

ABSTRACT
STAT1 phosphorylation in response to exogenous interferon (IFN) administration can be inhibited by rotaviral replication both in vitro and in vivo. In addition many rotavirus strains are resistant to the actions of different IFN types. The regulation by rotaviruses (RVs) of antiviral pathways mediated by multiple IFN types is not well understood. In this study, we find that during infection in vitro and in vivo, RVs significantly deplete IFN type I, II, and III receptors (IFNRs). Regulation of IFNRs occurred exclusively within RV-infected cells and could be abrogated by inhibiting the lysosomal-endosomal degradation pathway. In vitro, IFNR degradation was conserved across multiple RV strains that differ in their modes of regulating IFN induction. In suckling mice, exogenously administered type I, II, or III IFN induced phosphorylation of STAT1-Y701 within intestinal epithelial cells (IECs) of suckling mice. Murine EW strain RV infection transiently activated intestinal STAT1 at 1 day postinfection (dpi) but not subsequently at 2 to 3 dpi. In response to injection of purified IFN-α/β or -λ, IECs in EW-infected mice exhibited impaired STAT1-Y701 phosphorylation, correlating with depletion of different intestinal IFNRs and impaired IFN-mediated transcription. The ability of EW murine RV to inhibit multiple IFN types led us to test protection of suckling mice from endotoxin-mediated shock, an outcome that is dependent on the host IFN response. Compared to mortality in controls, mice infected with EW murine RV were substantially protected against mortality following parenteral endotoxin administration. These studies identify a novel mechanism of IFN subversion by RV and reveal an unexpected protective effect of RV infection on endotoxin-mediated shock in suckling mice.
IMPORTANCE Antiviral functions of types I, II, and III IFNs are mediated by receptor-dependent activation of STAT1. Here, we find that RV degrades the types I, II, and III IFN receptors (IFNRs) in vitro. In a suckling mouse model, RV effectively blocked STAT1 activation and transcription following injection of different purified IFNs. This correlated with significantly decreased protein expression of intestinal types I and II IFNRs. Recent studies demonstrate that in mice lipopolysaccharide (LPS)-induced lethality is prevented by genetic ablation of IFN signaling genes such as IFNAR1 and STAT1. When suckling mice were infected with RV, they were substantially protected from lethal exposure to endotoxin. These findings provide novel insights into the mechanisms underlying rotavirus regulation of different interferons and are likely to stimulate new research into both rotavirus pathogenesis and endotoxemia.
KEYWORDS: rotavirus, interferon, innate immunity, interferon receptor, endotoxin
INTRODUCTION
The remarkable infectivity and replicative ability of rotaviruses (RVs) in vitro and in vivo are largely determined by their ability to subvert the induction and receptor-mediated amplification of different interferon (IFN) types (1). Understanding RV regulation of innate immunity is important for the development of effective live attenuated RV vaccines to prevent the approximately 200,000 annual RV deaths worldwide. Previously, we found that a major determinant of successful RV replication in the mouse model is viral inhibition of the IFN response (2–4). Following RV infection in vivo, the secreted IFNs arising from several intestinal cell types transduce signaling from multiple types of IFN receptors (IFNRs), resulting in the phosphorylation of STAT1 and the expression of hundreds of antiviral genes (4–7). By comparing RV strains with distinct replication phenotypes in mice lacking combinations of IFN receptors, all three major intestinal IFN types (IFN type I [IFN-α/β], type II [IFN-γ], and type III [IFN-λ]) were found to be important regulators of RV replication (3, 4, 8, 9). How successfully replicating RV strains achieve resistance to the actions of different IFN types is largely unknown.
We previously reported that during infection in vitro, RV efficiently blocks the activation of STAT1 in response to saturating doses of exogenously applied IFN-β (7). In addition, RV also inhibits STAT1 by blocking its nuclear translocation (10, 11). At the single-cell level, all RV strains studied to date have inhibited activation of STAT1 phosphorylated at Y701 (STAT1-pY701) in response to exogenous IFN within RV-infected cells (7). In addition, the porcine RV SB1-A strain, but not several other RV strains, rendered uninfected bystander HT29 cells refractory to IFN-mediated STAT1 phosphorylation (7). The mechanisms by which RVs inhibit the action of IFN in infected or bystander cells have not been determined. In this study, we report that, in vitro, RV infection depletes multiple types of IFNRs in infected cells by directing their lysosomal-endosomal degradation. Furthermore, in suckling mice RV infection results in inhibition of IFN-mediated STAT1 activation, IFN-mediated transcription, and the depletion of multiple IFNR proteins in the intestine. Finally, in a murine model of endotoxin-mediated mortality which requires functional IFN responses (12–15), RV-infected suckling mice were significantly protected from the lethal effects of endotoxin. These findings provide new insights into how RV efficiently replicates despite the presence of an active host enteric innate immune response.
RESULTS
The expression of multiple interferon receptors is downregulated by RV infection.
Previously, we showed that various RV strains inhibit STAT1-Y701 phosphorylation in response to exogenous IFN (7), but the mechanisms responsible for this effect are not clear. Human intestinal epithelial HT29 cells phosphorylate STAT1-Y701 in response to stimulation with exogenous type I, II, or III IFN (Fig. 1A) (7), providing a tractable system in which to study the inhibition of STAT1. We found that infection of HT29 cells with increasing multiplicities of infection (MOIs) of either a porcine SB1-A or a simian RRV RV strain resulted in decreased expression of the IFN-γ receptor (IFNGR1) (Fig. 1B). A time course analysis revealed that the decrease in IFNGR1 occurs between 6 and 8 h postinfection (p.i.) (Fig. 1C). In order to examine whether these effects of RV infection extended to other IFN type receptors, we analyzed the expression of receptors for the two other major IFNR types (IFNAR1 for IFN-α and IFNLR1 for IFN-λ) by immunoblotting. Remarkably, in addition to decreased IFNGR1 expression, infection with either SB1-A or RRV also led to significantly lower levels of IFNAR1 and IFNLR1 (Fig. 1D).
FIG 1.

Rotavirus infection leads to decreased expression of different IFNRs. (A) HT29 cells express functional IFN-α/β, -γ, and -λ receptors. Cells were stimulated with type I, II, or III IFN (500 U/ml, 25 ng/ml, and 500 U/ml, respectively) for 6 h or 30 min and analyzed for STAT1-pY701 activation by flow cytometry. (B) Cells were infected with porcine SB1-A or simian RRV RV at the MOIs indicated and at 12 hpi analyzed by immunoblotting. (C) Cells infected with SB1-A RV or RRV were analyzed for IFNGR1 expression at different times postinfection. (D) Cells infected with SB1-A or RRV RV were examined for expression of different IFNRs at 12 hpi. Arrows indicate bands corresponding to IFNRs. Numbers refer to relative ratios of IFNR levels to the level of actin. Asterisks indicate nonspecific bands. All experiments were performed three or more times with similar results.
IFNR downregulation by RV occurs only in infected cells and is conserved among different viral strains.
In earlier studies, we observed that RV infection can perturb both the IFN induction and signaling pathways in discrete single-cell populations (4, 7). To better understand the regulation of IFNRs by RV, we carried out single-cell flow cytometry analysis of IFNGR1 and RV VP6 expression following infection with several RV strains. To precisely gate cells based on their IFNGR1 expression levels, we took advantage of the fact that HT29 cell treatment with cycloheximide (CHX) results in a substantial depletion of IFNGR1 (Fig. 2A), as is apparent by flow cytometry analysis (Fig. 2B).
FIG 2.

