCD, respectively, for 15 min at 37°C. The control cells remained untreated. The cells were then washed twice with DMEM or RPMI 1640 medium and incubated with RuV antigens at 4°C for 1 h. The cells were washed twice again with DMEM or RPMI 1640 medium. To analyze the effect of the E1 fusion loop mutations on the HA and cell-binding activities of RuV, LLC-MK2 and Jurkat cells were incubated with WT or mutant N88A/D136A RuV-VLPs at 4°C for 1 h in DMEM containing CaCl2 (2 mM) or in Ca2+-free DMEM. The cells were then washed twice with DMEM with or without Ca2+. The control cells remained untreated. After the treatment described above, the cells were fixed with 1% paraformaldehyde for 15 min and then washed with PBS. Before antibody labeling of Raji cells, the cells were incubated with FcR blocking reagent, human (Miltenyi Biotec, Bergisch Gladbach, Germany), at 4°C for 15 min. The bound RuV antigens were reacted with a MAb specific for the RuV E1 protein and a PE-conjugated secondary anti-mouse antibody and detected with a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). The geometric mean fluorescent intensity (MFI) was determined using BD CellQuest Pro software (Becton Dickinson).
Fusion assay.
293T cells were transfected with pcDNA3.1-E2E1, pcDNA3.1-CE2E1, pcDNA3.1-CE2E1-N88A/D136A, or the empty pcDNA3.1 vector together with an AcGFP-expressing plasmid, pAcGFP1 (TaKaRa Bio, Inc., Shiga, Japan). The cell surface expression of the E1 protein was analyzed by staining the cells with a MAb specific for the RuV E1 protein and a PE-conjugated secondary anti-mouse antibody, followed by detection with a FACSCalibur flow cytometer. The geometric MFI was determined using BD CellQuest Pro software. At 32 h posttransfection, the cells were left untreated or treated with SMase (150 mU/ml) for 60 min, incubated in a low-pH (pH 6.0) culture medium for 15 min, and then incubated in standard culture medium for 8 h at 37°C. The cells were then fixed with PBS containing 2.5% formaldehyde. The fixed cells were washed with PBS. Nuclear DNA was stained with 0.2 μg/ml 4′,6′-diamidino-2-phenylindole (DAPI; Nacalai Tesque) and observed with an FV1000D spectral-type confocal laser scanning microscope (Olympus, Tokyo, Japan). To quantify the fusion event, a DSP-based fusion assay was performed (27, 28). 293CD4/DSP1–7 and 293FT/DSP8–11 cells constitutively expressing DSP1–7 and DSP8–11, respectively, were mixed and cultured together and transfected with pcDNA3.1-E2E1 or the empty pcDNA3.1 vector together with pcDNA3.1-FLuc. At 32 h posttransfection, the cells were left untreated or treated with SMase (150 mU/ml) for 1 h or with MβCD (10 mM) for 30 min, incubated in low-pH (pH 6.0) culture medium for 15 min, and then incubated in standard culture medium for 8 h at 37°C. The Renilla luciferase activity derived from the DSP and the firefly luciferase activity derived from the control pcDNA3.1-FLuc plasmid in the cells were then measured with a Dual-Luciferase Reporter Assay System (Promega). The expression of Renilla luciferase was normalized to the expression of firefly luciferase.
Liposome flotation assay.
