

ABSTRACT
Increasing evidence suggests that repetitive elements may play a role in host gene regulation, particularly through the donation of alternative promoters, enhancers, splice sites, and termination signals. Elevated transcript expression of the endogenous retrovirus group HERV-K (HML-2) is seen in many human cancers, although the identities of the individual proviral loci contributing to this expression as well as their mechanisms of activation have been unclear. Using high-throughput next-generation sequencing techniques optimized for the capture of HML-2 expression, we characterized the HML-2 transcriptome and means of activation in an in vitro model of human mammary epithelial cell transformation. Our analysis showed significant expression originating from 15 HML-2 full-length proviruses, through four modes of transcription. The majority of expression was in the antisense orientation and from proviruses integrated within introns. We found two instances of long terminal repeat (LTR)-driven provirus transcription but no evidence to suggest that these active 5′ LTRs were influencing nearby host gene expression. Importantly, LTR-driven transcription was restricted to tumorigenic cells, suggesting that LTR promoter activity is dependent upon the transcriptional environment of a malignant cell.
IMPORTANCE Here, we use an in vitro model of human mammary epithelial cell transformation to assess how malignancy-associated shifts in the transcriptional milieu of a cell may impact HML-2 activity. We found 15 proviruses to be significantly expressed through four different mechanisms, with the majority of transcripts being antisense copies of proviruses located within introns. We saw active 5′ LTR use in tumorigenic cells only, suggesting that the cellular environment of a cancer cell is a critical component for induction of LTR promoter activity. These findings have implications for future studies investigating HML-2 as a target for immunotherapy or as a biomarker for disease.
KEYWORDS: human endogenous retrovirus, mammary cell transformation, provirus expression
INTRODUCTION
The hallmark characteristic of a retrovirus is the ability to reverse transcribe its RNA-based genome into DNA, which is then irreversibly integrated into the host genome. This integrated sequence, known as a provirus, is transcribed and translated in a way similar to that of any other host gene (1). Retroviruses encode a minimum of four genes (gag, pro, pol, and env) flanked by long terminal repeats (LTRs), which contain all elements necessary for driving and terminating transcription. Although transmission generally occurs horizontally, through infection of somatic cells, germ line infection can lead to genetic transmission from parent to offspring. Germ line proviruses are inherited in a Mendelian fashion and are subject to natural selection. Proviruses with detrimental effects are generally lost from the population, while those with advantageous or neutral effects can become fixed. Proviruses in the human genome are known as human endogenous retroviruses (HERVs) (2, 3).
LTR retrotransposon sequences, including HERVs, make up about 8% of the human genome, which is a large proportion compared to the 1 to 2% comprising protein-coding exons. HERV sequences are the result of bursts of integration events, which occurred in humans as recently as 100,000 years ago and can be traced back through our primate ancestors (4, 5). Although there are currently no known infectious HERVs due to the accumulation of mutations over time, likely combined with selection against proviruses capable of giving rise to infectious virus, many of these sequences are still able to produce viral mRNA, protein, and even retrovirus-like particles (6–9). Active LTRs are capable of influencing host gene regulation up to 100 kb away, generally through the donation of alternative promoters, enhancers, splice sites, and termination signals. Additionally, HERV sequences are largely silenced in normal tissue through epigenetic effects, including DNA and chromatin modifications (10, 11).
The HERV-K (HML-2) (Human mouse mammary tumor virus [MMTV]-Like group 2) group contains the most recently integrated as well as the most biologically active proviruses. Their activity is upregulated under several different kinds of conditions, including autoimmune diseases, neurological disorders, and cancer, although the role that their upregulation plays in such ailments is unclear (12–14). HML-2 was first detected due to its sequence similarity to MMTV, resulting in speculation of a causal role in human breast cancer development (15). Although there is no evidence to support this claim, HML-2 expression is, nonetheless, affiliated with malignant disease. HML-2 transcript abundance is elevated in many types of human cancers, including up to 85% of breast cancer samples (8, 16). Many groups have considered utilizing this expression as a biomarker for disease progression, and HML-2 proteins are being investigated as possible tumor-associated antigens and therapeutic targets (16–19). As the field stands, not much is known about the specifics of this phenomenon, including which individual HML-2 proviruses are contributing to this expression, what gene products they encode, how provirus transcription is regulated, or if HML-2 promoter activity is restricted to malignant cells.
We sought answers to these questions by characterizing the HML-2 transcriptome in an in vitro model of human mammary epithelial cell (HMEC) transformation, analyzing how malignant shifts in the cellular environment may affect HML-2 expression. We employed a transcriptome sequencing (RNA-Seq) protocol that focuses on long (301-bp), paired-end (PE) reads, with unique, stranded alignments to both the human reference genome (hg19) and an HML-2 reference genome containing all known HML-2 elements, 36 of which are nonreference (6). Through computational analysis of these alignments using the Integrative Genomics Viewer (IGV), we were able to characterize the mechanisms of HML-2 transcription before and after cellular transformation and determine if HML-2 activity was having an impact on nearby host gene regulation.
RESULTS
High-throughput analysis of HML-2 expression levels during HMEC transformation.
For this investigation, we utilized an in vitro model of HMEC transformation, as initially described by Elenbaas et al. (20). This model mirrors the transition of a primary cell to a tumorigenic one in three steps, producing transformed cell lines with significant variances in the transcriptional milieu. This method allows for the analysis of how malignant shifts in the cellular environment may affect HML-2 expression.
A schematic depiction of this model is shown in Fig. 1A. Immortalization of primary HMECs is achieved through telomere maintenance by hTERT (encoding human telomerase reverse transcriptase) overexpression. These HME cells are then transformed into HMLE cells via introduction of the simian virus 40 (SV40) early region, containing small and large T antigens. This procedure results in uncontrolled cellular division caused by inhibition of the tumor-suppressing p53 and pRB pathways (20–22). Lastly, overexpression of the commonly amplified breast cancer oncogenes HRAS(V12) and ERBB2 (also known as HER2/neu) produces the tumorigenic HMLE-Ras and HMLE-Her2 cells, respectively (23–25).
FIG 1.

Properties of cell lines used in this study. (A) Schematic of the HMEC transformation process in vitro, as initially described by Elenbaas et al. (20). Transformation steps are shown with arrows, and transformation stages are labeled in blue. Cell line names and overexpressed genes introduced by each transfection step are labeled in black. (B) Jensen-Shannon (JS) divergence matrix, showing pairwise dissimilarity in global gene expression of each cell line sequenced. Values are given as JS distances; increased JS distance signifies an increase in dissimilarity. (C) Bar graph showing the total unique HML-2 transcript levels in each cell line analyzed (red bars), including Tera-1 results from our previously published study (blue bar) (6). These are compared against the transcript expression levels of housekeeping genes (GAPDH [glyceraldehyde-3-phosphate dehydrogenase], NONO, RIF1, and TET2, green bars) in the Hcc1954 cell line.
Passage-matched samples of HME, HMLE-Ras, and HMLE-Her2 cells were subjected to RNA-Seq analysis alongside Hcc1954, an established tumorigenic breast cancer cell line (26). Statistical analysis using Jensen-Shannon divergence, which measures pairwise similarities among multiple conditions, was used to determine the degree of change in global gene expression within each cell line (Fig. 1B). A clear shift in the transcriptional environment was seen between the HME cells and their tumorigenic counterparts, suggesting that cellular transformation notably alters the global transcriptional milieu of a cell.
HML-2 expression was analyzed using an RNA-Seq protocol previously established by our laboratory (6). This method (see Materials and Methods) is optimized for capture of HML-2 expression and relies on long (301-bp), paired-end reads produced from stranded libraries, which are filtered for unique alignment only. Critically, this method has proved successful for capturing distinct transcripts from all identified proviruses, regardless of their age, with high sequence similarity, as well as proviruses incorrectly annotated in the human reference genome (GRCh37/hg19 build) (6, 27). In our current analysis, 96 to 99% of HML-2 reads were retained after filtering, and all aligned uniquely to an individual provirus (Table 1). The high levels of sensitivity and specificity exhibited by our results suggest that our protocol is stringent enough to bypass any multimapping issues and is able to accurately detect and quantitate transcript levels from all 96 HML-2 proviruses identified to date.