Single-cell analysis of IFNR expression following infection with selected RV strains. (A) Depletion of IFNGR1 in HT29 cells following cycloheximide (CHX) treatment (25 μg/ml) detected by immunoblot analysis. (B) Representative FACS plot showing gating strategy used to define IFNGR1-high and -low populations after CHX treatment. UT, untreated. (C) Flow cytometry analysis of IFNGR1 and RV VP6 antigen expression in RV-positive and -negative HT29 cells at 24 hpi. HT29 cells were infected for 12 h with the indicated RV strains. For each RV strain, control cells treated for 4 h with cycloheximide (CHX; + to deplete IFNGR1 or left untreated −) were used. Plots show MFI of IFNGR1 in VP6-positive (RV+) and VP6-negative (RV−) populations of cells. Vertical lines indicate receptor levels with and without CHX treatment. (D) The results from three independent experiments carried out as described for panel C are plotted. Significant differences between results in treated cells versus those in untreated cells are indicated on the figure. (E) Cells infected as described in panel C were analyzed for IFNAR1 and VP6. Byst, bystander cells. (F) Cells infected as described in panel C were analyzed for IFNGR1 and VP6 at 1 to 3 days p.i. UI, uninfected. Experiments were performed two or more times with similar results.
Different RV strains have evolved distinct strategies to inhibit the induction of IFN genes by targeting either β-TrCP and/or IFN regulatory factor 3 (IRF3) for degradation, as exemplified by porcine (targeting β-TrCP), simian (IRF3), and bovine (targeting β-TrCP and IRF3) RV strains (5, 16–23). HT29 cells were infected with different RV strains (porcine SB1-A, simian RRV and SA11, a simian SA11 mono-reassortant expressing porcine OSU NSP1 [SOF], and bovine UK) for 16 h and analyzed by flow cytometry (Fig. 2C). IFNGR1 levels (as mean fluorescence intensity [MFI]) from RV-positive (RV+) and RV-negative (RV−) cell populations (defined as VP6 positive or negative, respectively) were plotted separately (Fig. 2C and D). We found that all RV strains examined mediated significant decreases in IFNGR1 expression in only infected cells, not uninfected bystander cells (Fig. 2D), indicating that the decrease in IFNR expression in any given cell is a direct effect of RV infection in that cell. In addition, the RV-mediated IFNGR1 degradation in VP6+ cells was significant and comparable to depletion of receptor levels seen following 4 h of CHX treatment (Fig. 2D). These RV-specific effects on IFNGRs were also seen with the IFNAR1, which was also downregulated only in RV+ cells following infection with either RRV or SB1-A (Fig. 2E). To confirm that the RV-mediated IFNR decrease was restricted to infected cells, we prolonged HT29 cell infection with either RRV or SB1-A for up to 3 days and then analyzed levels of IFNGR1 and VP6 by flow cytometry (Fig. 2F). The results indicated that at 1 to 3 days postinfection (dpi), significant decreases in IFNRs are restricted to infected RV+ cells (Fig. 2F). Collectively, these results indicate that downregulation of IFNRs occurs only within infected RV+ cells and that this downregulation is conserved across various RV strains that inhibit IFN induction by promoting degradation of either β-TrCP or IRF3 or both.
Rotavirus depletes IFNRs by lysosomal-endosomal degradation.
To ascertain how RV infection downregulates IFNRs, we first examined the status of transcripts encoding IFNAR1 during RV replication in vitro. HT29 cells infected with the porcine SB1-A RV strain were analyzed for the levels of IFNAR1 and RV transcripts (Fig. 3A). Under these conditions a significant decrease in IFNAR1 mRNA was not detected (a specific IFNAR1 PCR product was detected at cycle 24 for both infected and control specimens), suggesting that receptor regulation is unlikely to be due to perturbation of IFNAR1 gene transcription. Analysis of IFNGR1 turnover using cycloheximide (CHX) chase experiments suggested that SB1-A RV accelerated the degradation of this receptor (Fig. 3B). Since IFNR degradation occurs by a proteasomal and/or lysosomal-endosomal degradation pathway (24–27), we next examined whether inhibitors of proteasomal degradation (MG132) or of lysosomal-endosomal degradation (NH4Cl and chloroquine) could inhibit IFNR depletion during RV infection. In uninfected HT29 cells, treatment with MG132 resulted in accumulation of IFNGR1 but not of IFNAR1 (Fig. 3C). In contrast, treatment with inhibitors of lysosomal-endosomal degradation resulted in the accumulation of both IFNAR1 and IFNGR1. Infection with porcine SB1-A or simian RRV led to reduced IFNGR1 and IFNAR1 levels compared to those of uninfected controls (Fig. 3C). Treatment with NH4Cl or chloroquine rescued virus-mediated IFNAR1 and IFNGR1 depletion, whereas MG132 treatment effectively rescued only IFNGR1 from RV-mediated degradation. By fluorescence-activated cell sorter (FACS) analysis of IFNGR1, we confirmed that inhibiting the lysosomal-endosomal degradative pathway rescued receptor expression within RV-infected cells (Fig. 3D). Together, these results indicate that RV-mediated decreases in the levels of IFNRs occur primarily, but not exclusively, via a lysosomal-endosomal degradation pathway.
FIG 3.

Rotavirus downregulates IFNRs via lysosomal-endosomal degradation. (A) HT29 cells were left uninfected (UI) or infected with RV for 12 h prior to analysis of IFNAR1 transcripts by semiquantitative RT-PCR. Detection of RV genes 5 and 7 (inset) was used to confirm infection. (B) HT29 cells infected with SB1-A (or uninfected controls) were treated for increasing durations with CHX and lysed at 10 hpi for the indicated immunoblotting analysis. (C) Effect of inhibitors of protein degradation on RV-mediated downregulation of the indicated IFNRs. Cells infected with SB1-A or RRV were treated at 4 hpi with 10 μM MG132, 25 μM chloroquine (ChlQ), or 12 mM NH4Cl and lysed 10 h later for analysis by immunoblotting. Numbers below lanes are the levels of IFNRs after normalization to actin relative to levels in the untreated controls. (D) Cells were infected with RRV and treated with 25 μM chloroquine (ChlQ) for 12 h before analysis of IFNGR1 and VP6 expression by FACS. All experiments were performed three or more times with similar results.
Multiple IFNRs can activate STAT1 in IECs and act in concert during RV infection in vivo.
Our results suggested that IFNR degradation is a potential mechanism to account for the resistance of RV to different types of IFNs in vivo (1). To test this hypothesis, we first evaluated the ability of several exogenously administered IFNs to stimulate STAT1-Y701 phosphorylation in the uninfected mouse intestine. Suckling mice were parenterally administered either IFN-I, -II, or -III, and their intestinal tissue was subjected to immunohistochemistry analysis (IHC) for the extent of STAT1-pY701 nuclear expression (Fig. 4A and B). Compared to activation in phosphate-buffered saline (PBS)-injected controls, which exhibited no detectable STAT1-pY701 staining, intraperitoneal (i.p.) administration of either IFN-λ or IFN-γ resulted in robust STAT1-Y701 activation, primarily within intestinal epithelial cells (IECs) (Fig. 4A and B). Remarkably IFN-γ, like IFN-λ, was an efficient STAT1 activator in the intestinal epithelium (Fig. 4B, insets 2 and 3), as was also the case in HT29 IECs in vitro (Fig. 1A). In contrast, IFN-α administration resulted in phospho-STAT1 (pSTAT1) expression in both IECs and within lamina propria lymphocytes (LPLs) (Fig. 4B, inset 4). Thus, all three major IFN subtypes elicit a STAT1 response in IECs (Fig. 4C) while IFN-α also activated STAT1 in LPLs. These results demonstrate that in suckling mice functional IFN-I/II/III receptors are all present on IECs, the primary sites of RV replication in vivo.
FIG 4.

Intestinal cell types responding to exogenously administered IFN-I, -II, and -III in suckling mice. (A) Three- to 5-day-old mice (n = 2 to 3 per group) were given 1 μg of purified IFN-λ, -γ, or -α i.p. as described in Materials and Methods. Intestines were collected 30 min later and stained for STAT1-pY701. (B) Magnification of boxed insets 1 to 4 from panel A. (C) Schematic summary of the ability of the three IFN types to stimulate STAT1 phosphorylation in intestinal epithelial cells (IECs) or lamina propria lymphocytes (LPLs).
