

ABSTRACT
Chronic hepatitis B affects over 300 million people who are at risk of developing liver cancer. The basis for the persistence of hepatitis B virus (HBV) in hepatocytes, even in the presence of available antiviral therapies, lies in the accumulation of covalently closed circular DNA (cccDNA) in nuclei of infected cells. While methods for cccDNA quantification from liver biopsy specimens and cell lines expressing the virus are known, information about cccDNA formation, stability, and turnover is lacking. In particular, little is known about the fate of cccDNA during cell division. To fill the gaps in knowledge concerning cccDNA biology, we have developed a fluorescence imaging in situ hybridization (FISH)-based assay for the detection of duck hepatitis B virus (DHBV) cccDNA and HBV nuclear DNA in established cell lines. Using FISH, we determined the distribution of cccDNA under conditions mimicking chronic infections with and without antiviral therapy, which prevents de novo viral replication. Our results showed that the copy numbers of viral nuclear DNA can vary by as much as 1.8 orders of magnitude among individual cells and that antiviral therapy leads to a reduction in nuclear DNA in a manner consistent with symmetrical distribution of viral DNA to daughter cells.
IMPORTANCE A mechanistic understanding of the stability of HBV cccDNA in the presence of antiviral therapy and during cell division induced by immune-mediated lysis of infected hepatocytes will be critical for the future design of curative antiviral therapies against chronic hepatitis B. Current knowledge about cccDNA stability was largely derived from quantitative analyses of cccDNA levels present in liver samples, and little was known about the fate of cccDNA in individual cells. The development of a FISH-based assay for cccDNA tracking provided the first insights into the fate of DHBV cccDNA and nuclear HBV DNA under conditions mimicking antiviral therapy.
KEYWORDS: hepatitis B virus, covalently closed circular DNA, antiviral, viral persistence, antivirals
INTRODUCTION
An estimated 300 million people are chronically infected with hepatitis B virus (HBV) (1). On the basis of the Global Burden of Disease Study, over 700,000 patients die each year of HBV-induced diseases; about half of those patients succumb to hepatocellular carcinoma (2). The current standard of care relies on therapies with nucleoside analogs (NA) which inhibit viral reverse transcriptase (RT) and hence prevent de novo virus production (3). NAs can control but cannot cure HBV infections. They inhibit de novo HBV DNA synthesis but do not reduce levels of preexisting covalently closed circular DNA (cccDNA), the template for transcription of viral RNAs. As a consequence, cccDNA persists in the nuclei of infected hepatocytes (3). So far, a therapeutic strategy to functionally inactivate or even destroy cccDNA within nondividing hepatocytes has been lacking. Because the stability of cccDNA is the reason for the chronic aspect of HBV infections, and due to its resilience in the face of current antiviral therapies, cccDNA has become a major focus of current HBV research.
The HBV genome is a ca. 3.2-kb-long relaxed circular DNA (rcDNA) genome that replicates by reverse transcription of an RNA intermediate, the pregenome (reviewed in reference 3). Following infection and uncoating, capsids migrate to nuclear pores, where they disassemble and deliver their cargo, rcDNA, into the nucleus. RcDNA is repaired by as-yet-unidentified cellular enzymes. This process yields cccDNA, the template for the transcription of the six viral mRNAs. The pregenome, a greater-than-genome-length mRNA, is packaged in the cytoplasm into core particles and reverse transcribed into rcDNA. Early in infection, cores are transported to the nucleus, leading to intracellular cccDNA amplification, until a certain copy number is reached and maintained. Later in infection, cores bind to envelope proteins, enter the secretory pathway, and exit the cells. CccDNA does not undergo semiconservative replication and hence is maintained entirely by the cytoplasmic reverse transcription pathway.
Major gaps in knowledge exist concerning cccDNA formation, stability, and turnover, including the fate of cccDNA during cell division. What we do know comes from quantification of cccDNA present in liver tissue of woodchucks and chimpanzees during recovery from acute infections (4–7). Those studies suggested that cccDNA is diluted and perhaps lost during cell division. However, almost nothing is known about the actual copy number of cccDNA in infected hepatocytes and how cccDNA is distributed to daughter cells following cell division. So far, only one report has provided data about cccDNA copy number in hepatocytes of ducks chronically infected with the HBV-related duck hepatitis B virus (DHBV) (8). It relied on PCR-based quantification of nuclear DHBV DNA and revealed that the copy number of cccDNA molecules varied among hepatocytes in a range of approximately 7 to 20 copies per nucleus. More-detailed information about the fate of cccDNA during cell division and, in particular, under conditions where de novo DNA replication is inhibited will be crucial for a better understanding of the impact of NA-based antiviral therapies on viral persistence. With this knowledge in hand, novel strategies with improved therapeutic outcomes can be developed. Thus, as a first step toward this goal, we sought to identify cccDNA using fluorescence in situ hybridization (FISH) in cell lines expressing either HBV or DHBV. Our results demonstrated that this approach has the potential to reveal the distribution of cccDNA in nondividing cells as well as in cells undergoing cell division in the presence of the antiviral drugs used in the treatment of chronic hepatitis B.
RESULTS
Detection of nuclear DNA in DHBV- and HBV-producing cell lines.