TABLE 1.
Retention rate of HML-2 reads after filtering for unique alignments only
| Cell line | Retention (%) of filtered HML-2 reads |
|---|---|
| Hcc1954 | 99.7 |
| HMLE-Ras | 98.5 |
| HMLE-Her2 | 96.7 |
| HME | 96.0 |
Transcript expression levels were calculated in FPKM (fragments per kilobase of transcript per million mapped reads). This approach takes the number of reads that align to the exons of each transcript and normalizes them against the length of the transcript as well as the total number of reads in the sequencing library. Total HML-2 expression levels were determined by summing the FPKM values of all individual proviruses. These values were compared to sequencing data from our previously published experiment that characterized the HML-2 expression profile in the human teratocarcinoma cell line Tera-1 (6), as this cell line is known to express HML-2 elements at exceptionally high levels (2, 28, 29). We detected HML-2 mRNA in all cell lines sequenced, with the highest levels seen in tumorigenic Hcc1954 and HMLE-Ras cells. Although these levels were approximately 10-fold lower than those seen in Tera-1 cells, they were comparable to those of some critical housekeeping genes, including RIF1 and TET2 (Fig. 1C).
HML-2 transcript expression is dominated by older proviruses producing antisense mRNAs.
It is well documented that tumorigenesis results in an increase in HML-2 expression in a large number of human cancers (2, 7, 8, 19, 30, 31). However, the nature of this expression, in particular, whether it is due to an increase in intensity of expression of the same proviruses or to an increase in diversity of expressed loci, is unclear. To investigate this issue further, we broke down the HML-2 expression profiles of each sequenced cell line by individual provirus as well as orientation of transcription. As is our convention, all proviruses mentioned in this study are listed by chromosomal location, with aliases and genomic coordinates shown in Table 2.
TABLE 2.
HML-2 proviruses with alternative names and genomic coordinatesa
| Provirus | Alternative name(s) | Chromosomal location (hg19) |
|---|---|---|
| 1q21.3 | chr1: 150,605,284-150,608,361 | |
| 1q22 | K102, K(C1b), K50a, ERVK-7 | chr1: 155,596,457-155,605,636 |
| 3q12.3 | KII, ERVK-5 | chr3: 101,410,737-101,419,859 |
| 3q21.2 | KI, ERVK-4 | chr3: 125,609,302-125,618,439 |
| 4p16.1b | K50c | chr4: 9,659,580-9,669,174 |
| 4p16.3a | chr4: 234,989-239,459 | |
| 7q34 | K(OLDAC004979), ERVK-15 | chr7: 141,451,918-141,455,938 |
| 8p23.1b | K27 | chr8: 8,054,700-8,064,221 |
| 8p23.1c | chr8: 12,073,970-12,083,497 | |
| 9q34.11 | K31 | chr9: 131,612,515-131,619,736 |
| 10p14 | K(C11a), K33, ERVK-16 | chr10: 6,866,141-6,875,603 |
| 12q24.11 | chr12: 111,007,843-111,009,325 | |
| 12q24.33 | K42 | chr12: 133,667,122-133,673,064 |
| 14q11.2 | K(OLDAL136419), K71 | chr14: 24,480,600-24,484,985 |
| 19q13.12b | K(OLDAC012309), KOLD12309, K50F | chr19: 37,597,549-37,607,066 |
We found only 15 proviruses to be appreciably transcribed (FPKM > 0.5) (Fig. 2A; Table 3). Surprisingly, only five of these proviruses were significantly sense transcribed, with 75% of that transcription stemming from three loci (1q21.3, 1q22, and 3q12.3) (Fig. 2B). In contrast, a more diverse set of proviruses were expressed in the antisense orientation (Fig. 2C), suggesting that the majority of HML-2 transcription is due to sequences outside the provirus that result in a variety of noncoding transcripts. Overall, we found that antisense transcription constituted the majority of HML-2 expression in these cell lines: 72% of the total in Hcc1954, 73% in HMLE-Ras, 49% in HMLE-Her2, and 82% in HME cells (Fig. 3A). Age analysis of each expressed provirus, as determined by the estimated time since integration, showed that all but one provirus (1q22) integrated at least 5 million years ago (Fig. 3B).
FIG 2.

Multiple proviruses contribute to HML-2 group expression. (A) Bar graphs depicting the FPKM values of each significantly expressed provirus. Significant expression is defined as proviruses with FPKM of >0.5. Proviruses with FPKM of <0.5 are grouped together as “Other.” Raw FPKM values are listed in Table 3. Data are further split to show the FPKM of proviruses transcribed in sense (B) and in antisense (C) orientation. All human-specific proviruses, as described by Subramanian et al. (4), are designated with a red triangle.
TABLE 3.
FPKM values of significantly transcribed provirusesa
| Provirus | Hcc1954 |
HMLE-Ras |
HMLE-Her2 |
HME |
||||
|---|---|---|---|---|---|---|---|---|
| FPKM | %b | FPKM | % | FPKM | % | FPKM | % | |
| 1q21.3 | 2.50 | 11.03 | 2.04 | 8.27 | 0.23 | 3.62 | 0.39 | 2.67 |
| 1q22 | 0.00 | 0.00 | 2.69 | 10.87 | 2.45 | 38.03 | 1.45 | 9.84 |
| 3q12.3 | 2.75 | 12.14 | 1.52 | 6.14 | 0.40 | 6.21 | 0.25 | 1.72 |
| 3q21.2 | 1.63 | 7.17 | 2.38 | 9.61 | 0.33 | 5.18 | 1.49 | 10.14 |
| 4p16.1b | 0.81 | 3.58 | 0.00 | 0.00 | 0.07 | 1.04 | 0.00 | 0.00 |
| 4p16.3a | 0.30 | 1.31 | 0.41 | 1.67 | 0.15 | 2.30 | 0.53 | 3.60 |
| 7q34 | 5.21 | 22.97 | 1.54 | 6.23 | 0.17 | 2.60 | 0.89 | 6.03 |
| 8p23.1b | 0.05 | 0.20 | 1.33 | 5.36 | 0.13 | 1.98 | 0.24 | 1.65 |
| 8p23.1c | 0.64 | 2.80 | 0.57 | 2.30 | 0.19 | 2.96 | 0.06 | 0.41 |
| 9q34.11 | 0.90 | 3.99 | 2.68 | 10.85 | 0.34 | 5.34 | 2.98 | 20.24 |
| 10p14 | 0.00 | 0.00 | 2.41 | 9.75 | 0.33 | 5.10 | 3.32 | 22.54 |
| 12q24.11 | 0.33 | 1.46 | 2.75 | 11.14 | 0.68 | 10.58 | 0.88 | 5.99 |
| 12q24.33 | 1.38 | 6.10 | 1.21 | 4.92 | 0.53 | 8.18 | 0.88 | 5.95 |
| 14q11.2 | 1.03 | 4.53 | 0.36 | 1.44 | 0.00 | 0.00 | 0.00 | 0.00 |
| 19q13.12b | 1.36 | 6.01 | 0.88 | 3.58 | 0.06 | 0.99 | 0.49 | 3.30 |
| Other (27 transcripts) | 3.79 | 16.69 | 1.94 | 7.86 | 0.38 | 5.91 | 0.87 | 5.94 |
Significant expression is defined as transcripts with FPKM of >0.5. Transcripts with FPKM of <0.5 are grouped together as “Other.”
Percentage of total HML-2 reads in a given cell line.
FIG 3.