All manipulations were conducted at room temperature or 25°C unless otherwise stated. Liposomes consisting of DOPC-SM-cholesterol-Rhod PE in a molar ratio of 8:2:3:0.05 were prepared as follows. An appropriate volume of a stock solution of each lipid dissolved in chloroform-methanol (19:1, vol/vol) was mixed in a glass tube to obtain the final amounts: 1.6 μmol of DOPC, 0.4 μmol of SM, 0.6 μmol of Chol, and 10 nmol of Rhod PE. For liposomes of other compositions, the amounts of lipid put into the glass tubes were altered appropriately. The lipid mixture was then dried under N2 gas at a bath temperature of 35 to 40°C for at least 20 min. The resultant lipid film was rehydrated with 1 ml of Tris-buffered saline (TBS) (25 mM Tris-HCl [pH 7.4], 137 mM NaCl, 2.68 mM KCl) containing 1 mM CaCl2 (TBS-Ca) and sonicated for 20 to 30 min with a probe-type UP50H ultrasonic processor (Hielscher Ultrasonics GmbH) at an amplitude of 80% and a duty cycle of 50%. The lipid suspension was centrifuged twice at 15,000 × g for 10 min to precipitate the lipid aggregates, and the resultant supernatant was collected containing the liposomes. The liposomes were stored at 4°C and used within 3 days. After they were returned to room temperature, the liposomes (150 μl) and the RuV antigens (30 μl) were mixed in a total volume of 300 μl of TBS-Ca, containing 1% (vol/vol) protease inhibitor cocktail, in a polycarbonate centrifuge tube (part number 343778; Beckman Coulter, Inc.). The mixture was incubated for 1 h and then subjected to a liposome flotation assay, as previously described (45), with some modifications. Briefly, the reaction mixture (300 μl) was mixed with 300 μl of 80% (wt/vol) Nycodenz and then overlaid with 300 μl of 30% (wt/vol) Nycodenz, followed by 300 μl of TBS-Ca. All of the Nycodenz solutions described above were made with TBS-Ca containing 1% (vol/vol) protease inhibitor cocktail. The samples were centrifuged at 55,000 rpm (at a maximum g [gmax] of 259,000) for 5 h in an Optima TLX ultracentrifuge (Beckman Coulter, Inc.) using a TLS-55 rotor (Beckman Coulter, Inc.). Twelve 100-μl aliquots were retrieved from the top of the gradient. When the volume of the last fraction was smaller than 100 μl, it was adjusted to ∼100 μl with TBS-Ca. To monitor the liposome distribution, the fluorescence intensity of Rhod PE in each fraction was measured at an excitation of 544 nm and emission of 590 nm, using a FLUOstar Optima microplate reader (BMG Labtech GmbH). To analyze the viral particle distribution, equal parts of the retrieved fractions were analyzed by Western blotting with a goat anti-RuV polyclonal antibody (1:4,000 dilution) and an HRP-conjugated mouse anti-goat/sheep IgG MAb (1:4,000 dilution) as the primary and secondary antibodies, respectively. The chemiluminescent pattern on the blot was visualized with Immobilon Western Chemiluminescent HRP substrate (Merck Millipore Co.) and detected with a WSE-6200H LuminoGraphII image analyzer (ATTO Co.).
Cell viability assay.
Monolayers of Vero cells in 96-well plates were incubated with DMEM containing SMase (30 and 150 mU/ml), HPA-12 (0.625, 1.25, 2.5, and 5 μM) or myriocin (1, 10, and 100 nM) for 2 days at 37°C. Vero cells were also incubated in DMEM as a control. After incubation, cell viability was measured with a CellTiter-Glo, version 2.0, luminescent cell viability assay (Promega) according to the manufacturer's instructions.
ACKNOWLEDGMENTS
We thank Z. Matsuda and C. K. Lim for providing the DSP system and SINV, respectively. We are also grateful to M. Nagai for her technical support. We thank Kate Fox from the Edanz Group (Fukuoka, Japan) for editing a draft of the manuscript.
This research was supported by AMED-CREST and JSPS KAKENHI (JP15K08508). We declare that we have no conflicts of interest.