Dstet5 cells were derived from the chicken LMH cell line and express DHBV conditionally from a tetracycline promoter (9). Initial efforts to detect DHBV cccDNA by FISH analysis of metaphase spreads prepared from Dstet5 cells failed, suggesting that cccDNA might have been lost when chromosomes were spread onto glass slides. This observation was consistent with the widely held but unproven view that cccDNA is not tethered to chromosomes. Therefore, we modified the FISH protocol using cells that were collected after trypsin treatment instead of the “mitotic shake-off” method normally used for FISH. Under those conditions, the majority of nuclei remained intact after spread on glass slides (see Materials and Methods and Fig. 1A). With this method, nuclear signals were detected in Dstet5 cells and also in HepAD38 cells expressing HBV in a conditional fashion (Fig. 1) (10). Under conditions in which viral replication was prevented by addition of doxycycline to the culture medium of Dstet5 and HepAD38 cells, only a few FISH signals could be detected. The small number of signals could have been due to nonspecific binding of the DNA probe to chromosomal DNA or could have been derived from binding of the probe to integrated DHBV and HBV DNA used to establish the cell lines. It should be noted that most of the nuclear DNA in HepAD38 cells accumulates as so-called protein-free (PF) relaxed circular DNA and not as cccDNA as in Dstet5 cells (see Fig. 5C) (11–14). PF DNA accumulates in HepAD38 cells for reasons that are not yet understood. To ensure that the FISH signal was derived from DNA in nuclei of HepAD38 cells, we combined FISH with immunofluorescence (IF) using an antibody specific for lamin A/C. The results showed that the nuclear envelope maintained structural features that preserved lamin and that HBV DNA colocalized with the nucleus (Fig. 1). Importantly, the results showed that the nuclei were separated from the cytoplasm, indicating that FISH detected nuclear DNA and not cytoplasmic HBV DNA present in core particles (Fig. 1A). This view was confirmed with confocal microscopy demonstrating that FISH signals were present within nuclei of both Dstet5 and HepAD38 cells (Fig. 1). Finally, analysis of metaphase spreads from HepAD38 cells treated with colcimide revealed the presence of integrated HBV DNA fused to the cytomegalovirus-tetracycline (CMV-tet) promoter on chromosome 2, further demonstrating the specificity of the probe used to detect HBV DNA (Fig. 1B).
FIG 1.
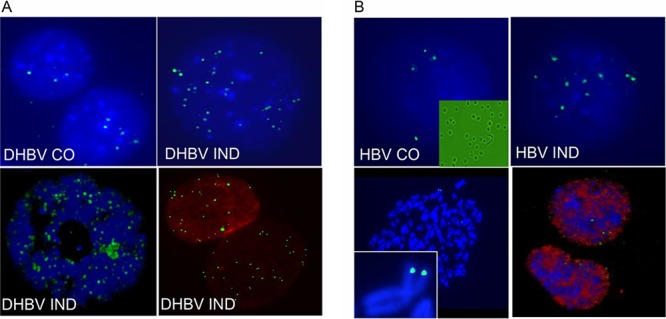
Detection of nuclear DNA by FISH. Representative examples of FISH signals were obtained with nuclear preparations from uninduced (CO) and induced (IND) DHBV and HBV replicating in Dstet5 (A) and HepAD38 (B) cells, respectively. (A) Viral replication in Dstet5 cells was induced by removing doxycycline from the medium for 6 to 7 days. Images were taken with a Zeiss fluorescence microscope through a 100× objective except for the bottom left image, which was taken with a Leica confocal microscope using a 60× objective. The bottom right image was derived from an experiment where FISH and IF were combined and where an antibody against lamin A/C was used as described in Materials and Methods. (B) Viral replication in HepAD38 cells was induced for 3 to 4 weeks. Images were taken as described for panel A. The top left image contains an inset derived from a bright-field image of nuclear preparations from HepAD38 cells. The bottom left image is derived from FISH analyses performed on metaphase spreads and includes an inset showing the signals on chromosome 2 derived from integrated HBV DNA used to establish the HepAD38 cell line.
FIG 5.

Dilution of HBV nuclear DNA. The figure shows the frequency distributions of FISH counts obtained from 200 nuclei derived from HepAD38 cells. The design of the experiment is shown in Fig. 4A. FISH analysis was performed at days 1, 4, 8, 12, 16, and 20 following addition of doxycycline (1 μg/ml) and TDF (20 μM) to the medium. Cells were passaged at a dilution of 1:5. (B) Cumulative distribution of the FISH counts shown in panel A (Experiment). The panel also shows simulations (Sim.) of symmetrical and asymmetrical distributions of cccDNA to daughter cells determined as described in the legend to Fig. 4C. (C) Southern blot analysis of HBV Hirt DNA from HepAD38 cells maintained under the conditions described for panel A. rc, relaxed circular DNA (representing PF [protein-free] DNA); dsl, double-stranded linear DNA; ccc, covalently closed circular DNA.
Distribution of cccDNA.
Next, we determined the distribution of DHBV cccDNA and HBV nuclear DNA by counting FISH signals present in 200 nuclei of Dstet5 and HepAD38 cells, respectively. Four independent experiments were conducted with each cell line. Uninduced control Dstet5 cells (containing DHBV) exhibited, on average, 3 to 6 FISH signals per nucleus, with medians ranging from 2 to 4 (Table 1 and Fig. 2A). Notably, we detected a few nuclei with relatively large numbers of FISH signals in each experiment, indicating that some Dstet5 cells replicated DHBV even in the presence of doxycycline (Fig. 2A). Interestingly, this and previous analyses of Dstet5 cells relying on IF with an antibody against the DHBV core antigen (cAg) also revealed a small fraction of cells expressing cAg, indicating some leakiness of the tetracycline promoter (J. A. Sohn and C. Seeger, unpublished results). We obtained similar results with HepAD38 cells maintained in the presence of doxycycline. Average counts among the four experiments ranged from 2 to 5 and medians from 1 to 4 (Fig. 2B and Table 2).
TABLE 1.
Distribution of DHBV cccDNAa
| Expt | FISH count |
|||||
|---|---|---|---|---|---|---|
| Control |
Induced |
|||||
| Avg | Median | min./max. | Avg | Median | min./max. | |
| 1 | 4 | 3 | 0/15 | 20 | 19 | 2/60 |
| 2 | 3 | 2 | 0/19 | 15 | 14 | 1/48 |
| 3 | 5 | 4 | 0/24 | 20 | 19 | 4/43 |
| 4 | 6 | 4 | 0/28 | 15 | 15 | 0/44 |
The table shows a summary of FISH counts from four independent experiments performed with Dstet5 cells as described in the Fig. 2A legend. Avg and Median, average and median of counts from 200 nuclei derived from uninduced (Control) and induced Dstet5 cells. min./max., minimum/maximum (range of FISH counts).
FIG 2.