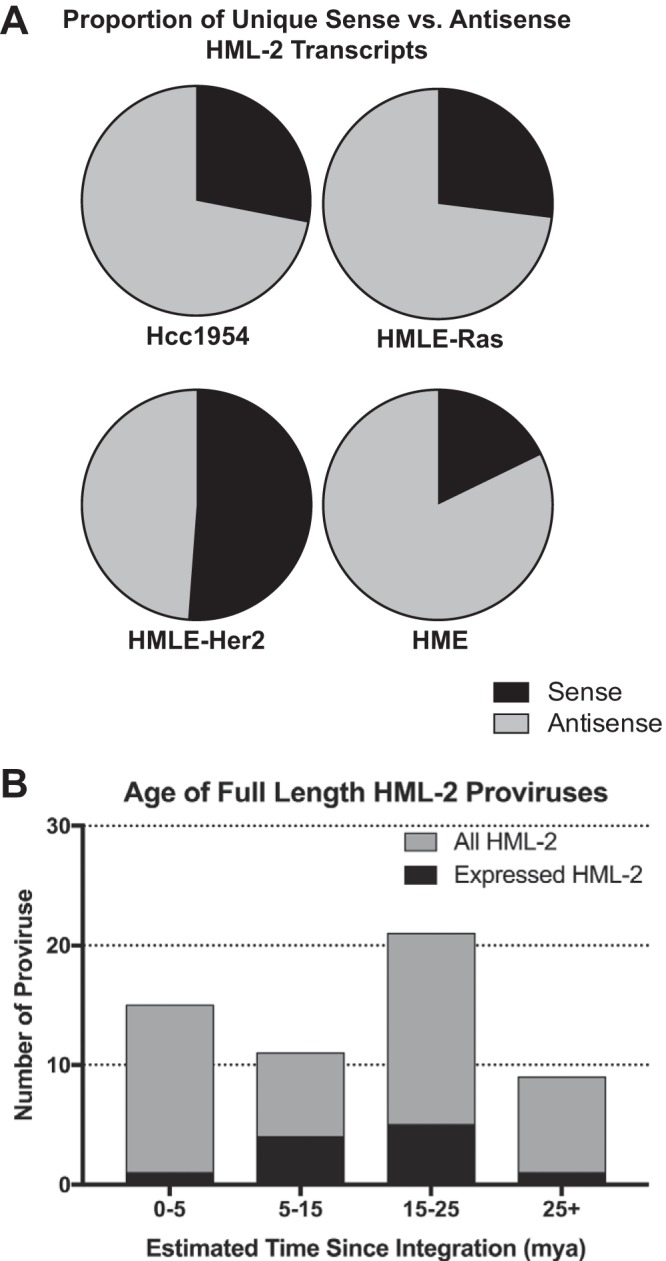
HML-2 expression is dominated by older proviruses producing antisense transcripts. (A) Pie charts comparing the percentage of total HML-2 expression that is due to sense (black) versus antisense (gray) transcription for each cell line sequenced. Sense was determined by the strandedness of the alignment. (B) Bar graph showing the ages, given as estimated time since integration in million years ago (mya), of full-length HML-2 proviruses. The total number of HML-2 proviruses within a given age range is shown in gray. Superimposed, in black, is the number of HML-2 proviruses with significant expression in this study. Age estimates are plotted as averages as determined by Subramanian et al. (4), and any proviruses with indeterminable ages were excluded.
HML-2 transcription occurs via four mechanisms.
We next wanted to investigate the mechanisms behind HML-2 transcription and determine whether these change with tumorigenicity. Through visual analysis of our alignments using the Integrative Genomics Viewer (32), we were able to detect HML-2 transcription patterns consistent with four different modes: initiation in the proviral LTR, read-through transcription, as part of a long noncoding RNA (lncRNA), and intronic transcription (Table 4).
TABLE 4.
Characterization and expression patterns of significantly expressed proviruses
| Provirus | LTRs | Mode of transcription | Impacting host gene encoding: | Cell line expression |
|||
|---|---|---|---|---|---|---|---|
| Hcc 1954 | HMLE-Ras | HMLE-Her2 | HME | ||||
| Antisense-transcribed proviruses | |||||||
| 3q21.2a | 5′/Δ3′ | Read-through | Repetitive element | + | + | + | |
| 7q34 | Δ5′ | Read-through | SSBP1 | + | + | + | |
| 8p23.1b | 5′/3′ | Read-through | Repetitive element | + | |||
| 8p23.1c | 5′/3′ | Read-through | Repetitive element | + | + | ||
| 9q34.11 | 5′/3′ | Intronic | CCBL1 | + | + | + | |
| 10p14 | 5′/3′ | Intronic | LINC00707 | + | + | ||
| 12q24.11a | 5′ | Intronic | PPTC7 | + | + | + | |
| 12q24.33 | 5′/3′ | Intronic | ZNF140 | + | + | + | + |
| 14q11.2 | Δ5′/3′ | Intronic | DHRS4L1 | + | |||
| 19q13.12b | 5′/3′ | Intronic | ZNF420 | + | + | ||
| Sense-transcribed proviruses | |||||||
| 1q21.3 | Δ5′/3′ | Read-through | Repetitive element | + | + | ||
| 1q22a | 5′/3′ | lncRNA | BC041646 | + | + | + | |
| 3q12.3 | 5′/3′ | LTR driven | + | + | |||
| 4p16.1b | 5′/Δ3′ | LTR driven | + | ||||
| 4p16.3a | 5′ | Intronic | ZNF876P | + | |||
Human-specific provirus.
We found the majority of HML-2 antisense expression to be associated with proviruses in introns (Fig. 4A, gray). A representative image of this mode of transcription is seen in Fig. 5A, in which proviral alignments are evident within an intronic region of a host gene. This correlation suggests that these proviruses were not self-transcribed, but rather that their expression was a consequence of being preserved within an incompletely removed intron. All other antisense HML-2 transcription was due to read-through (Fig. 4A, red), a consequence of being situated downstream of a transcribed host gene or repetitive element. A representative image of this mechanism is seen in Fig. 5B, where transcription is seen continuing past the last host gene exon and into the proviral sequence.
FIG 4.
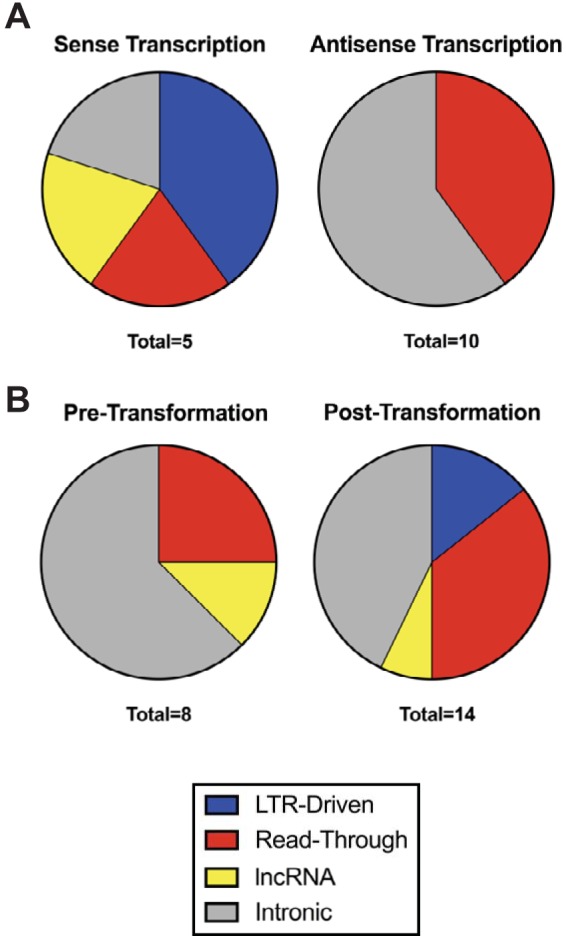
LTR-driven sense transcription is restricted to transformed cells. Pie charts showing the proportion of each mode of transcription identified through IGV to produce sense (left) and antisense (right) transcripts (A) as well as the proportion of each mode of transcription detected in cell lines pretransformation (left, includes HME cell line) and posttransformation (right, includes HMLE-Ras, HMLE-Her2, and Hcc1954 cell lines) (B). “Total” refers to the number of expressed proviruses in each group.