REFERENCES
- 1.No author listed 2013. Global vaccine action plan. Decade of vaccine collaboration. Vaccine 31(Suppl 2):B5–B31. [DOI] [PubMed] [Google Scholar]
- 2.Vynnycky E, Adams EJ, Cutts FT, Reef SE, Navar AM, Simons E, Yoshida LM, Brown DW, Jackson C, Strebel PM, Dabbagh AJ. 2016. Using seroprevalence and immunisation coverage data to estimate the global burden of congenital rubella syndrome, 1996–2010: a systematic review. PLoS One 11:e0149160. doi: 10.1371/journal.pone.0149160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lescar J, Roussel A, Wien MW, Navaza J, Fuller SD, Wengler G, Wengler G, Rey FA. 2001. The fusion glycoprotein shell of Semliki Forest virus: an icosahedral assembly primed for fusogenic activation at endosomal pH. Cell 105:137–148. doi: 10.1016/S0092-8674(01)00303-8. [DOI] [PubMed] [Google Scholar]
- 4.Rey FA, Heinz FX, Mandl C, Kunz C, Harrison SC. 1995. The envelope glycoprotein from tick-borne encephalitis virus at 2 Å resolution. Nature 375:291–298. doi: 10.1038/375291a0. [DOI] [PubMed] [Google Scholar]
- 5.Modis Y, Ogata S, Clements D, Harrison SC. 2003. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc Natl Acad Sci U S A 100:6986–6991. doi: 10.1073/pnas.0832193100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roussel A, Lescar J, Vaney MC, Wengler G, Wengler G, Rey FA. 2006. Structure and interactions at the viral surface of the envelope protein E1 of Semliki Forest virus. Structure 14:75–86. doi: 10.1016/j.str.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 7.Dube M, Etienne L, Fels M, Kielian M. 2016. Calcium-dependent rubella virus fusion occurs in early endosomes. J Virol 90:6303–6313. doi: 10.1128/JVI.00634-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mastromarino P, Rieti S, Cioe L, Orsi N. 1989. Binding sites for rubella virus on erythrocyte membrane. Arch Virol 107:15–26. doi: 10.1007/BF01313874. [DOI] [PubMed] [Google Scholar]
- 9.Mastromarino P, Cioe L, Rieti S, Orsi N. 1990. Role of membrane phospholipids and glycolipids in the Vero cell surface receptor for rubella virus. Med Microbiol Immunol 179:105–114. doi: 10.1007/BF00198531. [DOI] [PubMed] [Google Scholar]
- 10.Wilschut J, Corver J, Nieva JL, Bron R, Moesby L, Reddy KC, Bittman R. 1995. Fusion of Semliki Forest virus with cholesterol-containing liposomes at low pH: a specific requirement for sphingolipids. Mol Membr Biol 12:143–149. doi: 10.3109/09687689509038510. [DOI] [PubMed] [Google Scholar]
- 11.Nieva JL, Bron R, Corver J, Wilschut J. 1994. Membrane fusion of Semliki Forest virus requires sphingolipids in the target membrane. EMBO J 13:2797–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waarts BL, Bittman R, Wilschut J. 2002. Sphingolipid and cholesterol dependence of alphavirus membrane fusion. Lack of correlation with lipid raft formation in target liposomes. J Biol Chem 277:38141–38147. [DOI] [PubMed] [Google Scholar]
- 13.Samsonov AV, Chatterjee PK, Razinkov VI, Eng CH, Kielian M, Cohen FS. 2002. Effects of membrane potential and sphingolipid structures on fusion of Semliki Forest virus. J Virol 76:12691–12702. doi: 10.1128/JVI.76.24.12691-12702.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klimjack MR, Jeffrey S, Kielian M. 1994. Membrane and protein interactions of a soluble form of the Semliki Forest virus fusion protein. J Virol 68:6940–6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corver J, Moesby L, Erukulla RK, Reddy KC, Bittman R, Wilschut J. 1995. Sphingolipid-dependent fusion of Semliki Forest virus with cholesterol-containing liposomes requires both the 3-hydroxyl group and the double bond of the sphingolipid backbone. J Virol 69:3220–3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smit JM, Bittman R, Wilschut J. 