Nuclear DNA distribution of DHBV cccDNA and HBV nuclear DNA. The figure shows the frequency distributions (Freq.dist) of FISH counts from two independent experiments (experiment 1 [E.1] and E.2) performed with Dstet5 (A) (DHBV) and HepAD38 (B) (HBV) cells. Dstet5 and HepAD38 cells were maintained in the presence of doxycycline (Control) and without the antibiotic for 7 and 25 to 30 days (Induced), respectively. FISH counts were derived from a total of 200 nuclei. See also Tables 1 and 2 for a summary of results from a total of 4 experiments performed with Dstet5 and HepAD38 cells, respectively. ^, median; *, average (mean).
TABLE 2.
Distribution of HBV nuclear DNAa
| Expt | FISH count |
|||||
|---|---|---|---|---|---|---|
| Control |
Induced |
|||||
| Avg | Median | min./max. | Avg | Median | min./max. | |
| 1 | 2 | 2 | 0/15 | 8 | 8 | 0/22 |
| 2 | 2 | 1 | 0/8 | 7 | 5 | 0/35 |
| 3 | 4 | 4 | 0/19 | 11 | 10 | 0/45 |
| 4 | 5 | 4 | 0/16 | 8 | 7 | 0/21 |
The table shows a summary of FISH counts from four independent experiments performed with HepAD38 cells as described in the Fig. 2B legend. Avg and Median, average and median of counts from 200 nuclei derived from uninduced (Control) and induced HepAD38 cells. min./max., minimum/maximum (range of FISH counts).
The variation observed with uninduced (control) cells could have several explanations. First, FISH might detect integrated DNA in some but not all nuclei. This would most likely be a reflection of the variability in the physical structure of nuclei that could have occurred during the preparation of the slides. This view is underscored by the observed differences in the fractions of nuclei that exhibited 0 to 2 signals, ranging from 31% to 56% in Dstet5 cells (DHBV) and 28% to 77% in HepAD38 cells (HBV). Second, as alluded to above, accumulation of a greater number of signals in nuclear DNA can occur because the doxycycline promoter exhibits some leakiness, leading to the transcription and subsequent amplification of viral DNA. Finally, some FISH signals might represent nonspecific hybridization of the DNA probe to chromosomal DNA.
Dstet5 cells were induced in medium lacking doxycycline for 7 to 10 days, the time required for maximal accumulation of DHBV cccDNA (9). Averages and medians of the FISH signal values increased approximately 5-fold compared to the control values, ranging from 15 to 20 and from 14 to 19, respectively (Fig. 2A and Table 1). HepAD38 cells were induced much (up to 3 to 4 weeks) longer than Dstet5 cells to achieve maximal accumulation of HBV nuclear PF DNA. Averages and medians of the FISH signal values increased approximately 4-fold compared to the control values, ranging from 7 to 11 and from 5 to 10, respectively (Fig. 2B and Table 2). Overall, the levels of FISH signals in HepAD38 cells were 2 to 3 times lower than those in Dstet5 cells. However, the averages and medians of the FISH signals obtained with the two cell lines were very similar, suggesting that the distribution of nuclear DNA in both cases was similar to a normal distribution.
To validate the results obtained with Dstet5 and HepAD38 cells that were maintained over several weeks without doxycycline and to determine the kinetics of nuclear DNA accumulation, we performed a FISH analysis with HepAD38 cells at different time points following induction of HBV replication by removal of doxycycline from the medium (Fig. 3A and Table 3). The results showed that nuclear DNA slowly accumulated, with an increase in the average value from 3 to 6 by day 5 postinduction. Between days 5 and 10, the accumulation of nuclear DNA was more significant and reached nearly maximal levels, as averages and medians did not increase substantially between day 10 and day 15.
FIG 3.

Accumulation of nuclear HBV DNA. (A) The figure shows the frequency distributions of FISH counts derived from nuclear HBV DNA following removal of doxycycline from the medium of HepAD38 cells. FISH counts were derived from 200 nuclei isolated 1, 5, 10, and 15 days following induction of HBV replication. (B) Southern blot analysis of HBV DNA extracted by the Hirt procedure at the time points following HBV induction used for FISH as described for panel A. The positions of relaxed circular (rc) DNA, double-strand linear (dsl) DNA, and cccDNA are indicated. Note that rcDNA is also referred to as protein-free (PF) DNA in the text. Table 3 provides a summary of the FISH data shown in panel A. ^, median; *, average (mean).
TABLE 3.
Time course of HBV nuclear DNA accumulationa
| Day | FISH count |
% | ||||
|---|---|---|---|---|---|---|
| Avg | Median | min./max. | Total | Total − BG | ||
| 1 | 3 | 3 | 0/16 | 629 | 0 | |
| 5 | 6 | 5 | 0/17 | 1,142 | 513 | 22 |
| 10 | 14 | 13 | 0/39 | 2,721 | 2,092 | 89 |
| 15 | 15 | 15 | 3/27 | 2,985 | 2,356 | 100 |
The table shows a summary of FISH counts from the experiment described in the Fig. 3 legend. Avg, average of counts from 200 nuclei derived from HepAD38 cells harvested at the indicated days; Median, median of FISH counts; min./max., minimum/maximum (range of FISH counts); Total, total FISH count for all 200 nuclei; Total − BG (total minus background), FISH counts from day 1 were subtracted from the counts determined on days 5, 10, and 15; %, the values corresponding to the FISH counts minus the BG values from day 15 were set to 100% and the percentage values for days 5 and 10 calculated accordingly.
Southern blot analysis showed that HBV DNA could be detected at day 10 and 15, but not at day 5, although an increase in FISH counts could already be observed at that time (Fig. 3B). An explanation for these apparently inconsistent observations is that the FISH signals observed at day 1 represent background levels or were derived from signals of integrated HBV DNA (see Fig. 1B). In fact, an analysis of the sum of FISH counts derived from the 200 nuclei used for the experiment revealed a greater than 4-fold difference between the FISH counts at days 5 and 10 (Table 3). As a consequence, the overall HBV DNA levels determined by the Hirt procedure in DNA samples derived from day 5 were too low for detection by the Southern blot analysis.