Despite its IFN-resistant replication in suckling mice, murine RV infection triggers robust induction of multiple types of IFN genes in the intestine on days 1 and 2 postinfection (4, 9), as well as IFN-β secretion at day 4 (28), potentially leading to STAT1 activation. We directly examined this possibility by evaluating levels of intestinal STAT1-pY701 at different times following RV infection of suckling mice. Mice were infected with EW RV, and small intestines were collected on days 1, 2, and 3 p.i. for IHC analysis of STAT1-pY701 and RV VP6 antigen expression by peroxidase staining using serial tissue sections (Fig. 5A and data not shown). Intestines of RV-infected mice on day 1 p.i. showed nuclear STAT1-pY701 expression (Fig. 5A), which was most frequently observed in RV-negative villi. In comparison, in intestines of mice at 2 to 3 dpi, expression of STAT1-pY701 was not detected in either infected or uninfected cells (Fig. 5A) despite increased numbers of RV-infected IECs on days 2 and 3. Thus, although RV induces transcription of multiple intestinal IFN types (each capable of activating STAT1 in IECs) between 1 and 3 dpi (9), STAT1-pY701 expression occurs only transiently, on day 1 p.i.
FIG 5.

STAT1 activation in suckling mouse intestines during infection with RV. (A) Suckling mice (n = 2 to 3 per group) were infected with EW RV, and intestines were collected on days 1, 2, and 3 for analysis of RV antigen and STAT1-pY701 expression by immunohistochemistry. (B and C) Three- to 5-day-old WT or IFN-α/β/γ receptor-negative (IFN-αβγR−/−) mice (n = 2 to 3 per group) were infected with murine EW RV, and intestines were collected for analysis of STAT1-pY701 expression by immunohistochemistry at 24 hpi. The red arrow indicates STAT1 phosphorylation primarily within LPLs. The regions boxed in red are magnified in the insets. The percentage of villi staining positive for STAT1-pY701 in each group of mice was quantified (n = 200 villi) and is plotted in panel C. ***, P < 0.05, by a two-sided chi-square test.
Using Mx1 protein expression as an indirect marker for IFN responsiveness in individual cells, a previous study concluded that IFN-λ was primarily responsible for regulating the IEC anti-RV response, with type I IFN playing a minor role (29). Since EW RV infection results in detectable STAT1 activation in IECs (Fig. 5A, day 1), we examined whether this is dependent on IFN-λ. Suckling wild-type (WT) or IFN-α/β/γ receptor knockout mice (expressing intact IFN-λ receptor) were infected with RV, and small intestines were analyzed for expression of STAT1-pY701 at 1 dpi (Fig. 5B and C). We found that, as expected, infection resulted in expression of detectable STAT1-pY701 on day 1 p.i. in both IECs and LPLs of WT mice. This response was significantly diminished, but not eliminated, in the absence of receptors for IFN-α/β and IFN-γ (Fig. 5B and C). Thus, all three IFNRs appear to be responsible for the transient STAT1 activation in IECs following RV infection in suckling mice.
Inhibition of STAT1 activation, IFN-mediated transcription, and the depletion of different intestinal IFNRs occur during RV infection in vivo.
Despite the transient expression of STAT1-pY701 in IECs at 1 dpi (Fig. 5A), the sustained induction of different IFN genes (4, 9), accompanied by a lack of STAT1 activation, at later times during infection suggests that RV suppresses STAT1 phosphorylation in response to these IFNs. To directly test this hypothesis, we examined the ability of IECs from RV-infected mice to respond to ectopic stimulation with purified IFN. Mice were infected with RV (or mock infected) and at 24 h p.i. (hpi) administered purified IFN-α or IFN-λ to exogenously trigger STAT1-Y701 phosphorylation. Intestinal expression of STAT1-pY701 was examined by IHC (Fig. 6A and B), and the frequency of STAT1-pY701-positive cells was enumerated for IECs (Fig. 6C). We could not carry out similar STAT1-pY701 quantitation within LPLs due to their high density in the small intestine. Compared to levels in mock controls, STAT1 activation within IECs in response to both IFN-α/β and IFN-λ administration was considerably diminished in RV-infected pups at 1 dpi (Fig. 6A to C). Thus, RV infection impairs the ability of IECs to mediate ligand-dependent STAT1 activation by type I and III IFN receptors in vivo.
FIG 6.

Rotavirus actively blocks IFN-λ and IFN-α receptor-directed intestinal STAT1 phosphorylation in vivo. (A) Suckling mice (n = 2 to 3 per group) were infected with EW RV or mock infected and at 24 h p.i. challenged with purified IFN-λ or IFN-α for 30 min before analysis of STAT1-pY701 by immunohistochemistry. (B) Magnification of the regions boxed in red in panel A. (C) The fraction of IECs positive for STAT1-pY701 from the experiments shown in panel A were enumerated and are represented as a percentage of the total number of cells counted (n = 3,000 or more cells per group). ***, P < 0.05, by a two-sided chi-square test.
We next examined whether IFNR degradation occurs in vivo during murine RV infection of suckling mice. Levels of IFNAR1 and IFNGR1 were measured in intestinal tissue lysates from infected and uninfected suckling mice at 3 dpi by immunoblot analysis (Fig. 7A to C). A similar analysis for the IFNLR1 was not possible because we were unable to identify reliable antibodies for Western blot detection of murine intestinal IFNLR1. Acute RV infection led to significant decreases in the expression levels of both types I (Fig. 7A and C) and II IFN receptors (Fig. 7B and C). To extend these findings, we tested the ability of exogenously administered IFN-γ, which activates STAT1 in IECs (Fig. 4A), to induce the transcription of the IFNGR1-responsive gene Ido-1 (30, 31) (Fig. 7D). As expected, IFN-γ administration led to the transcriptional induction of Ido-1 in mock-infected pups, and this response was significantly inhibited by EW RV infection (Fig. 7D). In line with the substantial IFN resistance of homologous EW RV to the murine innate immune IFN response, exposure to IFN-γ for 16 h did not have a significant effect on RV replication (Fig. 7D). These findings agree with our results on RV-mediated dampening of STAT1 activation by 3 dpi (Fig. 5C) and the active inhibition of IFN-directed STAT1-Y701 phosphorylation by EW RV (Fig. 6A).
FIG 7.

Rotavirus infection leads to downregulation of IFNR protein- and receptor-mediated transcription in vivo. (A to C) Mice (n = 3 to 4 per group) were infected with EW or mock infected for 3 days prior to analysis of intestinal tissue by immunoblotting for expression of IFNAR1 (A) or IFNGR1 (B). Mean band intensities from individual mice calculated after normalization to the level of GAPDH are plotted in panel C. **, P < 0.005. (D) Suckling mice (n = 3 to 4 per group) were infected with EW murine RV for 16 h and then administered 1 μg of purified IFN-γ as described in Materials and Methods. Sixteen hours later, mice were sacrificed, and intestinal tissue was analyzed as indicated by qRT-PCR for Ido-1 or RV gene 11 transcripts. ***, P < 0.001.
Infection with murine RV protects suckling mice from endotoxin-mediated mortality.
The ability of endotoxin to cause severe shock and mortality in mice has been reported to be prevented by genetic ablation of components of IFN induction (32, 33), IFNAR1 (12, 34), and the JAK-Tyk2-STAT1 (13, 35–37) signal transduction pathway. Our results demonstrate that RVs degrade multiple IFNRs in infected cells and inhibit antiviral signaling downstream of different IFNRs. RV infection inhibited the IFN-mediated induction of Ido-1 (indoleamine 2,3-dioxygenase) in the intestine (Fig. 7D), which is a critical determinant of LPS-induced lethality in mice (38–40). Previously, we found that RV infection also leads to a significant inhibition of the IFN response and NF-κB signaling in uninfected bystander cells (4, 7). Based on these findings, we next examined whether RV infection could influence the outcome of LPS-induced lethality in mice.