1999. Low-pH-dependent fusion of Sindbis virus with receptor-free cholesterol- and sphingolipid-containing liposomes. J Virol 73:8476–8484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cong H, Jiang Y, Tien P. 2011. Identification of the myelin oligodendrocyte glycoprotein as a cellular receptor for rubella virus. J Virol 85:11038–11047. doi: 10.1128/JVI.05398-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heggie AD, Robbins FC. 1969. Natural rubella acquired after birth. Clinical features and complications. Am J Dis Child 118:12–17. doi: 10.1001/archpedi.1969.02100040014003. [DOI] [PubMed] [Google Scholar]
- 19.Johns TG, Bernard CC. 1999. The structure and function of myelin oligodendrocyte glycoprotein. J Neurochem 72:1–9. doi: 10.1046/j.1471-4159.1999.0720001.x. [DOI] [PubMed] [Google Scholar]
- 20.Hanada K. 2005. Sphingolipids in infectious diseases. Jpn J Infect Dis 58:131–148. [PubMed] [Google Scholar]
- 21.Heung LJ, Luberto C, Del Poeta M. 2006. Role of sphingolipids in microbial pathogenesis. Infect Immun 74:28–39. doi: 10.1128/IAI.74.1.28-39.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van der Meer-Janssen YP, van Galen J, Batenburg JJ, Helms JB. 2010. Lipids in host-pathogen interactions: pathogens exploit the complexity of the host cell lipidome. Prog Lipid Res 49:1–26. doi: 10.1016/j.plipres.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanada K. 2003. Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. Biochim Biophys Acta 1632:16–30. doi: 10.1016/S1388-1981(03)00059-3. [DOI] [PubMed] [Google Scholar]
- 24.Hanada K, Kumagai K, Yasuda S, Miura Y, Kawano M, Fukasawa M, Nishijima M. 2003. Molecular machinery for non-vesicular trafficking of ceramide. Nature 426:803–809. doi: 10.1038/nature02188. [DOI] [PubMed] [Google Scholar]
- 25.Yasuda S, Kitagawa H, Ueno M, Ishitani H, Fukasawa M, Nishijima M, Kobayashi S, Hanada K. 2001. A novel inhibitor of ceramide trafficking from the endoplasmic reticulum to the site of sphingomyelin synthesis. J Biol Chem 276:43994–44002. doi: 10.1074/jbc.M104884200. [DOI] [PubMed] [Google Scholar]
- 26.Koumanov KS, Tessier C, Momchilova AB, Rainteau D, Wolf C, Quinn PJ. 2005. Comparative lipid analysis and structure of detergent-resistant membrane raft fractions isolated from human and ruminant erythrocytes. Arch Biochem Biophys 434:150–158. doi: 10.1016/j.abb.2004.10.025. [DOI] [PubMed] [Google Scholar]
- 27.Ishikawa H, Meng F, Kondo N, Iwamoto A, Matsuda Z. 2012. Generation of a dual-functional split-reporter protein for monitoring membrane fusion using self-associating split GFP. Protein Eng Des Sel 25:813–820. doi: 10.1093/protein/gzs051. [DOI] [PubMed] [Google Scholar]
- 28.Wang H, Li X, Nakane S, Liu S, Ishikawa H, Iwamoto A, Matsuda Z. 2014. Co-expression of foreign proteins tethered to HIV-1 envelope glycoprotein on the cell surface by introducing an intervening second membrane-spanning domain. PLoS One 9:e96790. doi: 10.1371/journal.pone.0096790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakane S, Matsuda Z. 2015. Dual split protein (DSP) assay to monitor cell-cell membrane fusion. Methods Mol Biol 1313:229–236. doi: 10.1007/978-1-4939-2703-6_17. [DOI] [PubMed] [Google Scholar]
- 30.Dube M, Rey FA, Kielian M. 2014. Rubella virus: first calcium-requiring viral fusion protein. PLoS Pathog 10:e1004530. doi: 10.1371/journal.ppat.1004530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Katow S, Sugiura A. 1988. Low pH-induced conformational change of rubella virus envelope proteins. J Gen Virol 69:2797–2807. doi: 10.1099/0022-1317-69-11-2797. [DOI] [PubMed] [Google Scholar]