As with the HepAD38 cells maintained in the induced state for 25 to 30 days described above (Fig. 2), average and median values were approximating a normal distribution of nuclear DNA. The maximal range of the FISH signals was observed on day 10, where it stretched from 0 to 39. The cause for such a large variation is not yet understood (see Discussion).
Fate of nuclear DNA during cell division and antiviral therapy.
The fate of cccDNA during cell division is not yet known. It is possible that cccDNA is lost or distributed asymmetrically or symmetrically to daughter cells or both. To investigate this problem, we induced viral replication in Dstet5 and HepAD38 cells as described above. Doxycycline and the reverse transcriptase inhibitor tenofovir disoproxil fumarate (TDF) were then added to the culture medium to prevent de novo viral RNA synthesis and DNA replication, respectively. The cells were diluted 1:5 at 4-day intervals and harvested for FISH and Southern blot analyses (Fig. 4A). The results from the experiment performed with Dstet5 cells showed that FISH counts declined slowly between days 1 and 4 and then more rapidly between days 4, 8, and 12, when levels dropped to those observed within control cells (Fig. 4C and Table 4). The modest decline seen between days 1 and 4 could be explained by residual de novo cccDNA formation from viral particles containing mature rcDNA in the cytoplasm, because conversion of rcDNA to cccDNA is not inhibited by TDF. Alternatively, it is conceivable that intracellular TDF concentrations reached therapeutic levels only 3 to 4 days after addition to the culture medium. The observed decline of the FISH counts correlated well with the decline of cccDNA determined by Southern blotting (Fig. 4B). Averages and medians of the FISH counts obtained at the different time points were again very similar, indicating that the nature of the distribution of FISH counts did not change during the observation period (see Discussion). Finally, as with the uninduced (control) Dstet5 cells that were maintained in the presence of doxycycline, IF analysis revealed the presence of a few DHBV cAg-positive cells even after 12 to 17 days of incubation with TDF (M. Li and C. Seeger, unpublished observation). Whether accumulation of DHBV cccDNA in these cells was a result of cccDNA stability or was caused by incomplete inhibition of replication by TDF is not known. A comparison of the cumulative distributions of FISH counts obtained from the experiment with Dstet5 cells and simulated distributions representing symmetrical and asymmetrical distributions of cccDNA to daughter cells indicated that the distribution of FISH signals followed a pattern similar to that seen with the symmetrical distributions (see Fig. 4D and Discussion).
FIG 4.

Dilution of DHBV cccDNA. (A) The figure depicts the design of the experiment. Dstet5 cells were maintained in medium without doxycycline, permitting replication of DHBV. Doxycycline (1 μg/ml) and TFD (10 μM) were added to inhibit replication of DHBV. FISH analysis was performed at days 1, 4, 8, 12, and 17 following addition of the drugs. Cells were passaged at a dilution of 1:5. NUC, nucleoside analog. (B) Southern blot analysis of DHBV DNA in Dstet5 cells. DNA was extracted from confluent 60-mm-diameter plates by the Hirt procedure. Equal fractions of purified DNA derived from each plate were loaded onto each lane. cccDNA; covalently closed circular DNA. (C) The figure shows the frequency distributions of the FISH counts obtained from 200 nuclei from cells harvested at the indicated time points. Cells maintained in doxycycline served as a negative control (Control). (D) Cumulative distributions (Cum. Dist.) of the FISH counts shown in panel C (Experiment). Arrows point to the graphs for the indicated days. The panel also shows simulations (Sim.) of symmetrical (P = 0.5) and asymmetrical (P = 0.9) distributions of cccDNA to daughter cells. Input for the simulations was derived from experimental data for day 4. The lines marked 1D and 2D depict the calculated distributions of cccDNA after each cell division. The lines for day 8 (derived from the experiment) are indicated with dashed arrows. See also Table 4 for a summary of the data shown in panel C.
TABLE 4.
Dilution of DHBV cccDNAa
| Parameter | FISH count |
% reduction | |||
|---|---|---|---|---|---|
| Total | min./max. | Avg | Median | ||
| Day 1 | 3,502 | 0/54 | 18 | 15 | 0 (2,819)* |
| Day 4 | 2,955 | 0/58 | 15 | 12 | 19 |
| Day 8 | 1,452 | 0/38 | 7 | 6 | 73 |
| Day 12 | 734 | 0/32 | 4 | 2 | 98 |
| Day 17 | 780 | 0/31 | 4 | 3 | 97 |
| Day 25 | 844 | 0/29 | 4 | 3 | 94 |
| Control 1 | 801 | 0/15 | 4 | 3 | 100 (683)** |
| Control 2 | 564 | 0/9 | 3 | 2 | |
The table shows a summary of FISH counts from the experiment described in the Fig. 4 legend. Total, total FISH count from 200 nuclei derived from Dstet5 cells harvested at the indicated days; min./max., minimum/maximum (range of FISH counts); Avg, average of FISH counts; Median, median of FISH counts; Control, FISH counts from cells maintained in doxycycline; % reduction, percentage of reduction of FISH counts; *, the value in parentheses corresponds to the total count minus the control (background) value; **, the value in parentheses corresponds to the background value = the average of the control 1 and control 2 values.
Results obtained with Dstet5 cells were validated with an experiment using HepAD38 cells. While inhibition of HBV replication with doxycycline and TDF led to a reduction in FISH counts as observed with DHBV in Dstet5 cells, the rate of HBV PF DNA decline was lower than that observed with DHBV cccDNA (Fig. 5 and Table 5). For example, a comparison of the values obtained with DHBV at day 8 revealed a 73% reduction from day 1 values (Tables 4 and 5). In contrast, with HBV, the reduction was only 50%. The DHBV counts had declined by 97% by day 12, in contrast to the HBV counts, which had declined by 66%. As with DHBV, HBV DNA levels declined only minimally between days 1 and 4, and the averages and medians of the FISH counts remained at very similar levels, suggesting that the nature of the distribution of HBV PF DNA did not change during the observation period. As with the Dstet5 cells, the cumulative distributions of the FISH counts obtained with HepAD38 cells were similar to data from the simulation representing symmetrical distribution of nuclear DNA to daughter cells (Fig. 5C and Discussion).