FIG 5.

Modes of HML-2 transcription. Four modes of HML-2 transcription were identified from the 15 significantly expressed proviruses through IGV analysis of alignments: intronic (A), read-through (B), lncRNA associated (C), and LTR driven (D). Schematic drawings of each mode of transcription are shown on the upper part of each panel. Dark gray boxes depict host genes, and arrows correspond to the direction of transcription. Blue boxes depict proviral sequences, and arrows depict the direction of sense transcription. Black arrows designate transcription start sites. The ribbon on the top depicts RNA, with black sections signifying the transcripts of host genes and blue sections signifying provirus-derived sequences. Representative IGV visualizations of each mode of transcription are shown on the bottom. Light gray boxes show aligned sequencing reads, which range from 60 to 592 bp in length. Splicing is shown by light blue lines connecting reads. Locations of host and proviral genes are superimposed on top of alignments. Blue boxes depict LTR sequences, and double black lines depict internal proviral sequences.
Most interestingly, three of the sense-transcribed proviruses appear to have functional LTRs. 1q22 is expressed as part of an annotated lncRNA of unknown function (BC041646), known to be highly expressed in breast cells as well as white blood cells, placenta, lung, and lymph node (33–35). This lncRNA originates from an upstream simple repeat sequence and terminates using the polyadenylation signal found in the 1q22 3′ LTR (Fig. 5C). Two proviruses, 3q12.3 and 4p16.1b, appear to have LTR-driven transcription, whereby reads are seen originating from the transcription start site of the 5′ LTR. A representative image of LTR-driven transcription of 3q12.3 is shown in Fig. 5D. Interestingly, LTR-driven transcription was the only mechanism of expression strictly related to tumorigenicity, as it was not detected in the nontumorigenic cells (Fig. 4B, blue). These results suggest that the transcriptional milieu of a tumorigenic cell is critical for LTR promoter activity in this model.
3q12.3 and 4p16.1b have functional 5′ LTRs but do not affect host gene transcription.
Confirmation of the promoter activity of the 3q12.3 and 4p16.1b LTRs in the transformed cells was achieved using a dual-luciferase assay (36). As shown in Fig. 6, the 3q12.3 5′ LTR showed high promoter activity in all cell lines except for HME. This activity correlated with the transcript expression seen in Hcc1954 and HMLE-Ras cells. Interestingly, the LTR exhibited high promoter activity in HMLE-Her2 cells, despite the low level of 3q12.3 transcripts in that cell line (Fig. 6A). Since luciferase assays are based on transient transfection and are insensitive to epigenetic effects, it is possible that 3q12.3 is silenced epigenetically in the HMLE-Her2 cell line but capable of promoter activity under optimal transcriptional conditions. The 4p16.1b LTR showed low promoter activity in Hcc1954, which correlated with the low transcript expression also seen in only those cells (Fig. 6B), implying the absence of necessary trans-acting factors.
FIG 6.

Confirmation of active 5′ LTR activity. (A, B) Confirmation of 5′ LTR activity of two proviruses, 3q12.3 (A) and 4p16.1b (B), shown to have LTR-driven sense transcription in our data set. Promoter activity is given in relative light units (RLU, black) of the LTR-driven luciferase activity normalized to a cotransfected control of Renilla luciferase activity driven by an SV40 promoter. Luciferase activity is compared to the relative transcript expression given in FPKM (gray). FPKM values are from stranded Cuffdiff analyses and are normalized across all cell lines. All luciferase assays were conducted in triplicate, and their results are plotted as the means ± standard deviations. (C) Left, schematic showing the 3q12.3 locus including all genes found within 100 kb of either end of the provirus as annotated by RefSeq in the UCSC Genome Browser; right, heat map showing the expression levels of each gene within each cell line. Values are given as log (FPKM + 1) and are normalized through Cuffdiff analysis. Clustering is based on Euclidean distance.
Retroviral LTRs are capable of influencing host gene transcription up to 100 kb away (1). To investigate whether the active 3q12.3 or 4p16.1b LTRs were influencing host gene transcription, we compared the gene expression levels of all host genes, as annotated by RefSeq in the UCSC Genome Browser, within 100 kb in either direction of the provirus (34). We found no known genes in the vicinity of 4p16.1b, ruling out the possibility of its directly affecting host gene transcription. In contrast, there are seven genes (four upstream and three downstream) within 100 kb of the 3q12.3 provirus. However, we found no correlation between provirus expression and host gene expression in any cell line sequenced (Fig. 6C). From this result, we conclude that HML-2 LTR activity has no detectable effect on host gene transcription in these samples.
DISCUSSION
It has been postulated that nearly a quarter of all human cancers are of viral etiology (17, 37). Retroviruses are unique in that they can be transmitted endogenously through the germ line, in addition to exogenously from one individual to another. MMTV and Jaagsiekte sheep retrovirus (JSRV) are two examples of betaretroviruses that can be transmitted endogenously with pathogenic effects; MMTV causes mammary carcinoma in mice, and JSRV causes lung carcinoma in sheep (1, 38, 39). Since 8% of the human genome consists of ERV sequences, the potential role of these elements in driving or aiding tumorigenesis is of great concern.
Interest in HML-2 playing a role in human breast cancer began when it was first described due to its sequence similarity to MMTV (15). To date, several groups have reported HML-2 mRNA and protein as well as noninfectious virus-like particles in breast cancer cell lines and tumor samples that are at increased levels relative to surrounding nondiseased breast tissue and that decrease significantly with treatment (7, 13, 16). Recently, HML-2 activity has been investigated for use as a prognostic marker for disease onset and treatment efficacy (17, 40). Immunotherapy approaches, including HERV-K-specific monoclonal antibody and chimeric antigen receptor (CAR) T-cell treatment, are being tested based on the idea that expressed HML-2 proteins can be targeted as a tumor-associated antigen (18, 19, 41).
Although it is well established that HML-2 transcripts are increased posttransformation (8, 16, 18), information such as which individual loci are contributing to this expression, their mechanism(s) of activation, and whether they are directly altering host gene transcription, was lacking. The goal of this study was to fully characterize the HML-2 transcriptome during mammary epithelial cell transformation and to identify the methods of transcription involved in the expression of the different proviruses. For this purpose, we used an RNA-Seq approach previously developed by our lab (6). Past investigations have been conducted by looking at group expression, generally through DNA hybridization techniques or reverse transcription (RT)-PCR using LTR- or gene-specific primers (8, 13, 42). However, these techniques were not stringent enough to distinguish among individual proviruses, whose sequence similarity can be over 99% (4, 14). Additionally, unless strand-specific primers were used, they did not distinguish sense from antisense transcription, an obviously important factor to consider when determining the mechanism and consequences of HML-2 transcription.
We chose to focus this study on proviruses categorized as “full-length” (including those with internal deletions), as distinct from solo LTRs (4, 27), as some of these have the potential to code for proteins that might have functional consequences for the tumor cell, provide informative biomarkers, or be useful in immunotherapy. Solo LTRs, which are present at a 10-fold-higher frequency than full-length proviruses (4), may also play a role in altering host gene expression through the donation of alternative promoters, enhancers, and polyadenylation signals (1) and warrant a separate future investigation.
A potential concern with mapping RNA-Seq reads to individual proviruses is that their sequence similarity might preclude unique assignment, leading to loss of information regarding their expression, particularly for the younger proviruses. This problem was ameliorated by the use of a long-read sequencing protocol so that, overall, our data had a 96 to 99% retention rate after filtering for uniquely mapped reads (Table 1) and even the youngest proviruses were well represented despite their close similarity to one another. Those reads that mapped to more than one locus were short, averaging 112 bp in length, and were mostly located in the LTR regions of proviruses with solo LTRs of highly similar sequence. IGV analysis of these reads ensured that filtering them did not affect our transcriptome analysis, as the number of multimapped reads was low.