Suckling mice were infected with murine EW RV (or mock infected) for 3 days before administration of a single lethal dose of purified LPS. Mock-infected controls displayed symptoms of severe endotoxemia and exhibited 100% mortality within 24 h of LPS administration. In contrast, mice infected with RV for 3 days before LPS administration displayed increased survival, and >50% of infected mice did not succumb for up to 6 days post-LPS exposure (Fig. 8). These results reveal an unanticipated protective effect of RV infection against the undesirable host response to endotoxin and are in good agreement with both the essential role of an intact IFN response in endotoxin-mediated mortality (14, 41) and the ability of RV to inhibit the host response to different types of IFNs.
FIG 8.
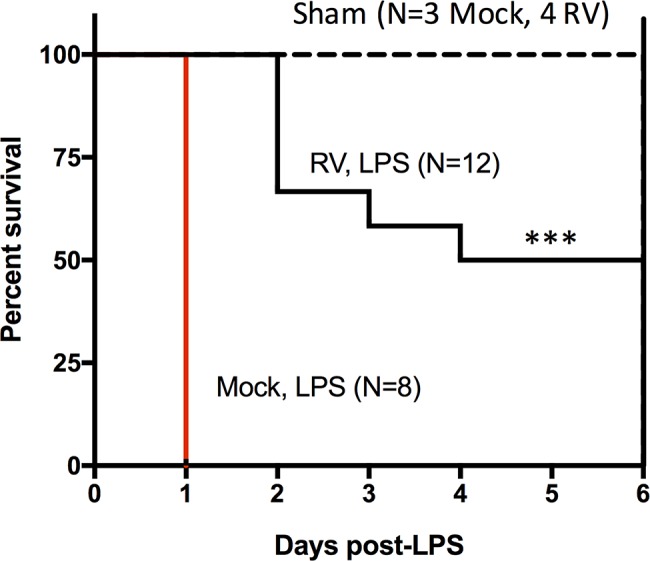
Suckling mice were infected for 3 days (or mock infected) with EW. Subsequently mice were challenged with 10 mg/kg LPS (or PBS as a sham control), and mortality was observed over 6 days. The number of mice (N) indicated for each group represents the total number of mice challenged in three separate experiments (***, P < 0.001; survival curves were compared by a Mantel-Cox test).
DISCUSSION
Intestinal replication of RV critically depends on its ability to subvert host innate responses mediated primarily by the types I, II, and III IFN receptors. In vitro, multiple RV strains inhibit STAT1 phosphorylation in response to exogenous IFN, a property intrinsic to the RV NSP1 protein (8). However, whether these STAT1-antagonistic functions also operate during RV infection in vivo and what underlying mechanisms mediate RV inhibition of STAT1 activation are not well understood. Since HT29 cells express functional IFNRs capable of triggering STAT1-Y701 phosphorylation in response to IFN-β, -γ, or -λ stimulation (Fig. 1A), they provided a tractable cell culture system to study the effects of RV infection on distinct IFNRs. Analysis of porcine SB1-A- and simian RRV-infected HT29 cells revealed a significant decrease in the levels of types I, II, and III IFNRs (Fig. 1C). The decrease in IFNRs occurred at 6 to 8 hpi, effectively preceding the previously observed inhibition of STAT1 phosphorylation in response to exogenous IFN (7). The decrease in IFNR levels mediated by RV infection is likely to be a direct effect of virus infection rather than a global negative-feedback mechanism potentiated by the secretion of IFNs since, after FACS analysis, the reduction in IFNR expression was observed only in the RV-infected cell population (Fig. 2C and D).
IFNR downregulation in vitro occurred during infection with multiple RV strains which use distinct strategies to block the induction phase of the IFN response by targeting either NF-κB or IRF3 or both (Fig. 1D and 2C) (1). Thus, regulation of IFNRs is conserved across RV strains from different species, in contrast to strain-specific strategies to inhibit induction of IFN expression (1). RV infection mediates the depletion of IFNRs by a process that accelerates their protein turnover rather than by inhibition of gene transcription (Fig. 3A and B). Normally, turnover of IFNAR1 and IFNGR1 occurs by a combination of proteasomal and lysosomal-endosomal pathways (25). In the context of RV infection, both IFNAR1 and IFNGR1 could be significantly rescued from depletion using chloroquine (Fig. 3C and D), consistent with the conclusion that RVs deplete IFNRs via lysosomal-endosomal protein degradation. A variety of viral pathogens have developed mechanisms to block IFN-mediated STAT1 activation, which is a major point of signaling convergence for the different IFNRs (42). The ability of RV to deplete multiple types of IFNRs is intriguing and, to our knowledge, a novel viral strategy to broadly negate an antiviral state in response to all three major IFN types.
Regulation of host innate responses by RV occurs within infected and RV-negative bystander cell populations, resulting in bulk (i.e., averaged) data that can be difficult to interpret without the aid of single-cell analytical approaches (4, 7). For several RV strains, significant downregulation of IFNAR1 and IFNGR1 occurred within infected cells, while bystander cells did not exhibit any significant reduction of receptor expression, even at 2 to 3 dpi (Fig. 2C, D, and F). Decreases in IFNRs in infected cells were substantial, approaching levels observed after treatment with CHX for 4 h (Fig. 2D), and could be rescued by treatment with chloroquine (Fig. 3D). Porcine SB1-A infection did not result in detectable degradation of IFNRs in RV-negative cells although these cells were previously shown to be refractory to saturating doses of exogenous IFN-mediated STAT1 activation (Fig. 2D) (7). Hence, degradation of IFNRs occurs only in the infected cell and does not appear to be responsible for the virally associated STAT1 inhibition that occurs in uninfected bystander cells, the mechanisms of which remain unknown. One possible mechanism by which RV could potentially regulate IFNR degradation is through PKR-mediated activation of the integrated stress response pathway. Activation of PKR, a sensor of viral double-stranded RNA (dsRNA), leads to phosphorylation of the translation factor eukaryotic initiation factor subunit 2α (eIF2α) and the mitogen-activated protein (MAP) kinase p38, which in turn results in IFNAR1 degradation (26, 43). RV infection does activate the PKR pathway, as sustained PKR-dependent eIF2α phosphorylation (44) and activation of p38 (45) have both been observed during RV infection in vitro. The exact mechanism of regulation of this signaling axis by RVs is still unclear and will be a focus of future studies.
RV replication in the intestine is accompanied by a significant induction of IFNs, including type I IFN originating primarily from hematopoietic cells and type III IFN arising primarily from epithelial cells (4, 46). Nevertheless, murine rotavirus replication in vivo is remarkably insensitive to the effects of these IFNs (4, 9), indicating that RV-encoded strategies to inhibit host cell signaling responses to different IFN types exist. To address this issue, we studied target cells in the small intestine that responded to types I, II, and III IFNs by directly detecting the phosphorylation status of STAT1 in response to exogenous IFN administration. Administration of both types I and III IFNs resulted in STAT1 activation in villus IECs, but type I IFN also stimulated STAT1 activation within LPLs in suckling mice (Fig. 4). These results reproduce and extend our previous findings to all three of the major IFN types (9). Surprisingly, IFN-γ resembled IFN-λ in its ability to induce STAT1 phosphorylation in IECs. Previous studies on intestinal epithelial antirotaviral responses have primarily focused on IFN types I and III (9, 29, 46). However, depletion of the IFN-γ receptor enhances replication of a simian RV strain (RRV) in the gut, and when combined with genetic ablation of the IFN-α/β receptor, results in a severe and frequently lethal disease, possibly due to the ability of RRV to spread to the biliary epithelium in the absence of IFN-γ receptor (3). Such IFN-γ-dependent effects on RV replication can be better understood in the context of the finding that IFN-γ activates epithelial STAT1 and thus may regulate the initial intestinal antiviral response to RVs and restrict its biliary replication. Furthermore, WT mice infected with EW RV at 1 dpi displayed STAT1-pY701 activation in both IECs and LPLs (Fig. 5A and C). In contrast, mice lacking type I/II IFNRs (and thus expressing only IFN-λ receptor) had a weaker STAT1 IEC response, with no detectable STAT1-pY701 in LPLs (Fig. 5B and C). Collectively, these results extend the conclusion that IFN-λ is unlikely to be the sole determinant of antiviral responses to RV and RV-mediated STAT1 activation in IECs and that such antiviral physiologically relevant responses are also elicited partly through the type I and II IFNRs.