TABLE 5.
Dilution of HBV nuclear DNAa
| Day | FISH count |
% reduction | |||
|---|---|---|---|---|---|
| Total | min./max. | Avg | Median | ||
| 1 | 3,278 | 0/39 | 16 | 16 | 0 (2,801)* |
| 4 | 2,965 | 0/35 | 15 | 16 | 11 |
| 8 | 1,871 | 0/37 | 9 | 9 | 50 |
| 12 | 1,427 | 0/30 | 7 | 6 | 66 |
| 16 | 1,053 | 0/28 | 5 | 4 | 79 |
| 20 | 477 | 0/16 | 2 | 2 | 100 (477)** |
The table shows a summary of FISH counts from the experiment described in the Fig. 5 legend. Total, total FISH count in 200 nuclei derived from HepAD38 cells harvested at the indicated days; Avg, average of FISH counts; Median, median of FISH counts; min./max., minimum/maximum (range of FISH counts); % reduction; percentage of reduction of FISH counts; *, the value in parentheses corresponds to the total count − the control (background) value; **, the value in parentheses corresponds to the background value = day 20 counts.
DISCUSSION
To date, quantitative analyses of HBV DNA have been performed with DNA extracted from infected tissue samples or from tissue culture cells that provided about the average of DNA copy numbers in a population of cells. Thus far, only one study reported DNA levels in individual DHBV-infected duck hepatocytes (8) (see below). Our study demonstrated that nuclear DHBV and HBV DNA can be detected in virus-producing cells using FISH. With DHBV, FISH primarily detected cccDNA. This conclusion is based on the fact that Dstet5 cells produce large amounts of cccDNA, because the DHBV genome used to establish these cells cannot express envelope proteins (9). As a consequence, core particles cannot enter the secretory pathway and instead deliver their rcDNA cargo to the nucleus, leading to (over)amplification of cccDNA (Fig. 4B) (15). In contrast, with HBV, FISH primarily detected nuclear PF DNA because PF DNA is the most abundant nuclear HBV DNA found in HepAD38 cells (Fig. 5C) (11–13).
A potential limitation of our study is that some of the FISH signals may have been derived from contaminating rcDNA in core particles present in the cytoplasm of Dstet5 and HepAD38 cells. However, for the following reasons, we concluded that the FISH signals were primarily derived from nuclear DHBV cccDNA and HBV PF DNA, respectively. First, the method used for the processing of Dstet5 and HepAD38 cells primarily yielded nuclei separated from the cytoplasm (Fig. 1B). Second, we demonstrated that HBV DNA is enclosed within the nucleus, as demarcated by the nuclear lamina network (Fig. 1). Third, FISH analysis performed with HBV-Met cells derived from hepatocytes of HBV transgenic mice (16), expressing exclusively cytoplasmic HBV DNA, did not yield any nuclear signals (Li and Seeger, unpublished). Finally, FISH signals could be detected inside nuclei from Dstet5 and HepAD38 cells using confocal microscopy imaging (Fig. 1).
Our results obtained with DHBV and HBV provided insights into the distribution of nuclear hepadnavirus DNA in cells replicating DHBV or HBV. The distribution in Dstet5 and HepAD38 cells exhibited a relatively large range, spanning from 0 to a maximum of 60 copies in Dstet5 cells (experiment 1 [E.1]) (Fig. 2 and Table 1) and 0 to 45 copies in HepAD38 cells (experiment 3) (Table 2). Differences in the accumulation of nuclear DNA among cells became apparent within a few days after induction of viral replication and were maintained within the observation period of up to several months with HepAD38 cells (Fig. 3 and Table 3). The reason for such variation is not known. It might be independent of viral protein expression and occur subsequently to DNA replication in core particles, because IF analysis of induced Dstet5 cells indicated that almost every cell expressed DHBV cAg (Sohn and Seeger, unpublished). CccDNA amplification depends on host factors controlling retrograde transport of core particles from the cytoplasm to the nucleus and perhaps also on the kinases and phosphatases responsible for the modification of the C-terminal region of the viral core proteins during reverse transcription (see, e.g., reference 17). Hence, it is conceivable that differences exist in the activity of one or several of these factors among individual cells that result in the differential accumulation of nuclear DNA observed in our study and previously by Zhang et al. (8). Alternatively, it is at least theoretically possible that some cells exhibit a very high rate of turnover of cccDNA, thus preventing its accumulation. However, there is so far no known activity in cells that could trigger the selective purging of viral DNA in an effective manner.
A caveat concerning our study is that it was performed with tissue culture cells. However, Zhang et al. (8) observed similar variations in cccDNA copy numbers in nuclei derived from livers of ducks chronically infected with DHBV. The numbers ranged from 1 to 17 copies in 90% of nuclei and were higher in the remaining 10%. In vivo studies performed with DHBV-infected ducks and woodchuck hepatitis virus (WHV)-infected woodchucks provided estimates of ca. 20 cccDNA copies per infected hepatocyte (5, 18), Hence, the broad distribution of cccDNA copy numbers and the average copy numbers observed in our study correlate well with observations made in vivo (Tables 1 to 5).