Although the high level of read retention postfiltering was partly due to the stringency of our protocol, which was optimized for read length and alignment specificity, it was also due in part to the individual proviruses that were expressed. Since the activity of HML-2 proviruses has been credited to their relatively young age compared to all other HERV groups, we were surprised to find that all the expressed proviruses but one (1q22) were ones that had integrated into the ancestral primate genome more than 5 million years ago (Fig. 3B). Further investigation of multimapped reads confirmed that no other young proviruses were expressed and inadvertently filtered out during our analysis. Since older proviruses have accumulated more mutations, and therefore more diversity, than younger ones, the low rate of multimapping that we saw is not surprising. Indeed, our previously published analysis (6) showed that our protocol was capable of detecting and distinguishing expression from every provirus, young or old, albeit with a modest loss of the most highly conserved regions of the more recently integrated ones.
We detected HML-2 transcripts in all cell lines sequenced, with the highest levels seen in the tumorigenic Hcc1954 and HMLE-Ras cell lines (Fig. 1C). Although the sample numbers are small, this correlation is in concordance with previous studies that suggest that HML-2 proviruses are expressed at low levels in normal tissue and that the activity of at least some increases with tumorigenicity (2, 30, 43). Surprisingly, we did not see increased HML-2 expression in the tumorigenic HMLE-Her2 cell line (Fig. 1C), which was the only tumorigenic cell line studied not to exhibit LTR-driven transcription (Table 4). It is possible that the overexpression of ERBB2 resulted in a transcriptional environment different from that of the other cell lines, one that does not support HML-2 activity. Although previous work suggests that the majority (∼85%) of breast cancer samples show increased HML-2 activity (8, 16), it is not found in all cases, and these cells will be subjected to further investigation. Additionally, HML-2 expression in breast cancer cell lines has been shown to drastically increase in response to treatment with female steroid hormones, specifically estradiol and progesterone (8, 44, 45). However, due to the absence of expression of these major hormone receptors in the cell lines that we analyzed (data not shown), it is unlikely that induced hormone expression would have had any effect on HML-2 transcription in this study. Due to the unavailability of primary HMEC and HMLE cell lines for testing (Fig. 1A), the individual influences of hTERT and SV40 overexpression in these cell lines were not examined.
We found 15 individual proviruses to be significantly expressed in our HMEC transformation model (Fig. 2). Using an HML-2 reference genome including all known proviruses in the hg19 reference genome plus 36 HML-2 elements not present in the reference sequence (27), we were able to confirm that no nonreference HML-2 proviruses were contributing to this expression (data not shown). Due to the age of the expressed proviruses, we did not expect many to still have functional LTRs. Indeed, we found that the majority of provirus expression was in the antisense direction and due to either intronic or read-through transcription (Fig. 4A). Although evidence suggests that LTRs can have bidirectional activity and that 3′ HML-2 LTRs are capable of producing antisense transcripts (1, 46), we did not see any transcripts originating from any 3′ LTR that were consistent with such activity. There is increasing belief that antisense and/or lncRNA HERV transcripts may play key roles in host gene expression, particularly through transcriptional interference, caused by two promoters competing for the use of RNA transcriptional machinery in cis, or interactions with their cRNA sequences. Complementary binding of antisense and sense RNA molecules can result in inhibition of translation of the sense RNA through induction of the RNA interference pathway or induce an innate immune response from the production of double-stranded RNA. However, the impact of noncoding HERV transcripts on host gene regulation is currently unknown (1, 6, 14, 31, 47).
We found three proviruses exhibiting active LTR use, as indicated by transcripts either initiating at the U3-R border or terminating at the R-U5 border. The 1q22 locus exhibited sense transcription driven by an upstream simple repeat element and is part of an annotated lncRNA (BC041646) of unknown function. This lncRNA is multiply spliced and utilizes the polyadenylation site in the 1q22 3′ LTR for termination (Fig. 5C). Two proviruses, 3q12.3 and 4p16.1b, were found to have LTR-driven transcripts produced from their 5′ LTRs. The retention of LTR activity for these proviruses was confirmed with dual-luciferase assays. This activity was restricted to tumorigenic cell lines (Hcc1954, HMLE-Ras, and HMLE-Her2), suggesting that HML-2 LTR activation is dependent upon some feature of malignant cellular environments (Fig. 6A and B). Exactly what cellular factors are required for this activity and how they interact with the LTR transcription factor binding sites are under investigation. These future results may shed light on why so few young proviruses were active in the cell lines used in this study.
Overall, we found that HML-2 proviruses expressed during HMEC transformation are transcribed by at least four mechanisms. The majority of expressed proviruses that we analyzed are older and found as antisense transcripts due to their presence in incompletely removed introns. Although our data suggest that five proviruses (1q21.3, 1q22, 3q12.3, 4p16.1b, and 4p16.3a) are significantly sense transcribed (Fig. 2B), in silico analysis of the five proviruses shows that none has any open reading frames for any of the main viral genes (gag, pro, pol, or env) (4). This analysis is in discordance with previous studies that suggest that many human breast cancer cell lines and patient samples are capable of producing HML-2 protein (9, 17, 18), indicating that there may be significant heterogeneity of HML-2 provirus expression among different breast cancer cell lines. Further investigations of more cell lines will need to be conducted to study the protein expression in these cells and conclude whether breast cancer is a viable target for immunotherapy. Two proviruses, at 3q12.3 and 4p16.1b, were expressed by LTR-driven transcription, and their activity was found to be restricted to tumorigenic cells (Fig. 4B), suggesting that the transcriptional environment of those cells is conducive for activation of their LTR promoters. Despite the LTR activity exhibited by 3q12.3 and 4p16.1b, we found no evidence to support the idea that they affected host gene transcription in the cell lines tested (Fig. 6C).
For this study, we looked at only a small number of cell lines; the consistency or variation in the expression patterns observed among normal and malignant cell lines and fresh tissue will be the subject of further study. Since HML-2 expression is known to vary among cancer types and even possibly among cancer samples (2, 8, 19, 31), the potential for some other HML-2 proviruses to be turned on posttransformation and contributing to the tumorigenic process cannot be ruled out.
MATERIALS AND METHODS
Cell culture.
The HME, HMLE-Her2, and HMLE-Ras cell lines were derived from nondiseased primary HMECs by infection with murine leukemia virus (MLV)-based vectors carrying the ERBB2 or HRAS(V12) genes and bulk selection for a drug resistance marker (20). They were maintained in bulk culture in mammary epithelial cell growth medium (MEGM) and supplemented as directed by the MEGM Bullet kit (catalog number CC-3150; Lonza, Walkersville, MD, USA). As per ATCC's recommendation for growing HMECs, complete MEGM was further supplemented with 0.1 μg/ml cholera toxin (catalog number C8052; Sigma, St. Louis, MO, USA). The tumorigenic human breast cancer cell line Hcc1954 was derived from a primary stage IIA, grade 3 invasive ductal carcinoma (catalog number CRL-2338; ATCC, Manassas, VA, USA). It was cultured in RPMI 1640 medium (catalog number 61870-036; Gibco, Carlsbad, CA, USA) supplemented with 100 μl/ml fetal bovine serum (FBS; catalog number S11195; Atlanta Biologicals, Norcross, GA, USA) and 10 μl/ml penicillin-streptomycin (Pen-Strep; catalog number 15140122; Gibco). The human teratocarcinoma cell line Tera-1 (catalog number HTB-105; ATCC) was cultured in McCoy's 5A medium (catalog number 16600082; Gibco) supplemented with 150 μl/ml FBS and 10 μl/ml Pen-Strep. All cell lines were incubated at 37°C with 5% CO2.