Although RV triggered intestinal STAT1 activation on day 1 p.i., ongoing infection at later times (2 to 3 dpi) was accompanied by an almost complete absence of phospho-STAT1, despite RV-mediated induction of IFN during these times (4, 9, 28). When STAT1 activation was examined in response to exogenously administered IFN-α or IFN-λ, significant impairment was observed in mice in IECs from 1 day postinfection compared to levels in uninfected controls (Fig. 6A to C). In intestinal lysates from RV-infected mice at 3 dpi, there was a profound decrease in the protein levels of receptors for types I and II IFNs (Fig. 7A to C). The depletion of IFNGR1 by RV correlated with transcriptional inhibition of the IFN-γ-responsive gene, Ido-1 (indoleamine 2,3-dioxygenase), in response to ectopically administered IFN-γ (Fig. 7D). The ability of murine RV to subvert IFN signaling in the mouse gut may also be the case for human RV strains infecting human intestine. A recent study (47) of human RV replication in human intestinal enteroids found that neither type I nor III IFN induced during infection significantly inhibited RV replication (47). Thus, inhibition of effector functions of multiple IFNs by homologous RV infection is likely conserved between mouse and human hosts.
The ability of purified endotoxin (LPS) to cause lethality is significantly reduced in mice lacking functional IFN induction (type I IFN, MyD88, and TLR4) and amplification (IFNAR1, Tyk2, and JAK1) pathways, indicating a critical role for host IFNs in this toxicity (12–15, 25, 33–37, 41). IFN-mediated induction of Ido-1, which is inhibited by EW RV in the intestine (Fig. 7D), has been shown to be a critical determinant of LPS-induced lethality in mice (30, 38–40, 48). Expression of Ido-1 occurs primarily in activated macrophages and dendritic cells and has profound effects on T cell and T regulatory (Treg) cell responses to inflammatory signals including LPS (38–40). Based on these prior findings, we chose to examine the effect of RV infection on LPS-induced lethality in suckling mice. Our results demonstrate that infection with EW RV resulted in significant protection from LPS lethality in suckling mice (Fig. 8). These findings reveal an unexpected protective effect of RV infection on endotoxemia, in contrast to results of prior studies on endotoxemia with several other viruses (49–56). Based on our data, it is not possible to conclude exactly how RV infection efficiently prevents LPS-induced mortality, which is a result of several complex host inflammatory responses (14). One possible explanation is that, in addition to infecting IECs, murine RV also infects specific host cell types at systemic sites and regulates their IFN-dependent responses to LPS. Specific host cell types, such as macrophages and dendritic cells, that are critical for the IFN responses to LPS and endotoxin tolerance can be infected by RV (12–14, 31–33, 36, 37, 39, 40). Dissemination of RV beyond the intestinal epithelial cell is supported by several previous reports of RV antigenemia and viremia in the serum and different extraintestinal sites in both humans and animals, including mice (57–63). Alternately, RV-infected cells may inhibit the IFN responses to LPS in uninfected bystander intestinal and systemic cells through the action of secreted factors. We have previously shown, both in vitro and in vivo, that certain RV strains can efficiently inhibit NF-κB-dependent transcription and exogenous IFN-directed activation of STAT1 in bystander cells that do not contain detectable viral RNA or antigen (4, 7). The nature of the secreted factor(s) that inhibits bystander IFN responses during RV infection is presently unknown. We are currently examining which of these possibilities could explain the effects of murine RV on LPS-induced lethality.
MATERIALS AND METHODS
Cells, viruses, and reagents.
Human intestinal epithelial HT29 and embryonic kidney HEK293 cells were purchased from the American Type Culture Collection (ATCC) and maintained in advanced Dulbecco's modified Eagle's medium (D-MEM)-Ham's F12 medium (Cellgro) or D-MEM (Cellgro) containing 10% fetal calf serum (Invitrogen) supplemented with nonessential amino acids, glutamine, penicillin, and streptomycin. Rotavirus strains RRV, OSU, SB1A, SA11, SOF, and UK were propagated in MA104 cells, and titers were determined by plaque assay as described previously (6). Murine RV EW strain was propagated in mice, and titers were determined as described previously (4). The following antibodies were used for immunoblotting: IFNAR1 (ab124764), IFNGR1 (ab134070), and IL28Ra (ab156378) from Abcam; Tyk2 (9312), pY701-STAT1 (D4A7), PCNA (D3H8P), glyceraldehyde-3-phosphate dehydrogenase (GAPDH; D1611), IκB-α (L35A5), and STAT1 (9H2) from Cell Signaling; IRF3 and RV VP6 (2B4) from Santa Cruz; tubulin and actin from Sigma; secondary horseradish peroxidase (HRP)-conjugated antibodies from Amersham. Antibodies were purchased for FACS analysis of IFNAR1 (MMHAR-3; PBL), IFNGR1 (558937; BD Biosciences), and pY701-STAT1 (612597; BD Biosciences). Antibody to RV VP6 (1E11) conjugated to Alexa 488 was described earlier (7). For immunoblot analysis of mouse tissue, the following antibodies were used: IFNAR1 (140904; US Biologicals), IFNGR1 (ab216642; AbCam), VP6 (2B4; Santa Cruz), and GAPDH (Cell Signaling). Antibodies to RV (B65110G; Meridian Life Sciences) and to pY701-STAT1 (D4A7; CST) were used for immunohistochemistry. Purified human IFN-β (PBL), human IFN-λ (PBL), and human IFN-γ (Millipore) were used for stimulation of cells. Purified universal IFN-A/D (PBL), murine IFN-γ (R&D Systems), and murine IFN-λ (PeproTech) were used in mouse experiments. The following inhibitors were used: MG132 (10 μM), chloroquine (25 μM), and ammonium chloride (12 mM) from Calbiochem and cycloheximide (μM) from Sigma.
Virus infections.
HT29 cells were plated in six-well or 24-well cluster plates, and completely confluent monolayers were infected 2 to 3 days later as described previously (5).
Immunoblotting, RT-PCR, and microscopy.
Cells were lysed in Laemmli buffer containing 2% SDS and 5% β-mercaptoethanol after two washes in phosphate-buffered saline (pH 7.0). Cell lysis was performed at room temperature for 20 min, and lysates were passed through a 25-gauge needle to reduce sample viscosity. Cell lysates were boiled for 5 min, and tubes were briefly spun at 10,000 × g and separated on SDS-PAGE gels. Following electrophoresis, proteins were transferred onto nitrocellulose membranes (Amersham Biosciences) and detected using the antibodies indicated on the figures. Blots were exposed to autoradiography film (Amersham) and developed using an enhanced chemiluminescence kit (GE Healthcare). Blots were subsequently stripped and reprobed using a ReBlot kit according to the manufacturer's instructions (Millipore). Protein band relative intensities from scanned blots were determined using the NIH Image program (http://rsb.info.nih.gov/nih-image/). For semiquantitative reverse transcription-PCR (RT-PCR), total RNA was extracted using Tri regent (Invitrogen) according to the manufacturer's instructions and treated with Turbo DNase (Invitrogen). Identical amounts of total RNA were reverse transcribed with oligo(dT) primer (Invitrogen), and the resulting cDNAs were amplified using gene-specific primers to IFNAR1 or GAPDH. The following primers were used: for IFNAR1, 5′-AGGCGGCGCGTGCGTAGAGGGGC-3′ and 5′-GGACCAATCTCTGAGCTTTGCG-3′; for GAPDH, 5′-ACCACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′. PCR products were collected at different cycles and analyzed on a 1% agarose gel.