Our report provides the first insights into the fate of DHBV cccDNA and nuclear HBV DNA under conditions mimicking antiviral therapy. Information about the stability of cccDNA in the presence of reverse transcriptase inhibitors, such as TDF, and during cell division induced by the killing of some infected hepatocytes by the adaptive immune response is paramount for the future design of curative antiviral therapies. Formally, we can envision at least three different scenarios resulting in cccDNA loss. The first predicts that cccDNA cannot survive mitosis because it is not tethered to chromosomes. The second involves asymmetric loss of cccDNA, a consequence of sequestration of cccDNA in infected hepatocytes favoring distribution to one of the two daughter cells. The third is based on symmetric distribution, a consequence of uniform distribution of cccDNA in the nuclei. The latter two possibilities could be associated with partial loss of cccDNA during cell division. On the basis of our analysis, we can reject the first possibility because complete loss of nuclear DNA after cell division should have resulted in a much more rapid decline of DHBV cccDNA and HBV PF DNA levels than was observed in our experiments. They showed that nuclear DNA loss occurred over a period of approximately 12 days, during which the cells were diluted (1:5) 3 times (Fig. 4 and 5). Between the first and second dilution of the cells (days 1 to 4), the loss of cccDNA was insignificant. It can be explained by de novo synthesis of cccDNA from rcDNA released from mature capsids present in the cytoplasm of Dstet5 cells, because cccDNA formation is not inhibited by TDF. Consistent with such an interpretation, we previously observed that cccDNA levels slightly increased following the first 3 to 4 days of incubation of Dstet5 cells with lamivudine (3TC), before they began to decline (9). Notably, this cccDNA increase correlated with a rapid decline of rcDNA levels in the presence of the RT inhibitor between days 1 and 8 (9). In our current study, the most significant loss of cccDNA levels occurred between days 4 and 8, resulting in reductions of 73% and 66% compared with day 1 and day 4, respectively. On the basis of the 1:5 dilution of the cells performed during each passage, a 75% reduction would be expected if cccDNA had been lost during each of the approximately two cell divisions that occurred between days 4 and 8. Comparison of the cumulative distributions of FISH counts obtained from Dstet5 cells at different time points following inhibition of viral DNA synthesis with computer-generated graphs simulating distribution probabilities for cccDNA to daughter cells of 0.5 and 0.9 favored a model akin to symmetrical distribution of DHBV cccDNA (Fig. 4D). Skewness in the cccDNA distribution to daughter cells intrinsic to asymmetrical distributions would have resulted in a significantly larger fraction of nuclei lacking cccDNA than was observed in our experiments. Interestingly, the cumulative distribution curve for day 8 is almost identical to the symmetrical (P = 0.5) distribution representing one cell division (1D in Fig. 4D) and not, as expected from the 1:5 dilution of the cells, with the graph for two cell doublings (2D). The reason for this discrepancy is not certain, but it could be due to the above-mentioned replenishing of the cccDNA pool from cytoplasmic rcDNA.
The results obtained with HepAD38 cells were more difficult to interpret. While PF DNA levels declined by about 50% between the second and third dilutions, the rate of loss that occurred between the third and fourth dilutions was much lower. As a consequence, the cumulative distributions of the observed FISH signals did not approximate the simulated graphs representing symmetrical distribution of PF DNA and also did not approximate those obtained with DHBV (Fig. 5B and Table 5). Information explaining the discrepancy between the results obtained with Dstet and HepAD38 cells is not yet available. An appealing possibility is that the growth rate of HepAD38 cells might be influenced by the load of HBV DNA, which, as shown in this study and discussed further below, varies significantly among cells.
A caveat concerning the interpretation of our results is appropriate with regard to the assumption that all tissue culture cells divide at the same rate. However, it is conceivable that some cells divide at a lower rate than others and hence lose cccDNA at a lower rate than the other dividing cells. Such a scenario, together with the aforementioned de novo cccDNA synthesis in the presence TDF, might explain the lower-than-expected decline of FISH counts observed in our experiments. Independently of this reservation, it is apparent from our study that cccDNA loss, consistent with the fact that current NA-based antiviral therapies are not curative, is a relatively slow process. Inferring from our results determined with tissue culture cells and those reported by Zhang et al. (8) with respect to duck hepatocytes, it is clear that some hepatocytes carry many copies of cccDNA. It is likely that these cells are responsible for the persistence of HBV in the face of even prolonged antiviral therapy exposure. It might be possible to accelerate the loss of functional cccDNA in hepatocytes with a clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9-based approach. The effectiveness of this strategy in targeting cccDNA has already been documented (19, 20).
Takin the data together, our study provided novel insights into the distribution of DHBV cccDNA and nuclear HBV DNA under conditions that mimic de novo infection and antiviral therapy. Since the method described in this report allows a combination of FISH and IF, it can be used for future investigations aimed at identifying the nuclear locales harboring cccDNA.
MATERIALS AND METHODS
Tissue culture.
HepAD38 and Dstet5 cells were maintained in Dulbecco's modified Eagle medium (DMEM)/F12 medium supplemented with 10% fetal bovine serum (FBS), sodium pyruvate, nonessential amino acids, l-glutamine, penicillin/streptomycin, 250 μg/ml of G418, and 1 μg/ml of doxycycline in 5% carbon dioxide at 37°C. The cells were maintained on tissue culture dishes coated with Matrigel (Corning, Tewksbury, MA; catalog no. 356234).
Preparation of nuclei for FISH.
For the preparation of nuclei for FISH, we followed the method for metaphase spreads described by Stehle et al. (21) with some modifications. HepAD38 and Dstet5 cells were grown in 60-mm-diameter dishes until they were 70% to 80% confluent. The cells were then subjected to trypsinization, resuspended in 5 ml of 0.56% KCl, and incubated at room temperature for 8 to 10 min. The cells were centrifuged for 5 min at 1,000 rpm, the supernatant was decanted, and 5 ml of ice-cold methanol-glacial acetic acid (3:1) was added. The pellets were briefly resuspended by shaking the tubes and were centrifuged as described above. The fixation procedure was repeated twice. After the final centrifugation, the cell pellet was resuspended in 1 to 1.5 ml ice-cold methanol-glacial acetic acid (3:1) with a P1000 Pipetman pipette. A Pasteur pipette was used to deliver 2 to 3 drops of the suspension onto glass slides (Colormark, Portsmouth, NH; catalog no. CM-4951) positioned over a beaker containing water at a temperature of 55°C to 65°C, resulting in the retention of nuclei on the glass slides (Fig. 1B). The slides were then dehydrated by incubation in 70%, 90%, and 100% ethanol for 5 min each at room temperature. The slides were stored at room temperature for up to 3 months. To obtain metaphase chromosomes, 70% to 80% confluent cells were incubated with fresh complete medium for 30 min and colcemid was added to the medium at a final concentration of 1 μg/ml. HepAD38 cells were incubated for 1 h and Dstet5 cells for 30 min at 37°C and then processed as described above.