RNA extraction and purification.
Approximately 1 to 2 million cells from the Hcc1954, HMLE-Ras, HMLE-Her2, and HME cell lines were used as input for the TRIzol-PureLink RNA minikit system for separate RNA extractions (catalog numbers 15596-026 and 1218301A; Ambion, Carlsbad, CA, USA). The RNA samples were treated with 2 U DNase (Turbo DNA-free kit, catalog number AM1907; Ambion) for 1 h at 37°C. Using a protocol described previously (48), we confirmed that all traces of DNA were removed by quantitative PCR (qPCR) amplification of the TM region of HML-2 env.
RNA-Seq library preparation.
Purified RNA was used to produce an Illumina RNA-Seq library using the TruSeq Stranded Total RNA kit with Ribo-Zero Gold (catalog number RS-122-2301; Illumina, San Diego, CA, USA), depleting all samples of rRNA. Amplified RNA was converted to cDNA, which was not sheared to keep the reads as long as possible. Samples were multiplexed so that 25% of each sequencing lane was occupied by each cell line. Sequencing was done on the MiSeq benchtop sequencer using the MiSeq Reagent kit v3 (catalog number MS-102-3001; Illumina), which produced approximately 24 million paired-end (PE) reads, all 301 bp in length. Samples were demultiplexed before analysis with CASAVA-1.8.2 (Illumina).
RNA-Seq analysis.
MiSeq reads were trimmed with the Trimmomatic program (49) to remove Illumina adapters and to filter out any reads that did not have an average quality score of at least 25, signifying a 0.3% probability of an incorrect base call. We used the Qualimap program (50) to generate a BamQC report to determine the median insert size of our reads. We found the average fragment length to be ∼320 bp for each cell line, suggesting that our PE reads overlapped by an average of 282 bp. We merged any overlapping PE reads using FLASh (Fast Length Adjustment of Short reads) (51). Our final read lengths used for alignment ranged from 60 to 592 bp.
Alignments were accomplished using TopHat v2.0.10, which is built on the short-read mapping program Bowtie v2.1.0 (52, 53) and allows for up to 2 mismatches per aligned read. Reads were aligned to two separate reference genomes: the human reference genome (GRCh37/hg19 build) and an HML-2 reference genome consisting of all known HML-2 elements, including those not annotated in hg19 (27, 54, 55). The HML-2 reference genome consists of 1,073 “chromosomes” that represent 96 full-length proviruses, 976 solo LTRs, and 1 prototype short interspersed nuclear element (SINE)-R (a SINE derived from an HML-2 provirus) (56). Both stranded and unstranded alignments were performed. Data generated from the stranded alignments contained all RNAs transcribed in the sense direction and are designated as such. Data generated from the unstranded alignments contain both antisense- and sense-transcribed RNAs and are designated “total RNA.” After alignment, all reads were filtered using SAMtools (57) for uniquely aligned reads only, by keeping only reads with a mapping quality score of 50. Unfiltered reads, which include sequences present in more than one provirus, are designated “unfiltered,” whereas filtered reads are designated “unique.”
Transcript abundance levels were generated using the Cuffdiff program (58), which normalizes read count against the length of the expressed gene. Values are given in units of FPKM. These FPKM values were further normalized across all four cell lines sequenced so that they could be compared against one another. An FPKM value of 1 corresponded to about 50 raw reads, depending on the cell line, resulting in 1.5× coverage of a 10-kb provirus. In analyses also involving the comparison of Tera-1 FPKM values, a separate Cuffdiff process was run. The RNA-Seq analysis used to generate the Tera-1 data was described in detail in a previous publication (6). Transcript expression levels are provided either as normalized FPKM values or as a percentage of total HML-2 abundance (determined by dividing the FPKM of one provirus by the summed FPKM of all HML-2 proviruses). Graphics were produced using Prism 6 (GraphPad Software, La Jolla, CA, USA), and heat maps were produced using the RStudio Pheatmap package (RStudio, Boston, MA, USA). Jensen-Shannon distance matrices and gene feature plots were produced from Cuffdiff output files using cummeRbund (58). Visualization of the aligned reads to determine the mode of transcription was performed using Integrative Genomics Viewer (Broad Institute, Cambridge, MA, USA) (32). Nearby host genes are as annotated by RefSeq on the UCSC Genome Browser (33, 34).
Dual-luciferase assay.
The sequences of the 3q12.3 and 4p16.1b proviruses were obtained from the UCSC Genome Browser (34), and primers flanking the 5′ LTR of each provirus were selected using the Primer3 program (59). Additional restriction enzyme cleavage site sequences were added to the 5′ ends of each primer to aid in the insertion of the LTR into the reporter construct. Primer sequences used for LTR amplification were 5′-ATTATAGGTACCAAGGAGGCTGAGCAGATGAG-3′ and 5′-ATTATTAAGCTTTTCCAGGGGCATCAGAAACT-3′ for the 3q12.3 5′ LTR and 5′-ATTATAAGATCTCCCTGGATTCCATAAGCAGA-3′ and 5′-ATTATTAAGCTTATAATGGCCCAATCATTCCA-3′ for the 4p16.1b 5′ LTR.
Genomic DNA from Tera-1 cells was purified using the DNeasy blood and tissue kit (catalog number 69504; Qiagen, Germantown, MD, USA) and used as the template for PCR amplification of LTRs using Taq DNA polymerase (catalog number 10342-020; Invitrogen, Carlsbad, CA, USA). The amplified sequences were directly cloned using basic molecular biology techniques into the multiple-cloning region of the pGL4.17[luc2/Neo] promoter-less firefly luciferase vector (catalog number E6721; Promega, Madison, WI, USA) found directly upstream of the luc2 gene. All constructs were fully sequenced to check for PCR-induced mutations before transfection.
Hcc1954, HMLE-Ras, HMLE-Her2, and HME cells were seeded at 100,000 cells/well in a 24-well plate for transfection. Cells were cotransfected with the LTR-containing pGL4 vector together with a pRL-SV40 internal control Renilla luciferase vector (catalog number E2231; Promega) at a 30:1 ratio. Transfections were carried out using the Opti-MEM reduced-serum medium (catalog number 31985-070; Gibco) and Lipofectamine 2000 (catalog number 11668-019; Thermo Fisher Technologies), as recommended by the manufacturers' protocols. Cells were transfected 24 h after seeding and incubated at 37°C for 48 h before lysis. Luminescence was quantified using the dual-luciferase assay system (catalog number E1910; Promega) and measured in relative light units (RLU) on a BioTek Synergy HT plate reader using Gen5 data analysis software (BioTek Instruments, Winooski, VT, USA). Empty vectors as well as nontransfected cells were used as controls to assess any background signal, which was subtracted from the luminescence measurements. LTR promoter activity was determined as luc2 activity normalized against that of the internal Renilla control.
Accession number(s).
All RNA-Seq data reported in this paper have been deposited in the NCBI Gene Expression Omnibus database under accession number GSE84275.
ACKNOWLEDGMENTS
We thank the Tufts University Genomics core facility for their RNA-Seq advice and help with library preparation as well as John Yoon for helpful discussions and editorial advice.
M.M., N.B., C.K., and J.M.C. conceived and designed the experiments. M.M. performed the experiments. M.M., N.B., Z.H.W., and J.M.C. analyzed the data. M.M. and J.M.C. wrote the paper, and all authors revised the final manuscript.
J.M.C. was a Research Professor of the American Cancer Society. This work was supported by Research Grant R35 CA 200421 from the National Cancer Institute.