FACS analysis.
Cells were plated in 24-well cluster plates and infected with RV. Infected cells were harvested at 6 hpi or 16 hpi, and transfected cells were harvested at 48 after transfection for flow cytometry analysis. Cells were washed in PBS and fixed at room temperature for 10 min using 1.6% (vol/vol) methanol-free paraformaldehyde (Electron Microscopy Services). Cells were washed in FACS staining buffer (PBS containing 0.5% [wt/vol] bovine serum albumin) and permeabilized in cold methanol at 4°C for 10 min. Cells were washed in FACS staining buffer, stained using conjugated antibodies at 4°C for 30 min, and washed as described previously prior to analysis by flow cytometry on an LSRII or FACSCalibur instrument (BD Biosciences). The flow data were analyzed using FlowJo software and Cytobank analysis routines.
Cycloheximide chase.
HT29 cells were infected with RV at an MOI of 3.0, and cycloheximide (CHX)-containing serum-free medium was added at different times p.i. Cells were lysed at 10 hpi for analysis by immunoblotting.
Interferon treatment and virus infection of mice.
Universal hybrid IFN-A/D (PBL Assay Science, Piscataway Township, NJ), murine IFN-γ (R&D Systems, Minneapolis, MN), and IFN-λ2 (PeproTech, Rocky Hill, NJ) were intraperitoneally injected into 3- to 5-day-old 129sv suckling mice (1 μg of each IFN type per animal), and small intestines were collected from IFN-treated and untreated mice 1 day later. For RV infection, 129sv 4-day-old suckling mice were orally inoculated by gavage with 104 50% diarrhea doses (DD50) of murine RV-EW, and small intestines were collected at 1 to 3 dpi. Mice were administered purified IFN-A/D, IFN-γ, or IFN-λ2 at 24 hpi and were sacrificed 30 min later for analysis of pY701-STAT1 expression in the small intestine by immunohistochemistry or 18 h later for analysis by quantitative RT-PCR (qRT-PCR). All animal studies were approved by the VA Palo Alto Health Care System (VAPAHCS) Institutional Animal Care and Use Committee (IACUC).
Mouse immunohistochemistry, qRT-PCR, and immunoblotting.
Paraformaldehyde-fixed and paraffin-embedded small intestine sections (5 μm) were deparaffinized and rehydrated. Antigen retrieval was performed on deparaffinized sections followed by 10 min of 3% H2O2 incubation. Polyclonal goat anti-RV IgG (1:500) (Meridian Life Science) and monoclonal rabbit anti-pY701-STAT1 (1:500) (Cell Signaling Technology) were used for detection. After three washes with PBS, slides were incubated with biotinylated horse anti-goat or anti-rabbit secondary antibody (Vector Laboratories, Inc., Burlingame, CA) for 30 min. Signal was developed using Vector streptavidin-horseradish peroxidase together with a diaminobenzidine (DAB) detection system and counterstained with hematoxylin. For analysis of transcripts, mice were sacrificed, and sections of the small intestine were collected on ice in TRIzol (Life Technologies). Total RNA was extracted according to the manufacturer's instructions and subjected to DNase digestion before use in qRT-PCR. Synthesis of cDNA and subsequent microfluidics PCR on a Fluidigm platform were done as described earlier (4). Serial 10-fold dilutions of mouse reference RNA (Agilent) were run in duplicate for each PCR run. Relative gene expression in infected and uninfected mouse intestinal samples was derived using the 2ΔCT (where CT is threshold cycle) method, with reference RNA serving as a calibrator and hypoxanthine phosphoribosyltransferase (HPRT) serving as a housekeeping control. For immunoblot analysis of mouse tissues, sections of small intestines were collected on ice in cold lysis buffer containing protease and phosphatase inhibitors. Tissues were homogenized using a handheld micropestle (Thermo) and incubated on ice for 20 min. Lysates were cleared by centrifugation at 10,000 × g for 20 min, and supernatants were mixed with an equal volume of Laemmli buffer, boiled for 5 min, and subjected to immunoblot analysis.
LPS administration to mice.
Three- to 5-day-old 129sv mice were orally inoculated by gavage with 104 DD50 of murine RV-EW. At 3 dpi, mice were intraperitoneally injected with PBS or purified LPS (10 mg/kg body weight; Sigma) and monitored for lethality for 6 days. Results were accumulated from three separate experiments.
ACKNOWLEDGMENTS
We acknowledge the technical assistance of Xibing Che in all aspects of this study. We are grateful to Nandini Sen and Sergei Kotenko for useful suggestions during the study, Lusijah Rott for help with flow cytometry studies, Linda Jacob for administrative assistance, and members of the Greenberg laboratory for useful discussions.
This work was supported by grants 1RO1 AI021362, R56 AI125249, and 1RO1 AI1125249 to H.B.G. from the National Institutes of Health and 1IO 1BX000158-01A1 to H.B.G. from Veterans Affairs.
REFERENCES
- 1.Arnold MM, Sen A, Greenberg HB, Patton JT. 2013. The battle between rotavirus and its host for control of the interferon signaling pathway. PLoS Pathog 9:e1003064. doi: 10.1371/journal.ppat.1003064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Broome RL, Vo PT, Ward RL, Clark HF, Greenberg HB. 1993. Murine rotavirus genes encoding outer capsid proteins VP4 and VP7 are not major determinants of host range restriction and virulence. J Virol 67:2448–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feng N, Kim B, Fenaux M, Nguyen H, Vo P, Omary MB, Greenberg HB. 2008. Role of interferon in homologous and heterologous rotavirus infection in the intestines and extraintestinal organs of suckling mice. J Virol 82:7578–7590. doi: 10.1128/JVI.00391-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sen A, Rothenberg ME, Mukherjee G, Feng N, Kalisky T, Nair N, Johnstone IM, Clarke MF, Greenberg HB. 2012. Innate immune response to homologous rotavirus infection in the small intestinal villous epithelium at single-cell resolution. Proc Natl Acad Sci U S A 109:20667–20672. doi: 10.1073/pnas.1212188109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sen A, Feng N, Ettayebi K, Hardy ME, Greenberg HB. 2009. IRF3 inhibition by rotavirus NSP1 is host cell and virus strain dependent but independent of NSP1 proteasomal degradation. J Virol 83:10322–10335. doi: 10.1128/JVI.01186-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sen A, Pruijssers AJ, Dermody TS, Garcia-Sastre A, Greenberg HB. 2011. The early interferon response to rotavirus is regulated by PKR and depends on MAVS/IPS-1, RIG-I, MDA-5, and IRF3. J Virol 85:3717–3732. doi: 10.1128/JVI.02634-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sen A, Rott L, Phan N, Mukherjee G, Greenberg HB. 2014. Rotavirus NSP1 protein inhibits interferon-mediated STAT1 activation. J Virol 88:41–53. doi: 10.1128/JVI.01501-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feng N, Yasukawa LL, Sen A, Greenberg HB. 2013. Permissive replication of homologous murine rotavirus in the mouse intestine is primarily regulated by VP4 and NSP1. J Virol 87:8307–8316. doi: 10.1128/JVI.00619-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin JD, Feng N, Sen A, Balan M, Tseng HC, McElrath C, Smirnov SV, Peng J, Yasukawa LL, Durbin RK, Durbin JE, Greenberg HB, Kotenko SV. 2016. Distinct roles of type I and type III interferons in intestinal immunity to homologous and heterologous rotavirus infections. PLoS Pathog 12:e1005600. doi: 10.1371/journal.ppat.1005600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holloway G, Dang VT, Jans DA, Coulson BS. 2014. Rotavirus inhibits IFN-induced STAT nuclear translocation by a mechanism that acts after STAT binding to importin-α. J Gen Virol 95:1723–1733. doi: 10.1099/vir.0.064063-0. [DOI] [PubMed] [Google Scholar]