Fluorescence in situ hybridization (FISH).
Biotinylated HBV- and DHBV-specific DNA probes were prepared with a Bioprime DNA labeling kit (InVitrogen, Carlsbad, CA; catalog no. 18094-011). Plasmids CMV-ayw and pSP65-Gal5.1 (300 to 400 ng) were used for HBV and DHBV probes, respectively (10, 22). The labeled DNA was precipitated with ethanol and resuspended in 20 to 30 μl Tris-EDTA (TE) buffer (50 mM Tris-HCl, 1 mM EDTA, pH 8.0) and stored at −20°C. For hybridization of four slides, 600 ng of the probe, 15 μg of human Cot-1 DNA (Invitrogen, Carlsbad, CA; catalog no. 15279-011), and a 1/10 volume of 3 M sodium acetate were combined and precipitated with 2 volumes of ethyl alcohol (EtOH) and incubated at −70°C for 30 min. After centrifugation in a microcentrifuge at 4°C for 15 min, the pellet was resuspended in 30 μl hybridization solution per slide (50% formamide–2× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate]), denatured for 5 min at 70 to 75°C, and incubated at 37°C for 15 min before hybridization.
Slides were prepared immediately prior to hybridization as follows. Each slide received 30 μl of a solution containing 200 μg/ml RNase A (Roche, Mannheim, Germany; catalog no. 11119915001) and was covered with a coverslip and incubated at 37°C for 1 h on a rack in a water bath. The slides were then washed in a Coplin jar four times in 2× SSC for 2 min each time. The slides were transferred to a Coplin jar containing a solution of 0.005% pepsin (Sigma, Saint Louis, MO; catalog no. P6887)–0.01 M prewarmed HCl for 5 min at 37°C, washed three times in phosphate-buffered saline (PBS) for 2 min each time, and dehydrated with 70%, 80%, and 100% ethanol for 2 min at room temperature and air dried. To denature DNA, the slides were incubated in a solution of 70% formamide–2× SSC at 72 to 74°C for exactly 2 min in a 50-ml tube and then dehydrated as described above. The time for air drying did not exceed 20 min. A 30-μl volume of the hybridization solution was added to each slide, and the slide was covered with a coverslip and incubated overnight on a rack placed in a 37°C water bath. The next day, the slides were washed twice in 50-ml tubes containing a solution of 50% formamide–2× SSC at 45°C for 10 min, followed by two washes in 2× SSC at 37°C for 4 min each time. Then, the slides were incubated in 1× PBD buffer (4× SSC [pH 7.0]–0.05% Tween 20) at room temperature for 5 min.
The slides were incubated with 30 μl of a suspension containing fluorescein-coupled avidin (Vector Laboratories, Burlingame, CA; catalog no. A-2001) (10 μg/ml) at 37°C for 20 min, followed by three washes in 1× PBD for 2 min each time. Then, the slides were incubated with 30 μl of a suspension containing biotinylated anti-avidin antibody (5 μg/ml) (Vector Laboratories, Burlingame, CA; catalog no. BA-0300) at 37°C for 20 min, followed by three washes in 1× PBD for 2 min each time. Then, the slides were incubated a second time with fluorescein-coupled avidin and washed as described above. The slides were mounted immediately after the final wash without drying (UltraCruz; Santa Cruz, Santa Cruz, CA; catalog no. sc-24941) and sealed with nail polish before viewing with a Zeiss fluorescence microscope or a Leica TCPSP8 laser scanning confocal microscope was performed. When slides were processed further for immunofluorescence, they were incubated with PBS following the last wash with PBD buffer. Then, the slides were processed essentially as described below.
Immunofluorescence.
For immunofluorescence, cells in 24-well plates were fixed in 4% paraformaldehyde–PBS for 10 min at room temperature followed by washing in PBS three times. Blocking was done in PBS containing 2% bovine serum albumin (BSA) for 40 min at room temperature. Antibodies were diluted in blocking solution and incubated for 1 h at room temperature. After application of primary and secondary antibodies, washing steps were performed in PBS three times for 1 min each time. Lamin was detected with mouse anti-lamin A/C antibodies (BD Biosciences, Franklin Lakes, NJ; catalog no. 612162) and then with goat anti-mouse antibody conjugated with Alexa Fluor 594 (Life Technologies, Rockford, IL; catalog no. A11032). DHBV core antigen was detected with anti-DHBV core antigen (a gift from William Mason, Fox Chase Cancer Center) and then with goat anti-rabbit antibody conjugated with Alexa Fluor 594 (Invitrogen Molecular Probes, Rockford, IL; catalog no. A11037).
Extraction of viral DNA.
Viral DNA was isolated using the Hirt procedure as described by Yang et al. (23). Briefly, cells from a 60-mm-diameter dish were lysed in 1 ml of ice-cold TE buffer (50 mM Tris-HCl, 1 mM EDTA, pH 8.0) and 1 ml of 4% sodium dodecyl sulfate (SDS). Then, the lysate was transferred to a 15-ml tube followed by addition of 0.25 ml of 2.5 M KCl and incubated on ice for 10 min. The lysate was transferred to two 1.5-ml Eppendorf tubes and centrifuged at 16,000 rpm for 5 min at 4°C. The supernatant was extracted twice with phenol and once with butanol-isopropanol (7:3). DNA was precipitated with two volumes of ethanol at room temperature and centrifuged, and the pellet in each tube was dissolved in 50 μl TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0).
Statistical analysis.
FISH data were collected as follows. Using a Nikon microscope with a 10× objective, an area of 30 to 100 mm2 on a slide was scanned for DAPI (4′,6-diamidino-2-phenylindole)-stained nuclei. In the selected area, 200 intact nuclei were acquired with Ikaros/Isis software. FISH signals on the imaged nuclei were counted by eye on the computer screen and recorded for statistical analysis. PRISM software was used for statistical analysis and generation of graphs.