REFERENCES
- 1.Jern P, Coffin JM. 2008. Effects of retroviruses on host genome function. Annu Rev Genet 42:709–732. doi: 10.1146/annurev.genet.42.110807.091501. [DOI] [PubMed] [Google Scholar]
- 2.Ruda VM, Akopov SB, Trubetskoy DO, Manuylov NL, Vetchinova AS, Zavalova LL, Nikolaev LG, Sverdlov ED. 2004. Tissue specificity of enhancer and promoter activities of a HERV-K(HML-2) LTR. Virus Res 104:11–16. doi: 10.1016/j.virusres.2004.02.036. [DOI] [PubMed] [Google Scholar]
- 3.Bannert N, Kurth R. 2006. The evolutionary dynamics of human endogenous retroviral families. Annu Rev Genomics Hum Genet 7:149–173. doi: 10.1146/annurev.genom.7.080505.115700. [DOI] [PubMed] [Google Scholar]
- 4.Subramanian RP, Wildschutte JH, Russo C, Coffin JM. 2011. Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology 8:90. doi: 10.1186/1742-4690-8-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jha AR, Nixon DF, Rosenberg MG, Martin JN, Deeks SG, Hudson RR, Garrison KE, Pillai SK. 2011. Human endogenous retrovirus K106 (HERV-K106) was infectious after the emergence of anatomically modern humans. PLoS One 6:e20234. doi: 10.1371/journal.pone.0020234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhardwaj N, Montesion M, Roy F, Coffin JM. 2015. Differential expression of HERV-K (HML-2) proviruses in cells and virions of the teratocarcinoma cell line Tera-1. Viruses 7:939–968. doi: 10.3390/v7030939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Contreras-Galindo R, Kaplan MH, Leissner P, Verjat T, Ferlenghi I, Bagnoli F, Giusti F, Dosik MH, Hayes DF, Gitlin SD, Markovitz DM. 2008. Human endogenous retrovirus K (HML-2) elements in the plasma of people with lymphoma and breast cancer. J Virol 82:9329–9336. doi: 10.1128/JVI.00646-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang-Johanning F, Frost AR, Jian B, Epp L, Lu DW, Johanning GL. 2003. Quantitation of HERV-K env gene expression and splicing in human breast cancer. Oncogene 22:1528–1535. doi: 10.1038/sj.onc.1206241. [DOI] [PubMed] [Google Scholar]
- 9.Wang-Johanning F, Li M, Esteva FJ, Hess KR, Yin B, Rycaj K, Plummer JB, Garza JG, Ambs S, Johanning GL. 2014. Human endogenous retrovirus type K antibodies and mRNA as serum biomarkers of early-stage breast cancer. Int J Cancer 134:587–595. doi: 10.1002/ijc.28389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suntsova M, Gogvadze EV, Salozhin S, Gaifullin N, Eroshkin F, Dmitriev SE, Martynova N, Kulikov K, Malakhova G, Tukhbatova G, Bolshakov AP, Ghilarov D, Garazha A, Aliper A, Cantor CR, Solokhin Y, Roumiantsev S, Balaban P, Zhavoronkov A, Buzdin A. 2013. Human-specific endogenous retroviral insert serves as an enhancer for the schizophrenia-linked gene PRODH. Proc Natl Acad Sci U S A 110:19472–19477. doi: 10.1073/pnas.1318172110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van de Lagemaat LN, Medstrand P, Mager DL. 2006. Multiple effects govern endogenous retrovirus survival patterns in human gene introns. Genome Biol 7:R86. doi: 10.1186/gb-2006-7-9-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Douville R, Liu J, Rothstein J, Nath A. 2011. Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann Neurol 69:141–151. doi: 10.1002/ana.22149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang-Johanning F, Frost AR, Johanning GL, Khazaeli MB, LoBuglio AF, Shaw DR, Strong TV. 2001. Expression of human endogenous retrovirus k envelope transcripts in human breast cancer. Clin Cancer Res 7:1553–1560. [PubMed] [Google Scholar]
- 14.Gonzalez-Hernandez MJ, Cavalcoli JD, Sartor MA, Contreras-Galindo R, Meng F, Dai M, Dube D, Saha AK, Gitlin SD, Omenn GS, Kaplan MH, Markovitz DM. 2014. Regulation of the human endogenous retrovirus K (HML-2) transcriptome by the HIV-1 Tat protein. J Virol 88:8924–8935. doi: 10.1128/JVI.00556-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ono M, Yasunaga T, Miyata T, Ushikubo H. 1986. Nucleotide sequence of human endogenous retrovirus genome related to the mouse mammary tumor virus genome. J Virol 60:589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang-Johanning F, Radvanyi L, Rycaj K, Plummer JB, Yan P, Sastry KJ, Piyathilake CJ, Hunt KK, Johanning GL. 2008. Human endogenous retrovirus K triggers an antigen-specific immune response in breast cancer patients. Cancer Res 68:5869–5877. doi: 10.1158/0008-5472.CAN-07-6838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao J, Rycaj K, Geng S, Li M, Plummer JB, Yin B, Liu H, Xu X, Zhang Y, Yan Y, Glynn SA, Dorsey TH, Ambs S, Johanning GL, Gu L, Wang-Johanning F. 2011. Expression of human endogenous retrovirus type K envelope protein is a novel candidate prognostic marker for human breast cancer. Genes Cancer 2:914–922. doi: 10.1177/1947601911431841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang-Johanning F, Rycaj K, Plummer JB, Li M, Yin B, Frerich K, Garza JG, Shen J, Lin K, Yan P, Glynn SA, Dorsey TH, Hunt KK, Ambs S, Johanning GL. 2012. Immunotherapeutic potential of anti-human endogenous retrovirus-K envelope protein antibodies in targeting breast tumors. J Natl Cancer Inst 104:189–210. doi: 10.1093/jnci/djr540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krishnamurthy J, Rabinovich BA, Mi T, Switzer KC, Olivares S, Maiti SN, Plummer JB, Singh H, Kumaresan PR, Huls HM, Wang-Johanning F, Cooper LJ. 2015. Genetic engineering of T cells to target HERV-K, an ancient retrovirus on melanoma. Clin Cancer Res 21:3241–3251. doi: 10.1158/1078-0432.CCR-14-3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, Popescu NC, Hahn WC, Weinberg RA. 2001. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev 15:50–65. doi: 10.1101/gad.828901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dimri G, Band H, Band V. 2005. Mammary epithelial cell transformation: insights from cell culture and mouse models. Breast Cancer Res 7:171–179. doi: 10.1186/bcr1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rangarajan A, Hong SJ, Gifford A, Weinberg RA. 2004. Species- and cell type-specific requirements for cellular transformation. Cancer Cell 6:171–183. doi: 10.1016/j.ccr.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 23.Victorino VJ, Campos FC, Herrera AC, Colado Simao AN, Cecchini AL, Panis C, Cecchini R. 2014. Overexpression of HER-2/neu protein attenuates the oxidative systemic profile in women diagnosed with breast cancer. Tumour Biol 35:3025–3034. doi: 10.1007/s13277-013-1391-x. [DOI] [PubMed] [Google Scholar]
- 24.Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ. 2006. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol 3:269–280. doi: 10.1038/ncponc0509. [DOI] [PubMed] [Google Scholar]
- 25.Downward J. 2003. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 26.Olsson E, Winter C, George A, Chen Y, Torngren T, Bendahl PO, Borg A, Gruvberger-Saal SK, Saal LH. 2015. Mutation screening of 1,237 cancer genes across six model cell lines of basal-like breast cancer. PLoS One 10:e0144528. doi: 10.1371/journal.pone.0144528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wildschutte JH, Williams ZH, Montesion M, Subramanian RP, Kidd JM, Coffin JM. 2016. Discovery of unfixed endogenous retrovirus insertions in diverse human populations. Proc Natl Acad Sci U S A 113:E2326–E2334. doi: 10.1073/pnas.1602336113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruprecht K, Ferreira H, Flockerzi A, Wahl S, Sauter M, Mayer J, Mueller-Lantzsch N. 2008. Human endogenous retrovirus family HERV-K(HML-2) RNA transcripts are selectively packaged into retroviral particles produced by the human germ cell tumor line Tera-1 and originate mainly from a provirus on chromosome 22q11.21. J Virol 82:10008–10016. doi: 10.1128/JVI.01016-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gotzinger N, Sauter M, Roemer K, Mueller-Lantzsch N. 1996. Regulation of human endogenous retrovirus-K Gag expression in teratocarcinoma cell lines and human tumours. J Gen Virol 77(Part 12):2983–2990. doi: 10.1099/0022-1317-77-12-2983. [DOI] [PubMed] [Google Scholar]