- 11.Holloway G, Truong TT, Coulson BS. 2009. Rotavirus antagonizes cellular antiviral responses by inhibiting the nuclear accumulation of STAT1, STAT2, and NF-κB. J Virol 83:4942–4951. doi: 10.1128/JVI.01450-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dejager L, Vandevyver S, Ballegeer M, Van Wonterghem E, An LL, Riggs J, Kolbeck R, Libert C. 2014. Pharmacological inhibition of type I interferon signaling protects mice against lethal sepsis. J Infect Dis 209:960–970. doi: 10.1093/infdis/jit600. [DOI] [PubMed] [Google Scholar]
- 13.Karaghiosoff M, Steinborn R, Kovarik P, Kriegshauser G, Baccarini M, Donabauer B, Reichart U, Kolbe T, Bogdan C, Leanderson T, Levy D, Decker T, Muller M. 2003. Central role for type I interferons and Tyk2 in lipopolysaccharide-induced endotoxin shock. Nat Immunol 4:471–477. doi: 10.1038/ni910. [DOI] [PubMed] [Google Scholar]
- 14.Mahieu T, Libert C. 2007. Should we inhibit type I interferons in sepsis? Infect Immun 75:22–29. doi: 10.1128/IAI.00829-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mahieu T, Park JM, Revets H, Pasche B, Lengeling A, Staelens J, Wullaert A, Vanlaere I, Hochepied T, van Roy F, Karin M, Libert C. 2006. The wild-derived inbred mouse strain SPRET/Ei is resistant to LPS and defective in IFN-beta production. Proc Natl Acad Sci U S A 103:2292–2297. doi: 10.1073/pnas.0510874103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arnold MM, Barro M, Patton JT. 2013. Rotavirus NSP1 mediates degradation of interferon regulatory factors through targeting of the dimerization domain. J Virol 87:9813–9821. doi: 10.1128/JVI.01146-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arnold MM, Patton JT. 2009. Rotavirus antagonism of the innate immune response. Viruses 1:1035–1056. doi: 10.3390/v1031035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arnold MM, Patton JT. 2011. Diversity of interferon antagonist activities mediated by NSP1 proteins of different rotavirus strains. J Virol 85:1970–1979. doi: 10.1128/JVI.01801-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barro M, Patton JT. 2005. Rotavirus nonstructural protein 1 subverts innate immune response by inducing degradation of IFN regulatory factor 3. Proc Natl Acad Sci U S A 102:4114–4119. doi: 10.1073/pnas.0408376102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barro M, Patton JT. 2007. Rotavirus NSP1 inhibits expression of type I interferon by antagonizing the function of interferon regulatory factors IRF3, IRF5, and IRF7. J Virol 81:4473–4481. doi: 10.1128/JVI.02498-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Graff JW, Ettayebi K, Hardy ME. 2009. Rotavirus NSP1 inhibits NFκB activation by inducing proteasome-dependent degradation of beta-TrCP: a novel mechanism of IFN antagonism. PLoS Pathog 5:e1000280. doi: 10.1371/journal.ppat.1000280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Graff JW, Ewen J, Ettayebi K, Hardy ME. 2007. Zinc-binding domain of rotavirus NSP1 is required for proteasome-dependent degradation of IRF3 and autoregulatory NSP1 stability. J Gen Virol 88:613–620. doi: 10.1099/vir.0.82255-0. [DOI] [PubMed] [Google Scholar]
- 23.Graff JW, Mitzel DN, Weisend CM, Flenniken ML, Hardy ME. 2002. Interferon regulatory factor 3 is a cellular partner of rotavirus NSP1. J Virol 76:9545–9550. doi: 10.1128/JVI.76.18.9545-9550.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhattacharya S, HuangFu WC, Liu J, Veeranki S, Baker DP, Koumenis C, Diehl JA, Fuchs SY. 2010. Inducible priming phosphorylation promotes ligand-independent degradation of the IFNAR1 chain of type I interferon receptor. J Biol Chem 285:2318–2325. doi: 10.1074/jbc.M109.071498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuchs SY. 2013. Hope and fear for interferon: the receptor-centric outlook on the future of interferon therapy. J Interferon Cytokine Res 33:211–225. doi: 10.1089/jir.2012.0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu J, HuangFu WC, Kumar KG, Qian J, Casey JP, Hamanaka RB, Grigoriadou C, Aldabe R, Diehl JA, Fuchs SY. 2009. Virus-induced unfolded protein response attenuates antiviral defenses via phosphorylation-dependent degradation of the type I interferon receptor. Cell Host Microbe 5:72–83. doi: 10.1016/j.chom.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu J, Plotnikov A, Banerjee A, Suresh Kumar KG, Ragimbeau J, Marijanovic Z, Baker DP, Pellegrini S, Fuchs SY. 2008. Ligand-independent pathway that controls stability of interferon alpha receptor. Biochem Biophys Res Commun 367:388–393. doi: 10.1016/j.bbrc.2007.12.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broquet AH, Hirata Y, McAllister CS, Kagnoff MF. 2011. RIG-I/MDA5/MAVS are required to signal a protective IFN response in rotavirus-infected intestinal epithelium. J Immunol 186:1618–1626. doi: 10.4049/jimmunol.1002862. [DOI] [PubMed] [Google Scholar]
- 29.Pott J, Mahlakoiv T, Mordstein M, Duerr CU, Michiels T, Stockinger S, Staeheli P, Hornef MW. 2011. IFN-λ determines the intestinal epithelial antiviral host defense. Proc Natl Acad Sci U S A 108:7944–7949. doi: 10.1073/pnas.1100552108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sarkar SA, Wong R, Hackl SI, Moua O, Gill RG, Wiseman A, Davidson HW, Hutton JC. 2007. Induction of indoleamine 2,3-dioxygenase by interferon-gamma in human islets. Diabetes 56:72–79. doi: 10.2337/db06-0617. [DOI] [PubMed] [Google Scholar]
- 31.Robinson CM, Hale PT, Carlin JM. 2006. NF-kappa B activation contributes to indoleamine dioxygenase transcriptional synergy induced by IFN-gamma and tumor necrosis factor-alpha. Cytokine 35:53–61. doi: 10.1016/j.cyto.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 32.Thomas KE, Galligan CL, Newman RD, Fish EN, Vogel SN. 2006. Contribution of interferon-beta to the murine macrophage response to the Toll-like receptor 4 agonist, lipopolysaccharide. J Biol Chem 281:31119–31130. doi: 10.1074/jbc.M604958200. [DOI] [PubMed] [Google Scholar]
- 33.Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, Williams BR, Major J, Hamilton TA, Fenton MJ, Vogel SN. 2002. TLR4, but not TLR2, mediates IFN-β-induced STAT1α/β-dependent gene expression in macrophages. Nat Immunol 3:392–398. doi: 10.1038/ni774. [DOI] [PubMed] [Google Scholar]
- 34.Bhattacharya S, Katlinski KV, Reichert M, Takano S, Brice A, Zhao B, Yu Q, Zheng H, Carbone CJ, Katlinskaya YV, Leu NA, McCorkell KA, Srinivasan S, Girondo M, Rui H, May MJ, Avadhani NG, Rustgi AK, Fuchs SY. 2014. Triggering ubiquitination of IFNAR1 protects tissues from inflammatory injury. EMBO Mol Med 6:384–397. doi: 10.1002/emmm.201303236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bosmann M, Strobl B, Kichler N, Rigler D, Grailer JJ, Pache F, Murray PJ, Muller M, Ward PA. 2014. Tyrosine kinase 2 promotes sepsis-associated lethality by facilitating production of interleukin-27. J Leukoc Biol 96:123–131. doi: 10.1189/jlb.3A1013-541R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herzig D, Fang G, Toliver-Kinsky TE, Guo Y, Bohannon J, Sherwood ER. 2012. STAT1-deficient mice are resistant to cecal ligation and puncture-induced septic shock. Shock 38:395–402. doi: 10.1097/SHK.0b013e318265a2ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kamezaki K, Shimoda K, Numata A, Matsuda T, Nakayama K, Harada M. 2004. The role of Tyk2, Stat1 and Stat4 in LPS-induced endotoxin signals. Int Immunol 16:1173–1179. doi: 10.1093/intimm/dxh118. [DOI] [PubMed] [Google Scholar]