ACKNOWLEDGMENTS
C.S. acknowledges the many generous contributions by Stan Basickes for statistical analyses. We are grateful to Frank Chisari for sharing HBV-Met cells. We thank Glenn Rall and Jianming Hu for helpful comments on the manuscript.
C.S. acknowledges support from NIH. M.L. was supported by a fellowship from the National Science Foundation of China (NSFC).
C.S. designed the experiments. M.L. developed the FISH method and executed the FISH experiments. J.A.S. performed Southern blot analyses. C.S. wrote the manuscript.
REFERENCES
- 1.Ott JJ, Stevens GA, Groeger J, Wiersma ST. 2012. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 30:2212–2219. doi: 10.1016/j.vaccine.2011.12.116. [DOI] [PubMed] [Google Scholar]
- 2.Lavanchy D. 2004. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J Viral Hepat 11:97–107. doi: 10.1046/j.1365-2893.2003.00487.x. [DOI] [PubMed] [Google Scholar]
- 3.Seeger C, Zoulim F, Mason WS. 2013. Hepadnaviruses, 6 ed Lippincott, Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 4.Guidotti LG, Rochford R, Chung J, Shapiro M, Purcell R, Chisari FV. 1999. Viral clearance without destruction of infected cells during acute HBV infection. Science 284:825–829. doi: 10.1126/science.284.5415.825. [DOI] [PubMed] [Google Scholar]
- 5.Kajino K, Jilbert AR, Saputelli J, Aldrich CE, Cullen J, Mason WS. 1994. Woodchuck hepatitis virus infections: very rapid recovery after a prolonged viremia and infection of virtually every hepatocyte. J Virol 68:5792–5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo JT, Zhou H, Liu C, Aldrich C, Saputelli J, Whitaker T, Barrasa MI, Mason WS, Seeger C. 2000. Apoptosis and regeneration of hepatocytes during recovery from transient hepadnavirus infections. J Virol 74:1495–1505. doi: 10.1128/JVI.74.3.1495-1505.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu Y, Yamamoto T, Cullen J, Saputelli J, Aldrich CE, Miller DS, Litwin S, Furman PA, Jilbert AR, Mason WS. 2001. Kinetics of hepadnavirus loss from the liver during inhibition of viral DNA synthesis. J Virol 75:311–322. doi: 10.1128/JVI.75.1.311-322.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang YY, Zhang BH, Theele D, Litwin S, Toll E, Summers J. 2003. Single-cell analysis of covalently closed circular DNA copy numbers in a hepadnavirus-infected liver. Proc Natl Acad Sci U S A 100:12372–12377. doi: 10.1073/pnas.2033898100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo JT, Pryce M, Wang X, Barrasa MI, Hu J, Seeger C. 2003. Conditional replication of duck hepatitis B virus in hepatoma cells. J Virol 77:1885–1893. doi: 10.1128/JVI.77.3.1885-1893.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, King RW. 1997. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother 41:1715–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Köck J, Rösler C, Zhang JJ, Blum HE, Nassal M, Thoma C. 2010. Generation of covalently closed circular DNA of hepatitis B viruses via intracellular recycling is regulated in a virus specific manner. PLoS Pathog 6:e1001082. doi: 10.1371/journal.ppat.1001082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao W, Hu J. 2007. Formation of hepatitis B virus covalently closed circular DNA: removal of genome-linked protein. J Virol 81:6164–6174. doi: 10.1128/JVI.02721-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT. 2007. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol 81:12472–12484. doi: 10.1128/JVI.01123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raney AK, Eggers CM, Kline EF, Guidotti LG, Pontoglio M, Yaniv M, McLachlan A. 2001. Nuclear covalently closed circular viral genomic DNA in the liver of hepatocyte nuclear factor 1 alpha-null hepatitis B virus transgenic mice. J Virol 75:2900–2911. doi: 10.1128/JVI.75.6.2900-2911.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Summers J, Smith PM, Horwich AL. 1990. Hepadnavirus envelope proteins regulate covalently closed circular DNA amplification. J Virol 64:2819–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pasquetto V, Wieland SF, Uprichard SL, Tripodi M, Chisari FV. 2002. Cytokine-sensitive replication of hepatitis B virus in immortalized mouse hepatocyte cultures. J Virol 76:5646–5653. doi: 10.1128/JVI.76.11.5646-5653.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perlman DH, Berg EA, O'Connor PB, Costello CE, Hu J. 10 June 2005. Reverse transcription-associated dephosphorylation of hepadnavirus nucleocapsids. Proc Natl Acad Sci U S A doi: 10.1073/pnas.0502138102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jilbert AR, Wu T-T, England JM, de la Hall MP, Carp NZ, O'Connell AP, Mason WS. 1992. Rapid resolution of duck hepatitis B virus infections occurs after massive hepatocellular involvement. J Virol 66:1377–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seeger C, Sohn JA. 2014. Targeting hepatitis B virus with CRISPR/Cas9. Mol Ther Nucleic Acids 3:e216. doi: 10.1038/mtna.2014.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seeger C, Sohn JA. 16 May 2016. Complete spectrum of CRISPR/Cas9-induced mutations on HBV cccDNA. Mol Ther doi: 10.1038/mt.2016.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stehle IM, Postberg J, Rupprecht S, Cremer T, Jackson DA, Lipps HJ. 2007. Establishment and mitotic stability of an extra-chromosomal mammalian replicon. BMC Cell Biol 8:33. doi: 10.1186/1471-2121-8-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang GH, Seeger C. 1992. The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell 71:663–670. doi: 10.1016/0092-8674(92)90599-8. [DOI] [PubMed] [Google Scholar]
- 23.Yang W, Mason WS, Summers J. 1996. Covalently closed circular viral DNA formed from two types of linear DNA in woodchuck hepatitis virus-infected liver. J Virol 70:4567–4575. [DOI] [PMC free article] [PubMed] [Google Scholar]