- 30.Rycaj K, Plummer JB, Yin B, Li M, Garza J, Radvanyi L, Ramondetta LM, Lin K, Johanning GL, Tang DG, Wang-Johanning F. 2015. Cytotoxicity of human endogenous retrovirus K-specific T cells toward autologous ovarian cancer cells. Clin Cancer Res 21:471–483. doi: 10.1158/1078-0432.CCR-14-0388. [DOI] [PubMed] [Google Scholar]
- 31.Schmitt K, Reichrath J, Roesch A, Meese E, Mayer J. 2013. Transcriptional profiling of human endogenous retrovirus group HERV-K(HML-2) loci in melanoma. Genome Biol Evol 5:307–328. doi: 10.1093/gbe/evt010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thorvaldsdottir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. 2002. The human genome browser at UCSC. Genome Res 12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ. 2004. The UCSC Table Browser data retrieval tool. Nucleic Acids Res 32:D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Strausberg RL, Feingold EA, Grouse LH, Derge JG, Klausner RD, Collins FS, Wagner L, Shenmen CM, Schuler GD, Altschul SF, Zeeberg B, Buetow KH, Schaefer CF, Bhat NK, Hopkins RF, Jordan H, Moore T, Max SI, Wang J, Hsieh F, Diatchenko L, Marusina K, Farmer AA, Rubin GM, Hong L, Stapleton M, Soares MB, Bonaldo MF, Casavant TL, Scheetz TE, Brownstein MJ, Usdin TB, Toshiyuki S, Carninci P, Prange C, Raha SS, Loquellano NA, Peters GJ, Abramson RD, Mullahy SJ, Bosak SA, McEwan PJ, McKernan KJ, Malek JA, Gunaratne PH, Richards S, Worley KC, Hale S, Garcia AM, Gay LJ, Hulyk SW, Villalon DK, Muzny DM, Sodergren EJ, Lu X, Gibbs RA, Fahey J, Helton E, Ketteman M, Madan A, Rodrigues S, Sanchez A, Whiting M, Madan A, Young AC, Shevchenko Y, Bouffard GG, Blakesley RW, Touchman JW, Green ED, Dickson MC, Rodriguez AC, Grimwood J, Schmutz J, Myers RM, Butterfield YS, Krzywinski MI, Skalska U, Smailus DE, Schnerch A, Schein JE, Jones SJ, Marra MA, Mammalian Gene Collection Program Team . 2002. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc Natl Acad Sci U S A 99:16899–16903. doi: 10.1073/pnas.242603899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Allard STM, Kopish K. 2008. Luciferase reporter assays: powerful, adaptable tools for cell biology research. Cell Notes 21:23–26. [Google Scholar]
- 37.Zur Hausen H. 2009. The search for infectious causes of human cancers: where and why. Virology 392:1–10. doi: 10.1016/j.virol.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 38.Lower R, Lower J, Kurth R. 1996. The viruses in all of us: characteristics and biological significance of human endogenous retrovirus sequences. Proc Natl Acad Sci U S A 93:5177–5184. doi: 10.1073/pnas.93.11.5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wildschutte JH, Ram D, Subramanian R, Stevens VL, Coffin JM. 2014. The distribution of insertionally polymorphic endogenous retroviruses in breast cancer patients and cancer-free controls. Retrovirology 11:62. doi: 10.1186/s12977-014-0062-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Golan M, Hizi A, Resau JH, Yaal-Hahoshen N, Reichman H, Keydar I, Tsarfaty I. 2008. Human endogenous retrovirus (HERV-K) reverse transcriptase as a breast cancer prognostic marker. Neoplasia 10:521–533. doi: 10.1593/neo.07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou F, Krishnamurthy J, Wei Y, Li M, Hunt K, Johanning GL, Cooper LJ, Wang-Johanning F. 2015. Chimeric antigen receptor T cells targeting HERV-K inhibit breast cancer and its metastasis through downregulation of Ras. Oncoimmunology 4:e1047582. doi: 10.1080/2162402X.2015.1047582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lower R, Lower J, Tondera-Koch C, Kurth R. 1993. A general method for the identification of transcribed retrovirus sequences (R-U5 PCR) reveals the expression of the human endogenous retrovirus loci HERV-H and HERV-K in teratocarcinoma cells. Virology 192:501–511. doi: 10.1006/viro.1993.1066. [DOI] [PubMed] [Google Scholar]
- 43.Schmitt K, Heyne K, Roemer K, Meese E, Mayer J. 2015. HERV-K(HML-2) rec and np9 transcripts not restricted to disease but present in many normal human tissues. Mob DNA 6:4. doi: 10.1186/s13100-015-0035-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ono M, Kawakami M, Ushikubo H. 1987. Stimulation of expression of the human endogenous retrovirus genome by female steroid hormones in human breast cancer cell line T47D. J Virol 61:2059–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Etkind PR, Lumb K, Du J, Racevskis J. 1997. Type 1 HERV-K genome is spliced into subgenomic transcripts in the human breast tumor cell line T47D. Virology 234:304–308. doi: 10.1006/viro.1997.8670. [DOI] [PubMed] [Google Scholar]
- 46.Domansky AN, Kopantzev EP, Snezhkov EV, Lebedev YB, Leib-Mosch C, Sverdlov ED. 2000. Solitary HERV-K LTRs possess bi-directional promoter activity and contain a negative regulatory element in the U5 region. FEBS Lett 472:191–195. doi: 10.1016/S0014-5793(00)01460-5. [DOI] [PubMed] [Google Scholar]
- 47.Pelechano V, Steinmetz LM. 2013. Gene regulation by antisense transcription. Nat Rev Genet 14:880–893. doi: 10.1038/nrg3594. [DOI] [PubMed] [Google Scholar]
- 48.Bhardwaj N, Maldarelli F, Mellors J, Coffin JM. 2014. HIV-1 infection leads to increased transcription of human endogenous retrovirus HERV-K (HML-2) proviruses in vivo but not to increased virion production. J Virol 88:11108–11120. doi: 10.1128/JVI.01623-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garcia-Alcalde F, Okonechnikov K, Carbonell J, Cruz LM, Gotz S, Tarazona S, Dopazo J, Meyer TF, Conesa A. 2012. Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics 28:2678–2679. doi: 10.1093/bioinformatics/bts503. [DOI] [PubMed] [Google Scholar]
- 51.Magoc T, Salzberg SL. 2011. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Turner G, Barbulescu M, Su M, Jensen-Seaman MI, Kidd KK, Lenz J. 2001. Insertional polymorphisms of full-length endogenous retroviruses in humans. Curr Biol 11:1531–1535. doi: 10.1016/S0960-9822(01)00455-9. [DOI] [PubMed] [Google Scholar]
- 55.Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ, Slobodeniuc V, Kutter C, Watt S, Colak R, Kim T, Misquitta-Ali CM, Wilson MD, Kim PM, Odom DT, Frey BJ, Blencowe BJ. 2012. The evolutionary landscape of alternative splicing in vertebrate species. Science 338:1587–1593. doi: 10.1126/science.1230612. [DOI] [PubMed] [Google Scholar]
- 56.Ono M, Kawakami M, Takezawa T. 1987. A novel human nonviral retroposon derived from an endogenous retrovirus. Nucleic Acids Res 15:8725–8737. doi: 10.1093/nar/15.21.8725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing Subgroup . 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JA. 2007. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 35:W71–W74. doi: 10.1093/nar/gkm306. [DOI] [PMC free article] [PubMed] [Google Scholar]