- 38.Jung ID, Lee MG, Chang JH, Lee JS, Jeong YI, Lee CM, Park WS, Han J, Seo SK, Lee SY, Park YM. 2009. Blockade of indoleamine 2,3-dioxygenase protects mice against lipopolysaccharide-induced endotoxin shock. J Immunol 182:3146–3154. doi: 10.4049/jimmunol.0803104. [DOI] [PubMed] [Google Scholar]
- 39.Salazar F, Awuah D, Negm OH, Shakib F, Ghaemmaghami AM. 2017. The role of indoleamine 2,3-dioxygenase-aryl hydrocarbon receptor pathway in the TLR4-induced tolerogenic phenotype in human DCs. Sci Rep 7:43337. doi: 10.1038/srep43337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoshi M, Osawa Y, Ito H, Ohtaki H, Ando T, Takamatsu M, Hara A, Saito K, Seishima M. 2014. Blockade of indoleamine 2,3-dioxygenase reduces mortality from peritonitis and sepsis in mice by regulating functions of CD11b+ peritoneal cells. Infect Immun 82:4487–4495. doi: 10.1128/IAI.02113-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rackov G, Shokri R, De Mon MA, Martinez AC, Balomenos D. 2017. The role of IFN-beta during the course of sepsis progression and its therapeutic potential. Front Immunol 8:493. doi: 10.3389/fimmu.2017.00493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. 2015. Type I interferons in infectious disease. Nat Rev Immunol 15:87–103. doi: 10.1038/nri3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bhattacharya S, HuangFu WC, Dong G, Qian J, Baker DP, Karar J, Koumenis C, Diehl JA, Fuchs SY. 2013. Anti-tumorigenic effects of Type 1 interferon are subdued by integrated stress responses. Oncogene 32:4214–4221. doi: 10.1038/onc.2012.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rojas M, Arias CF, Lopez S. 2010. Protein kinase R is responsible for the phosphorylation of eIF2α in rotavirus infection. J Virol 84:10457–10466. doi: 10.1128/JVI.00625-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holloway G, Coulson BS. 2006. Rotavirus activates JNK and p38 signaling pathways in intestinal cells, leading to AP-1-driven transcriptional responses and enhanced virus replication. J Virol 80:10624–10633. doi: 10.1128/JVI.00390-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hernandez PP, Mahlakoiv T, Yang I, Schwierzeck V, Nguyen N, Guendel F, Gronke K, Ryffel B, Holscher C, Dumoutier L, Renauld JC, Suerbaum S, Staeheli P, Diefenbach A. 2015. Interferon-lambda and interleukin 22 act synergistically for the induction of interferon-stimulated genes and control of rotavirus infection. Nat Immunol 16:698–707. doi: 10.1038/ni.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saxena K, Simon LM, Zeng XL, Blutt SE, Crawford SE, Sastri NP, Karandikar UC, Ajami NJ, Zachos NC, Kovbasnjuk O, Donowitz M, Conner ME, Shaw CA, Estes MK. 2017. A paradox of transcriptional and functional innate interferon responses of human intestinal enteroids to enteric virus infection. Proc Natl Acad Sci U S A 114:E570–E579. doi: 10.1073/pnas.1615422114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mbongue JC, Nicholas DA, Torrez TW, Kim NS, Firek AF, Langridge WH. 2015. The role of indoleamine 2, 3-dioxygenase in immune suppression and autoimmunity. Vaccines (Basel) 3:703–729. doi: 10.3390/vaccines3030703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cardenas I, Mor G, Aldo P, Lang SM, Stabach P, Sharp A, Romero R, Mazaki-Tovi S, Gervasi M, Means RE. 2011. Placental viral infection sensitizes to endotoxin-induced pre-term labor: a double hit hypothesis. Am J Reprod Immunol 65:110–117. doi: 10.1111/j.1600-0897.2010.00908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen YC, Wang SY. 2002. Activation of terminally differentiated human monocytes/macrophages by dengue virus: productive infection, hierarchical production of innate cytokines and chemokines, and the synergistic effect of lipopolysaccharide. J Virol 76:9877–9887. doi: 10.1128/JVI.76.19.9877-9887.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Doughty L, Nguyen K, Durbin J, Biron C. 2001. A role for IFN-alpha beta in virus infection-induced sensitization to endotoxin. J Immunol 166:2658–2664. doi: 10.4049/jimmunol.166.4.2658. [DOI] [PubMed] [Google Scholar]
- 52.Lee LN, Dias P, Han D, Yoon S, Shea A, Zakharov V, Parham D, Sarawar SR. 2010. A mouse model of lethal synergism between influenza virus and Haemophilus influenzae. Am J Pathol 176:800–811. doi: 10.2353/ajpath.2010.090596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nansen A, Randrup Thomsen A. 2001. Viral infection causes rapid sensitization to lipopolysaccharide: central role of IFN-αβ. J Immunol 166:982–988. doi: 10.4049/jimmunol.166.2.982. [DOI] [PubMed] [Google Scholar]
- 54.Nguyen KB, Biron CA. 1999. Synergism for cytokine-mediated disease during concurrent endotoxin and viral challenges: roles for NK and T cell IFN-gamma production. J Immunol 162:5238–5246. [PubMed] [Google Scholar]
- 55.Ohtaki H, Ito H, Hoshi M, Osawa Y, Takamatsu M, Hara A, Ishikawa T, Moriwaki H, Saito K, Seishima M. 2012. High susceptibility to lipopolysaccharide-induced lethal shock in encephalomyocarditis virus-infected mice. Sci Rep 2:367. doi: 10.1038/srep00367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Gucht S, van Reeth K, Pensaert M. 2003. Interaction between porcine reproductive-respiratory syndrome virus and bacterial endotoxin in the lungs of pigs: potentiation of cytokine production and respiratory disease. J Clin Microbiol 41:960–966. doi: 10.1128/JCM.41.3.960-966.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Blutt SE, Fenaux M, Warfield KL, Greenberg HB, Conner ME. 2006. Active viremia in rotavirus-infected mice. J Virol 80:6702–6705. doi: 10.1128/JVI.00329-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blutt SE, Kirkwood CD, Parreno V, Warfield KL, Ciarlet M, Estes MK, Bok K, Bishop RF, Conner ME. 2003. Rotavirus antigenaemia and viraemia: a common event? Lancet 362:1445–1449. doi: 10.1016/S0140-6736(03)14687-9. [DOI] [PubMed] [Google Scholar]
- 59.Blutt SE, Matson DO, Crawford SE, Staat MA, Azimi P, Bennett BL, Piedra PA, Conner ME. 2007. Rotavirus antigenemia in children is associated with viremia. PLoS Med 4:e121. doi: 10.1371/journal.pmed.0040121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Candy DC. 2007. Rotavirus infection: a systemic illness? PLoS Med 4:e117. doi: 10.1371/journal.pmed.0040117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moon S, Wang Y, Dennehy P, Simonsen KA, Zhang J, Jiang B. 2012. Antigenemia, RNAemia, and innate immunity in children with acute rotavirus diarrhea. FEMS Immunol Med Microbiol 64:382–391. doi: 10.1111/j.1574-695X.2011.00923.x. [DOI] [PubMed] [Google Scholar]
- 62.Fenaux M, Cuadras MA, Feng N, Jaimes M, Greenberg HB. 2006. Extraintestinal spread and replication of a homologous EC rotavirus strain and a heterologous rhesus rotavirus in BALB/c mice. J Virol 80:5219–5232. doi: 10.1128/JVI.02664-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Uhnoo I, Riepenhoff-Talty M, Dharakul T, Chegas P, Fisher JE, Greenberg HB, Ogra PL. 1990. Extramucosal spread and development of hepatitis in immunodeficient and normal mice infected with rhesus rotavirus. J Virol 64:361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]