- 32.Wahlberg JM, Bron R, Wilschut J, Garoff H. 1992. Membrane fusion of Semliki Forest virus involves homotrimers of the fusion protein. J Virol 66:7309–7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DuBois RM, Vaney MC, Tortorici MA, Kurdi RA, Barba-Spaeth G, Krey T, Rey FA. 2013. Functional and evolutionary insight from the crystal structure of rubella virus protein E1. Nature 493:552–556. doi: 10.1038/nature11741. [DOI] [PubMed] [Google Scholar]
- 34.Sakata M, Tani H, Anraku M, Kataoka M, Nagata N, Seki F, Tahara M, Otsuki N, Okamoto K, Takeda M, Mori Y. 2017. Analysis of VSV pseudotype virus infection mediated by rubella virus envelope proteins. Sci Rep 7:11607. doi: 10.1038/s41598-017-10865-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bron R, Wahlberg JM, Garoff H, Wilschut J. 1993. Membrane fusion of Semliki Forest virus in a model system: correlation between fusion kinetics and structural changes in the envelope glycoprotein. EMBO J 12:693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.White J, Helenius A. 1980. pH-dependent fusion between the Semliki Forest virus membrane and liposomes. Proc Natl Acad Sci U S A 77:3273–3277. doi: 10.1073/pnas.77.6.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kielian M, Chanel-Vos C, Liao M. 2010. Alphavirus entry and membrane fusion. Viruses 2:796–825. doi: 10.3390/v2040796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rose PP, Hanna SL, Spiridigliozzi A, Wannissorn N, Beiting DP, Ross SR, Hardy RW, Bambina SA, Heise MT, Cherry S. 2011. Natural resistance-associated macrophage protein is a cellular receptor for Sindbis virus in both insect and mammalian hosts. Cell Host Microbe 10:97–104. doi: 10.1016/j.chom.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sakata M, Otsuki N, Okamoto K, Anraku M, Nagai M, Takeda M, Mori Y. 2014. Short self-interacting N-terminal region of rubella virus capsid protein is essential for cooperative actions of capsid and nonstructural p150 proteins. J Virol 88:11187–11198. doi: 10.1128/JVI.01758-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Otsuki N, Abo H, Kubota T, Mori Y, Umino Y, Okamoto K, Takeda M, Komase K. 2011. Elucidation of the full genetic information of Japanese rubella vaccines and the genetic changes associated with in vitro and in vivo vaccine virus phenotypes. Vaccine 29:1863–1873. doi: 10.1016/j.vaccine.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 41.Olson K, Trent DW. 1985. Genetic and antigenic variations among geographical isolates of Sindbis virus. J Gen Virol 66:797–810. doi: 10.1099/0022-1317-66-4-797. [DOI] [PubMed] [Google Scholar]
- 42.Seki F, Takeda M, Minagawa H, Yanagi Y. 2006. Recombinant wild-type measles virus containing a single N481Y substitution in its haemagglutinin cannot use receptor CD46 as efficiently as that having the haemagglutinin of the Edmonston laboratory strain. J Gen Virol 87:1643–1648. doi: 10.1099/vir.0.81682-0. [DOI] [PubMed] [Google Scholar]
- 43.Shirogane Y, Takeda M, Iwasaki M, Ishiguro N, Takeuchi H, Nakatsu Y, Tahara M, Kikuta H, Yanagi Y. 2008. Efficient multiplication of human metapneumovirus in Vero cells expressing the transmembrane serine protease TMPRSS2. J Virol 82:8942–8946. doi: 10.1128/JVI.00676-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ono N, Tatsuo H, Hidaka Y, Aoki T, Minagawa H, Yanagi Y. 2001. Measles viruses on throat swabs from measles patients use signaling lymphocytic activation molecule (CDw150) but not CD46 as a cellular receptor. J Virol 75:4399–4401. doi: 10.1128/JVI.75.9.4399-4401.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Busse RA, Scacioc A, Hernandez JM, Krick R, Stephan M, Janshoff A, Thumm M, Kuhnel K. 2013. Qualitative and quantitative characterization of protein-phosphoinositide interactions with liposome-based methods. Autophagy 9:770–777. doi: 10.4161/auto.23978. [DOI] [PMC free article] [PubMed] [Google Scholar]